Cancer Stem Cell-Inducing Media Activates Senescence Reprogramming in Fibroblasts


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
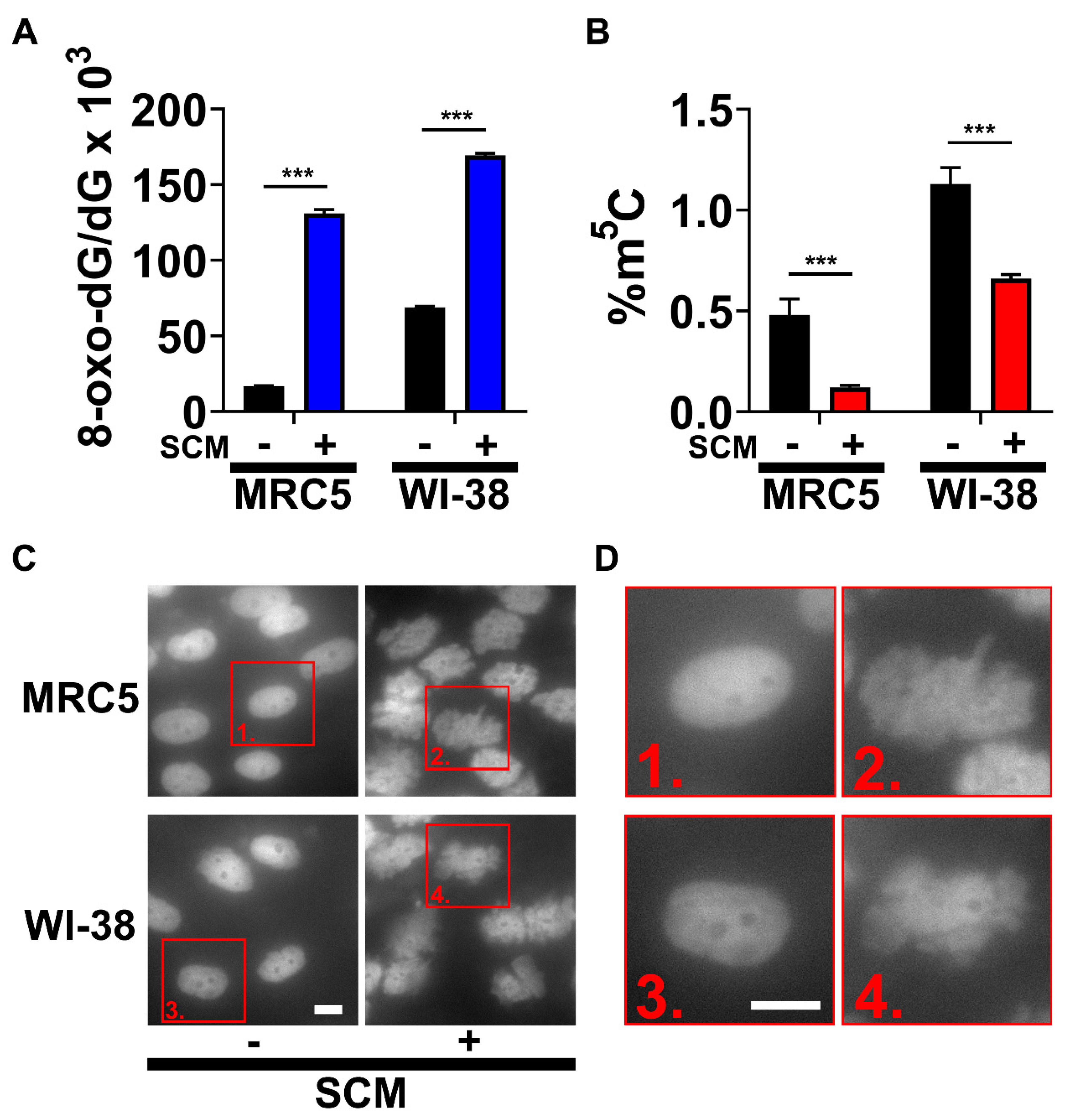
2.1. SCM Triggers Spontaneous Oxidation of DNA and Nuclear Blebbing
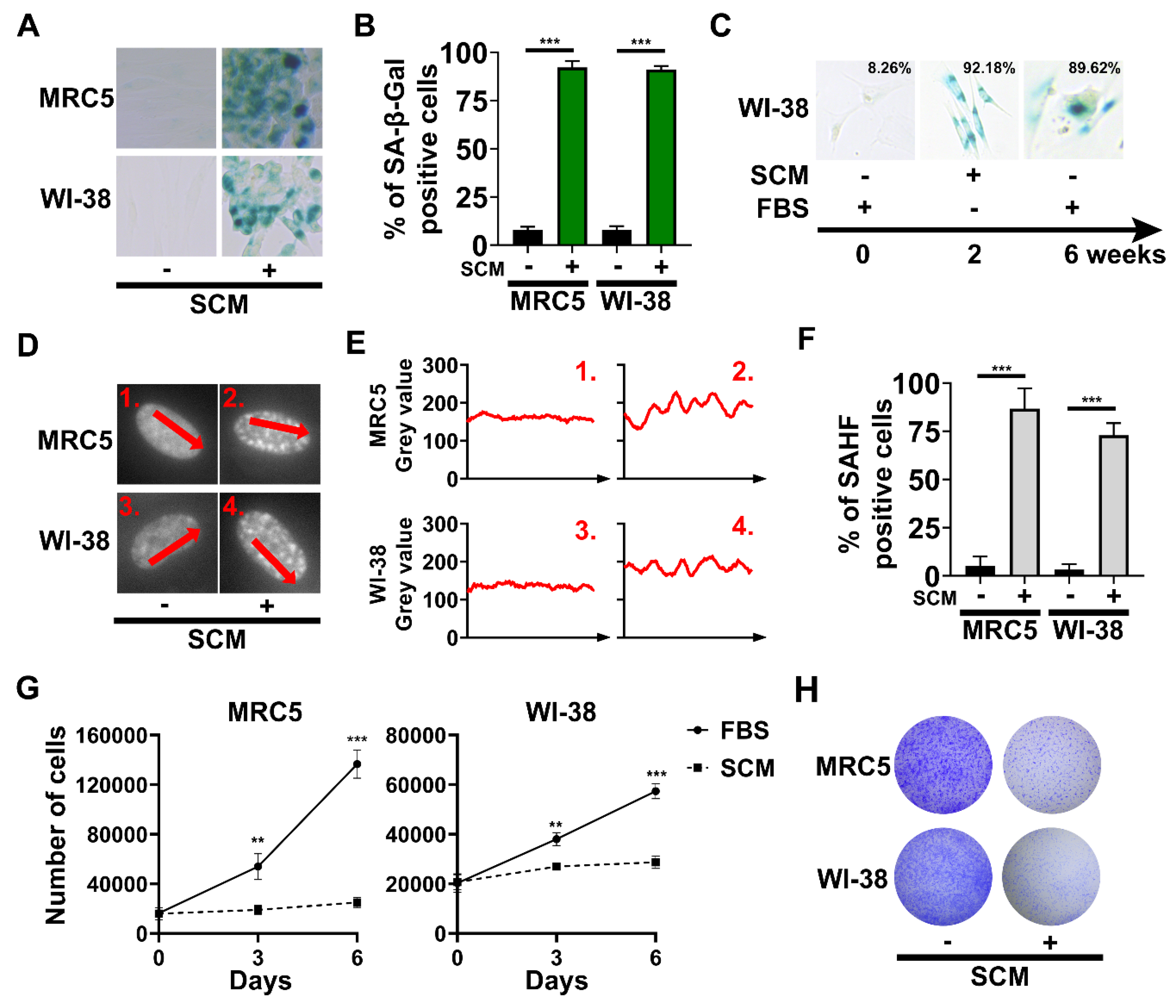
