Cotargeting CHK1 and PI3K Synergistically Suppresses Tumor Growth of Oral Cavity Squamous Cell Carcinoma in Patient-Derived Xenografts

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
2.1. Establishment and Characterization of the PDX Models
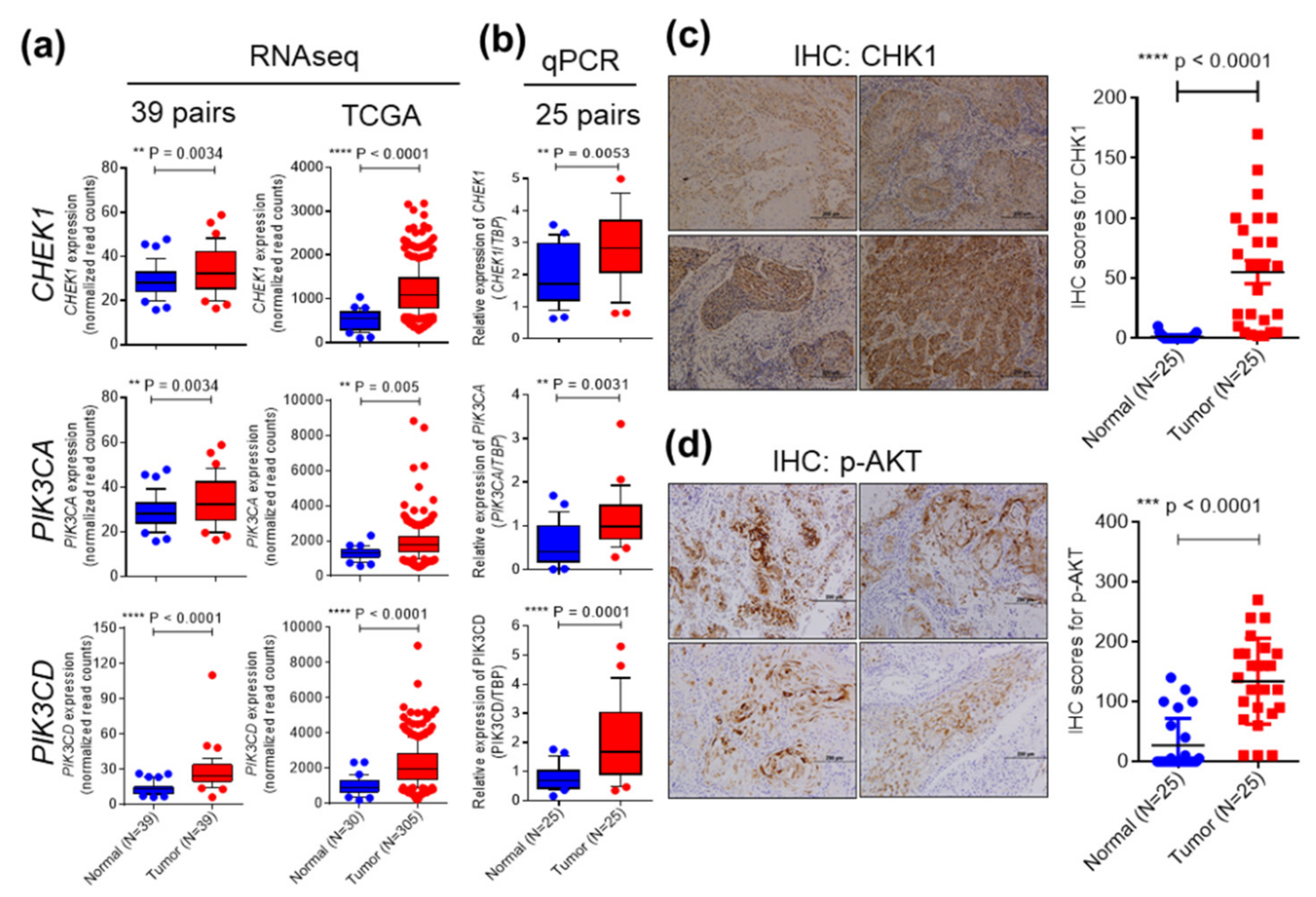
2.2. CHEK1 and PIK3CA/PIK3CD are Overexpressed in OSCC