2.2. The Emergence of Senescence-Associated Biomarkers in SCM-Treated Fibroblasts
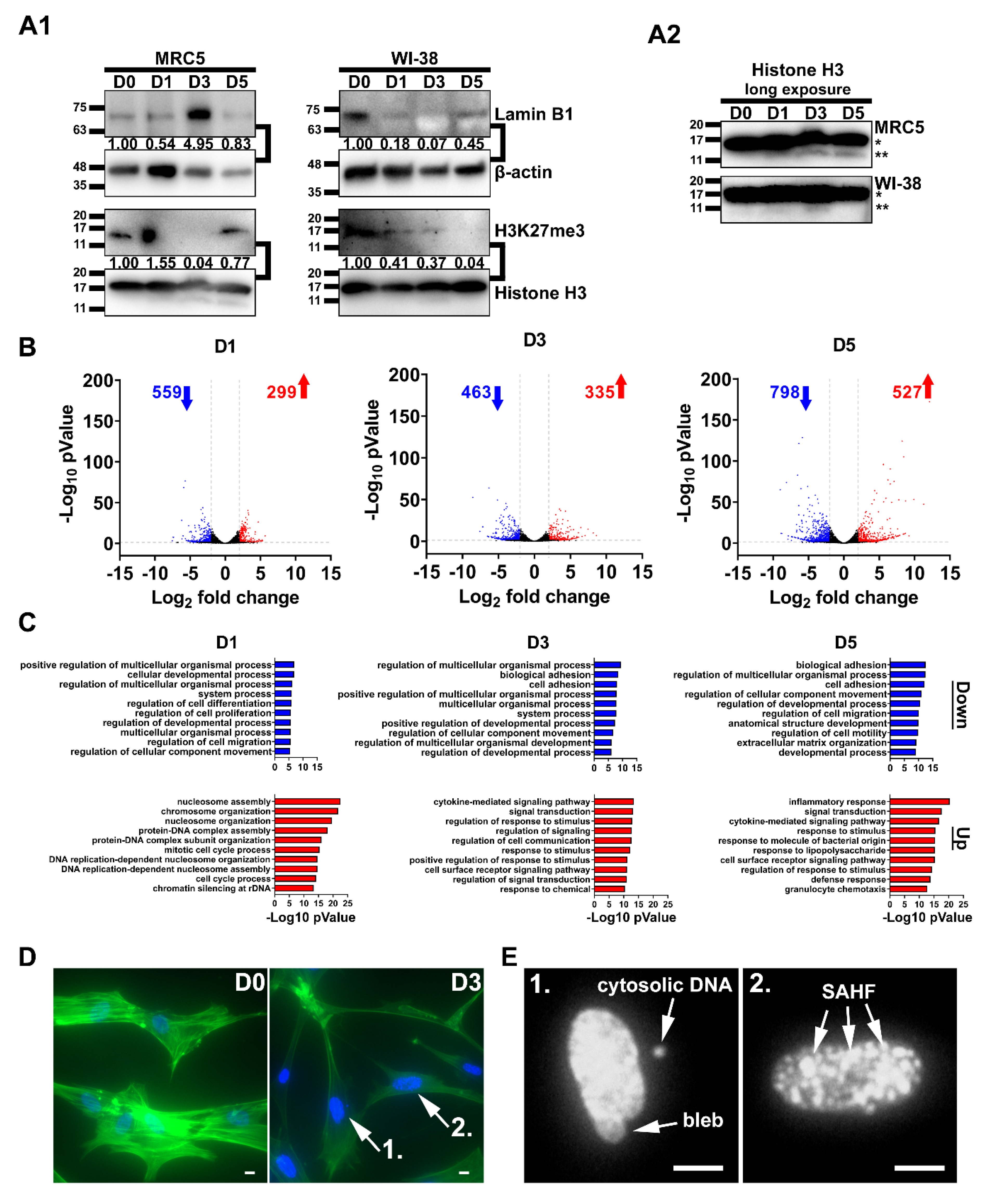
2.3. Chromatin Remodeling and Robust Changes in Gene Expression during SCM-Induced Senescence
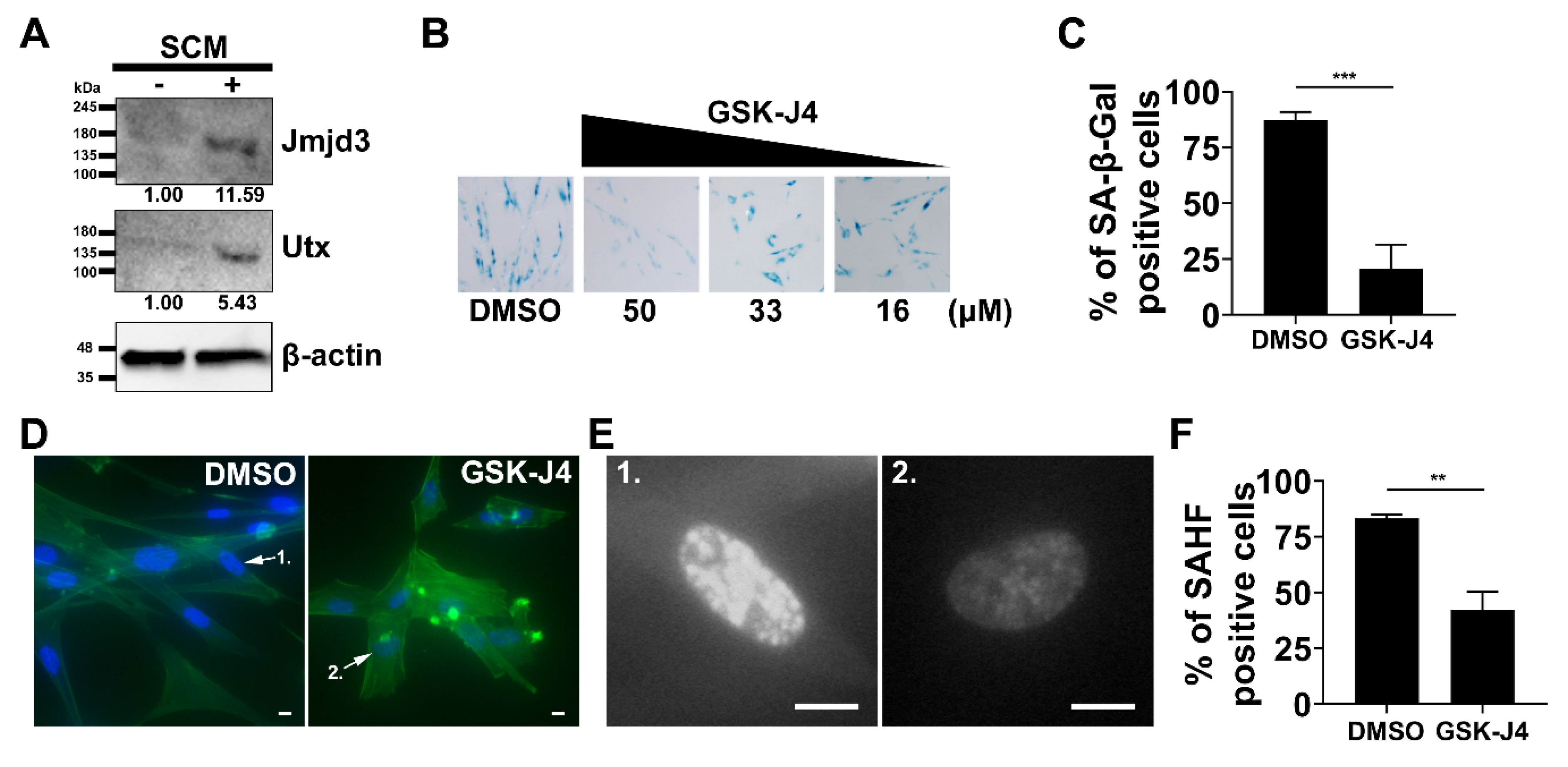
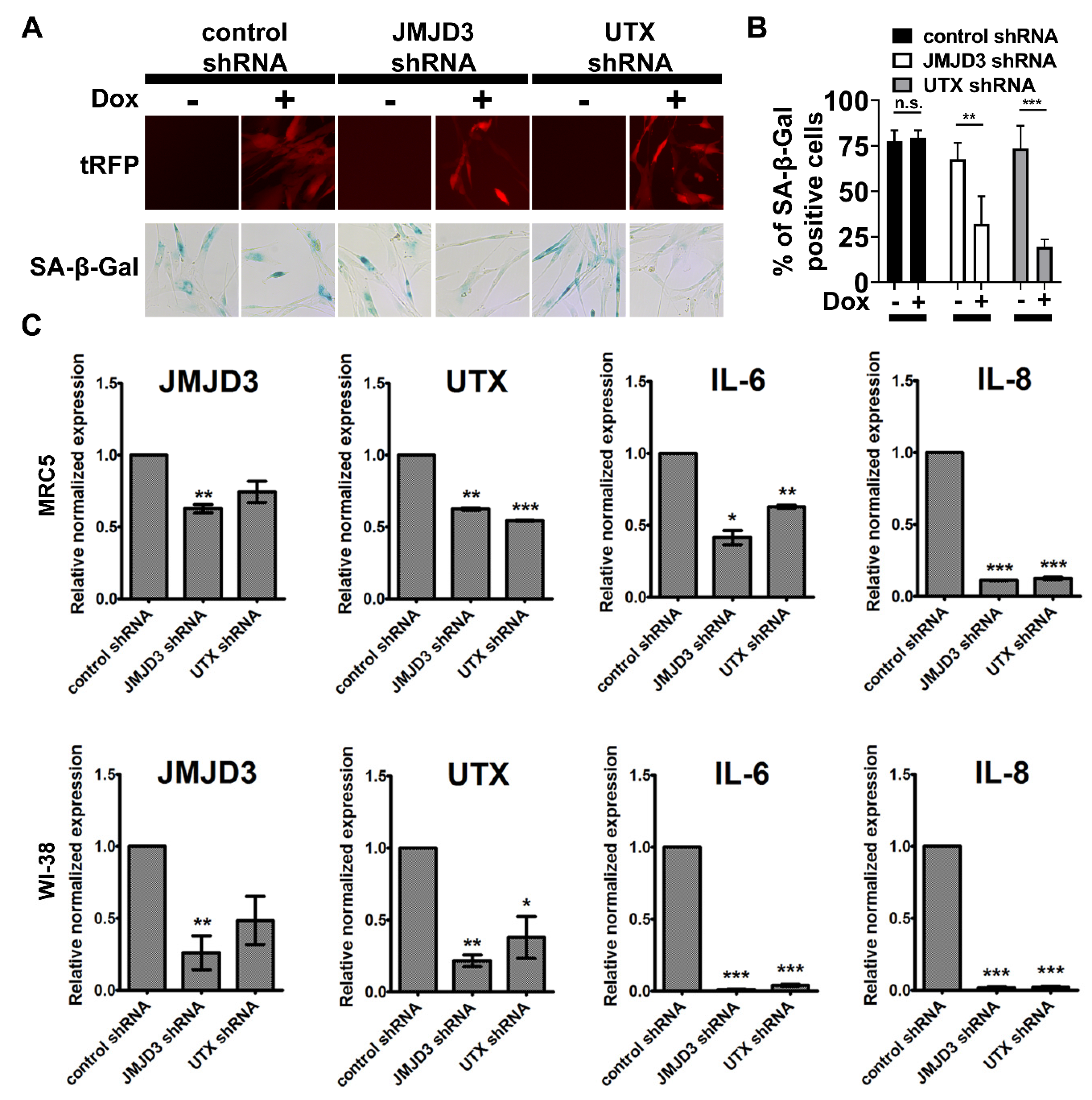
2.4. Inhibition of H3K27me3 Demethylases Prevents SCM-induced Senescence in Fibroblasts
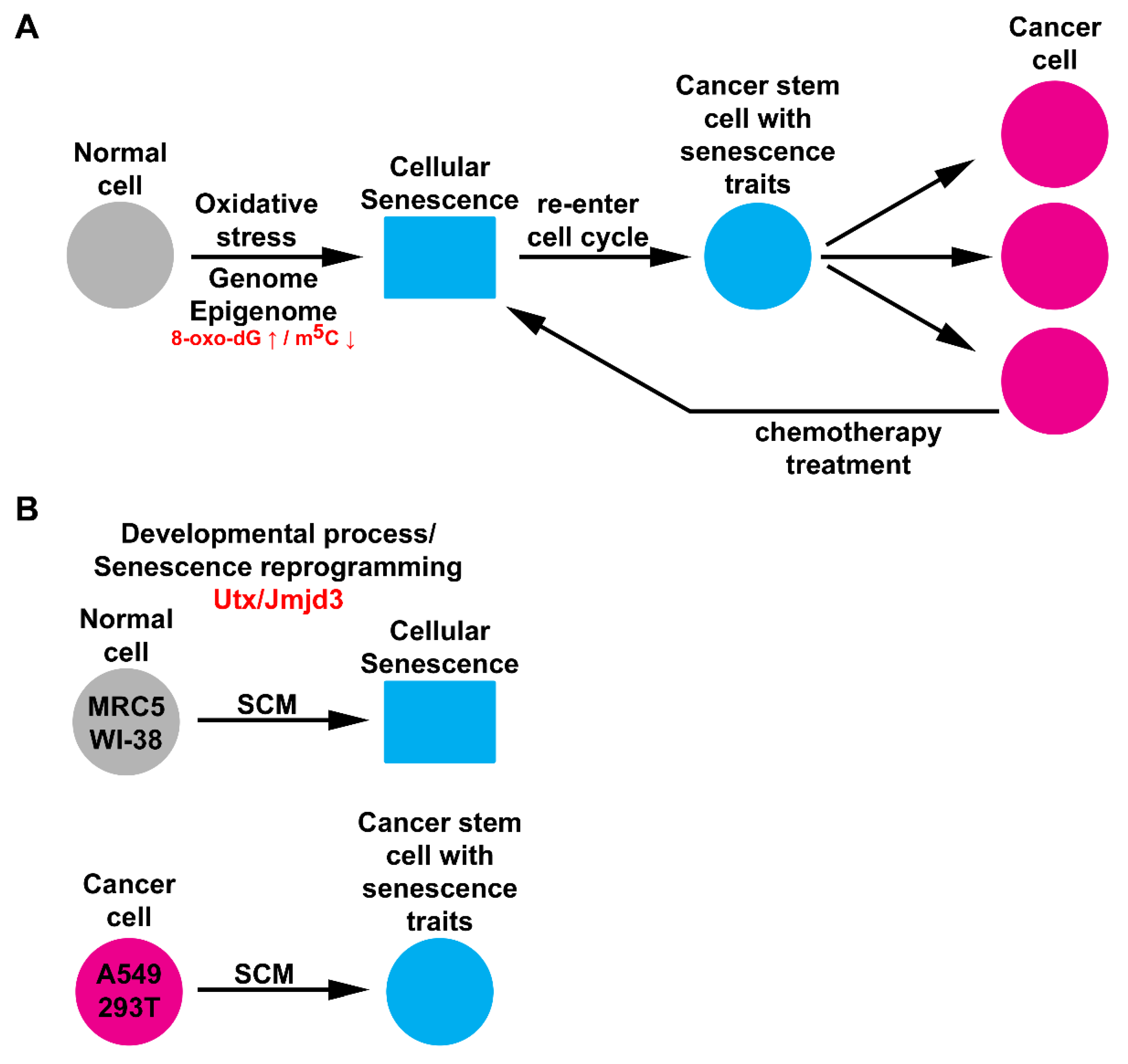
2.5. Chromatin Machinery for Senescence Reprogramming is Activated in Cancer Stem Cell Formation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Analysis of 8-oxo-dG and m5C
4.3. SA-β-gal and Crystal Violet Staining
4.4. Western Blotting
4.5. shRNA Knockdown and GSK-J4 Inhibitor Treatment
4.6. RNA Isolation, cDNA Synthesis, and qRT-PCR
- JMJD3: 5’TGCTCCGTCAACATCAACATTGGC and 5’TGCACGAAGCGGTACACAGGAATA;
- UTX: 5’GAACAGCTCCGCGCAAATAG and 5’CGTACCTGTGCAACTCCTGT;
- IL6: 5’CCAGGAGAAGATTCCAAAGATGTA and 5’CGTCGAGGATGTACCGAATTT;
- IL8: 5’CACAACCCTCTGCACCCAGTTT and 5’GAGAGTGATTGAGAGTGGACCAC;
- HPRT: 5’CTTGAGCACACAGAGGGCTACA and 5’CATTATGCTGAGGATTTGGAAAGG;
- 18S: 5’CGGCTACCACATCCAAGGAA and 5’GCTGGAATTACCGCGGCT.
4.7. DNA and Immunofluorescence Staining
4.8. DNA Constructs and Transfection
4.9. RNA-Sequencing and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef]
- Banito, A.; Rashid, S.T.; Acosta, J.C.; Li, S.; Pereira, C.F.; Geti, I.; Pinho, S.; Silva, J.C.; Azuara, V.; Walsh, M.; et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009, 23, 2134–2139. [Google Scholar] [CrossRef]
- Robin, J.D.; Magdinier, F. Physiological and pathological aging affects chromatin dynamics, structure and function at the nuclear edge. Front. Genet. 2016, 7, 153. [Google Scholar] [CrossRef]
- Rodier, F.; Coppé, J.P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Shah, P.P.; Donahue, G.; Otte, G.L.; Capell, B.C.; Nelson, D.M.; Cao, K.; Aggarwala, V.; Cruickshanks, H.A.; Rai, T.S.; McBryan, T.; et al. Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev. 2013, 27, 1787–1799. [Google Scholar] [CrossRef]
- Agger, K.; Cloos, P.A.C.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef]
- Zhao, W.; Li, Q.; Ayers, S.; Gu, Y.; Shi, Z.; Zhu, Q.; Chen, Y.; Wang, H.Y.; Wang, R.F. Jmjd3 inhibits reprogramming by upregulating expression of INK4a/Arf and targeting PHF20 for ubiquitination. Cell 2013, 152, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A.; Gafni, O.; Weinberger, L.; Zviran, A.; Ayyash, M.; Rais, Y.; Krupalnik, V.; Zerbib, M.; Amann-Zalcenstein, D.; Maza, I.; et al. The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature 2012, 488, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Barradas, M.; Anderton, E.; Acosta, J.C.; Li, S.; Banito, A.; Rodriguez-Niedenführ, M.; Maertens, G.; Banck, M.; Zhou, M.M.; Walsh, M.J.; et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009, 23, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Li, J.; Green, C.D.; Yu, X.; Tang, X.; Han, D.; Xian, B.; Wang, D.; Huang, X.; Cao, X.; et al. Histone demethylase UTX-1 regulates C. elegans life span by targeting the insulin/IGF-1 signaling pathway. Cell Metab. 2011, 14, 161–172. [Google Scholar] [CrossRef]
- Wang, J.K.; Tsai, M.C.; Poulin, G.; Adler, A.S.; Chen, S.; Liu, H.; Shi, Y.; Chang, H.Y. The histone demethylase UTX enables RB-dependent cell fate control. Genes Dev. 2010, 24, 327–332. [Google Scholar] [CrossRef]
- Wachs, F.P.; Couillard-Despres, S.; Engelhardt, M.; Wilhelm, D.; Ploetz, S.; Vroemen, M.; Kaesbauer, J.; Uyanik, G.; Klucken, J.; Karl, C.; et al. High efficacy of clonal growth and expansion of adult neural stem cells. Lab. Investig. 2003, 83, 949–962. [Google Scholar] [CrossRef]
- Qiang, L.; Yang, Y.; Ma, Y.J.; Chen, F.H.; Zhang, L.B.; Liu, W.; Qi, Q.; Lu, N.; Tao, L.; Wang, X.T.; et al. Isolation and characterization of cancer stem like cells in human glioblastoma cell lines. Cancer Lett. 2009, 279, 13–21. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef]
- Fraga, C.G.; Shigenaga, M.K.; Park, J.W.; Degan, P.; Ames, B.N. Oxidative damage to DNA during aging: 8-Hydroxy-2’-deoxyguanosine in rat organ DNA and urine. Proc. Natl. Acad. Sci. USA 1990, 87, 4533–4537. [Google Scholar] [CrossRef]
- Breiling, A.; Lyko, F. Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond. Epigenetics and Chromatin 2015, 8, 24. [Google Scholar] [CrossRef]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Singh Rai, T.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.F.; Young, A.R.J.; Wang, Z.; Wu, H.A.; Panda, T.; Kou, Y.; Kapoor, A.; Hasson, D.; Mills, N.R.; Ma’ayan, A.; et al. Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat. Commun. 2014, 5, 5210. [Google Scholar] [CrossRef]
- Shaik, S.; Liu, P.; Wang, Z.; Wei, W. Loss of Cdh1 Triggers Premature Senescence in Part via Activation of Both the RB/E2F1 and the CLASPIN/CHK1/P53 Tumor Suppressor Pathways. In Tumor Dormancy, Quiescence, and Senescence, Volume 2; Springer: Dordrecht, The Netherlands, 2014; pp. 207–217. ISBN 9789400777262. [Google Scholar]
- Sang, L.; Roberts, J.M.; Coller, H.A. Hijacking HES1: How tumors co-opt the anti-differentiation strategies of quiescent cells. Trends Mol. Med. 2010, 16, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Weins, A.; Schwarz, K.; Faul, C.; Barisoni, L.; Linke, W.A.; Mundel, P. Differentiation- and stress-dependent nuclear cytoplasmic redistribution of myopodin, a novel actin-bundling protein. J. Cell Biol. 2001, 155, 393–403. [Google Scholar] [CrossRef]
- Kruidenier, L.; Chung, C.W.; Cheng, Z.; Liddle, J.; Che, K.; Joberty, G.; Bantscheff, M.; Bountra, C.; Bridges, A.; Diallo, H.; et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012, 488, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Halim, N.H.A.; Zakaria, N.; Satar, N.A.; Yahaya, B.H. Isolation and characterization of cancer stem cells of the non-small-cell lung cancer (A549) cell line. Met. Mol. Biol. 2016, 1516, 371–388. [Google Scholar]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Schuettengruber, B.; Bourbon, H.M.; Di Croce, L.; Cavalli, G. Genome Regulation by Polycomb and Trithorax: 70 Years and Counting. Cell 2017, 171, 34–57. [Google Scholar] [CrossRef]
- Zhou, X.; Zhuang, Z.; Wang, W.; He, L.; Wu, H.; Cao, Y.; Pan, F.; Zhao, J.; Hu, Z.; Sekhar, C.; et al. OGG1 is essential in oxidative stress induced DNA demethylation. Cell. Signal. 2016, 28, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy mediates degradation of nuclear lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Shumaker, D.K.; Dechat, T.; Kohlmaier, A.; Adam, S.A.; Bozovsky, M.R.; Erdos, M.R.; Eriksson, M.; Goldman, A.E.; Khuon, S.; Collins, F.S.; et al. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc. Natl. Acad. Sci. USA 2006, 103, 8703–8708. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Laberge, R.M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef] [PubMed]
- Dreesen, O.; Ong, P.F.; Chojnowski, A.; Colman, A. The contrasting roles of lamin B1 in cellular aging and human disease. Nucleus 2013, 4, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Teo, Y.V.; Evans, S.A.; Neretti, N.; Sedivy, J.M. Regulation of Cellular Senescence by Polycomb Chromatin Modifiers through Distinct DNA Damage- and Histone Methylation-Dependent Pathways. Cell Rep. 2018, 22, 3480–3492. [Google Scholar] [CrossRef]
- Chandra, T.; Kirschner, K.; Thuret, J.Y.; Pope, B.D.; Ryba, T.; Newman, S.; Ahmed, K.; Samarajiwa, S.A.; Salama, R.; Carroll, T.; et al. Independence of Repressive Histone Marks and Chromatin Compaction during Senescent Heterochromatic Layer Formation. Mol. Cell 2012, 47, 203–214. [Google Scholar] [CrossRef]
- Shan, Y.; Zhang, Y.; Zhao, Y.; Wang, T.; Zhang, J.; Yao, J.; Ma, N.; Liang, Z.; Huang, W.; Huang, K.; et al. JMJD3 and UTX determine fidelity and lineage specification of human neural progenitor cells. Nat. Commun. 2020, 11, 382. [Google Scholar] [CrossRef]
- Akdemir, K.C.; Jain, A.K.; Allton, K.; Aronow, B.; Xu, X.; Cooney, A.J.; Li, W.; Barton, M.C. Genome-wide profiling reveals stimulus-specific functions of p53 during differentiation and DNA damage of human embryonic stem cells. Nucleic Acids Res. 2014, 42, 205–223. [Google Scholar] [CrossRef]
- Williams, K.; Christensen, J.; Rappsilber, J.; Nielsen, A.L.; Johansen, J.V.; Helin, K. The histone lysine demethylase JMJD3/KDM6B Is recruited to p53 bound promoters and enhancer elements in a p53 dependent manner. PLoS ONE 2014, 9, e96545. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, Y.; Gao, Y.; Geng, P.; Lu, Y.; Liu, X.; Yao, R.; Hou, P.; Liu, D.; Lu, J.; et al. JMJD3 promotes SAHF formation in senescent WI38 cells by triggering an interplay between demethylation and phosphorylation of RB protein. Cell Death Differ. 2015, 22, 1630–1640. [Google Scholar] [CrossRef]
- Perrigue, P.M.; Silva, M.E.; Warden, C.D.; Feng, N.L.; Reid, M.A.; Mota, D.J.; Joseph, L.P.; Tian, Y.I.; Glackin, C.A.; Gutova, M.; et al. The histone demethylase Jumonji coordinates cellular senescence including secretion of neural stem cell-attracting cytokines. Mol. Cancer Res. 2015, 13, 636–650. [Google Scholar] [CrossRef]
- Li, C.; Chai, Y.; Wang, L.; Gao, B.; Chen, H.; Gao, P.; Zhou, F.Q.; Luo, X.; Crane, J.L.; Yu, B.; et al. Programmed cell senescence in skeleton during late puberty. Nat. Commun. 2017, 8, 1312. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Xu, L.; Wu, X.; Zhang, L.; Fei, X.; Cao, Y.; Zhang, F. GSKJ4, an H3K27me3 demethylase inhibitor, effectively suppresses the breast cancer stem cells. Exp. Cell Res. 2017, 359, 405–414. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013, 155, 1119. [Google Scholar] [CrossRef] [PubMed]
- White, R.R.; Vijg, J. Do DNA Double-Strand Breaks Drive Aging? Mol. Cell 2016, 63, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Cruickshanks, H.A.; McBryan, T.; Nelson, D.M.; Vanderkraats, N.D.; Shah, P.P.; Van Tuyn, J.; Singh Rai, T.; Brock, C.; Donahue, G.; Dunican, D.S.; et al. Senescent Cells Harbour Features of the Cancer Epigenome. Nat. Cell Biol. 2013, 15, 1495–1506. [Google Scholar] [CrossRef] [PubMed]
- Ziv, Y.; Bielopolski, D.; Galanty, Y.; Lukas, C.; Taya, Y.; Schultz, D.C.; Lukas, J.; Bekker-Jensen, S.; Bartek, J.; Shiloh, Y. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM-and KAP-1 dependent pathway. Nat. Cell Biol. 2006, 8, 870–876. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Murga, M.; Gutierrez-Martinez, P.; Soria, R.; Fernandez-Capetillo, O. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev. 2008, 22, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Barciszewska, A.-M.; Giel-Pietraszuk, M.; Perrigue, P.M.; Naskręt-Barciszewska, M. Total DNA Methylation Changes Reflect Random Oxidative DNA Damage in Gliomas. Cells 2019, 8, 1065. [Google Scholar] [CrossRef]
- Barciszewska, M.Z.; Barciszewska, A.M.; Rattan, S.I.S. TLC-based detection of methylated cytosine: Application to aging epigenetics. Biogerontology 2007, 8, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perrigue, P.M.; Rakoczy, M.; Pawlicka, K.P.; Belter, A.; Giel-Pietraszuk, M.; Naskręt-Barciszewska, M.; Barciszewski, J.; Figlerowicz, M. Cancer Stem Cell-Inducing Media Activates Senescence Reprogramming in Fibroblasts. Cancers 2020, 12, 1745. https://doi.org/10.3390/cancers12071745
Perrigue PM, Rakoczy M, Pawlicka KP, Belter A, Giel-Pietraszuk M, Naskręt-Barciszewska M, Barciszewski J, Figlerowicz M. Cancer Stem Cell-Inducing Media Activates Senescence Reprogramming in Fibroblasts. Cancers. 2020; 12(7):1745. https://doi.org/10.3390/cancers12071745
Chicago/Turabian StylePerrigue, Patrick M., Magdalena Rakoczy, Kamila P. Pawlicka, Agnieszka Belter, Małgorzata Giel-Pietraszuk, Mirosława Naskręt-Barciszewska, Jan Barciszewski, and Marek Figlerowicz. 2020. "Cancer Stem Cell-Inducing Media Activates Senescence Reprogramming in Fibroblasts" Cancers 12, no. 7: 1745. https://doi.org/10.3390/cancers12071745
APA StylePerrigue, P. M., Rakoczy, M., Pawlicka, K. P., Belter, A., Giel-Pietraszuk, M., Naskręt-Barciszewska, M., Barciszewski, J., & Figlerowicz, M. (2020). Cancer Stem Cell-Inducing Media Activates Senescence Reprogramming in Fibroblasts. Cancers, 12(7), 1745. https://doi.org/10.3390/cancers12071745