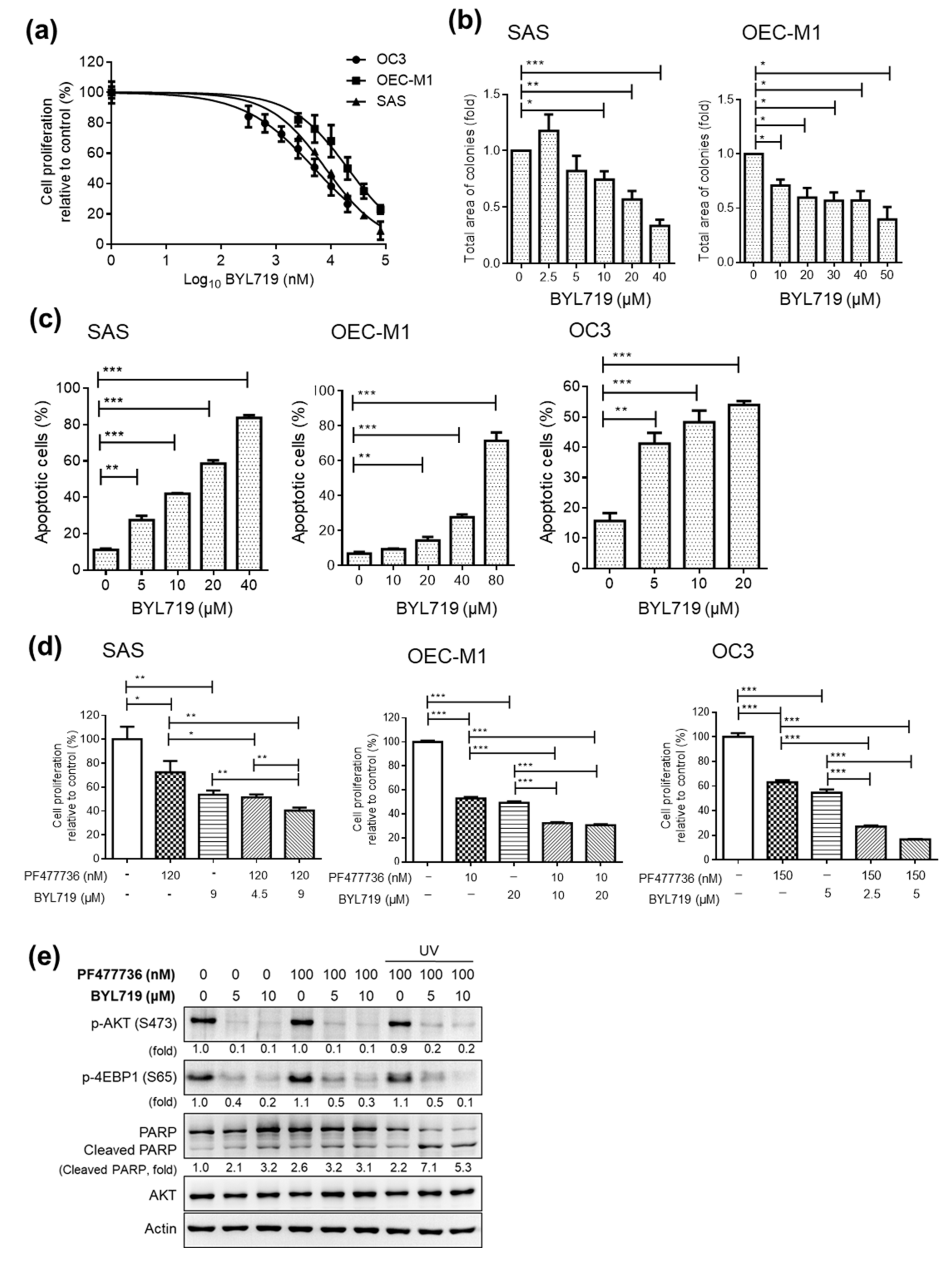
2.3. Targeting CHK1 and PI3K Decreases Cell Proliferation and Enhances Cell Death in OSCC Cell Lines
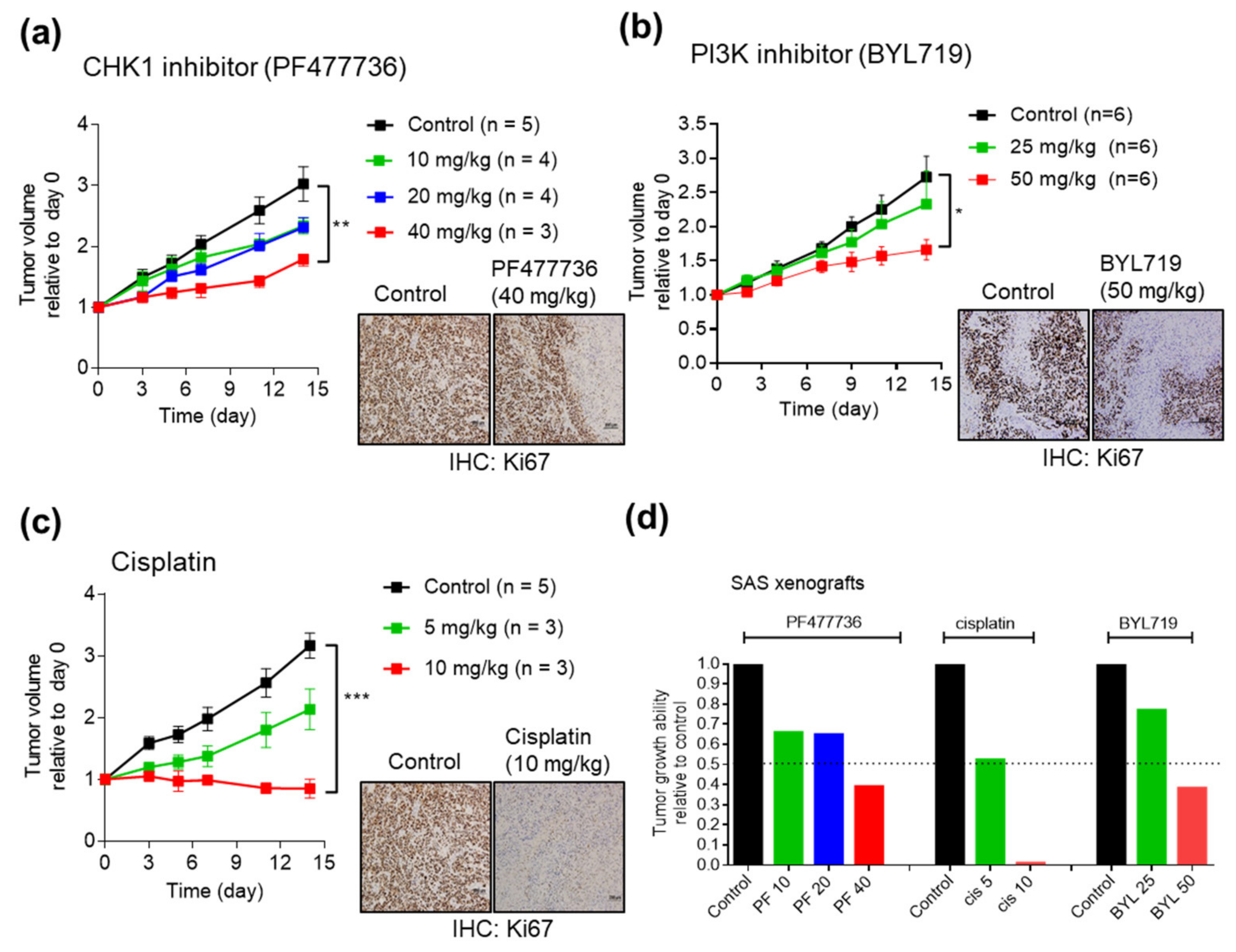
2.4. Antitumor Efficacy of CHK1 Inhibitors and PI3K Inhibitors in SAS Cell Line Xenografts
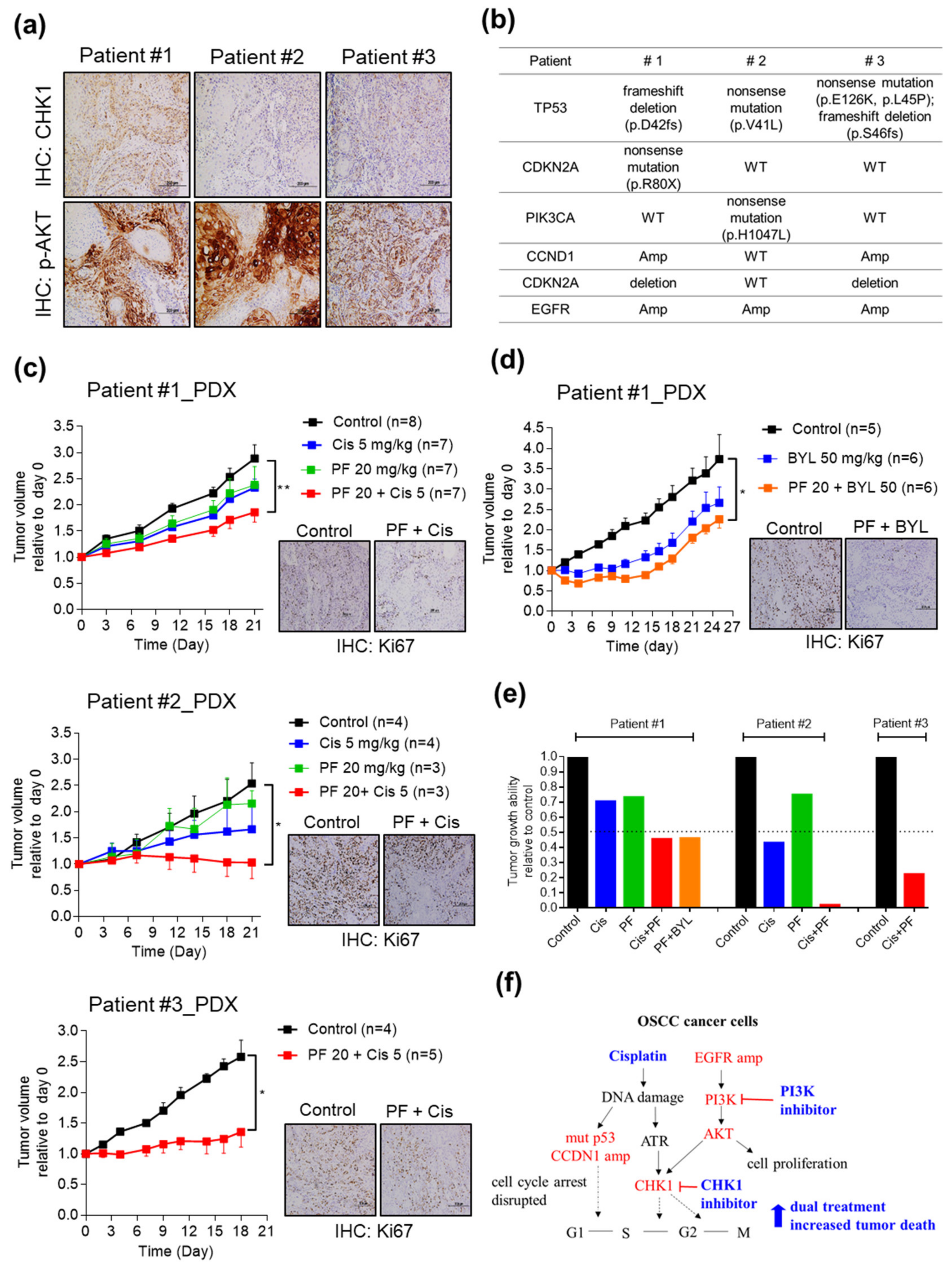
2.5. Combining CHK1 Targeting with Cisplatin or PI3K Inhibitor Treatment as a Novel Therapeutic Strategy in OSCC PDX Models
3. Discussion
4. Materials and Methods
4.1. Patient Characteristics and Clinical Specimens
4.2. Cell Culture
4.3. Drugs, Chemicals, and Antibodies
4.4. RNA-Seq and Transcriptome Analysis
4.5. Whole-Exome Sequencing
4.6. Quantitative Real-Time PCR (qRT-PCR)
4.7. Cell Cycle Analysis and Cell Viability Assay
4.8. Immunoblotting
4.9. IHC Staining
4.10. PDX Establishment and Dug Sensitivity Tests in Vivo
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Scully, C.; Bagan, J. Oral squamous cell carcinoma overview. Oral. Oncol. 2009, 45, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef]
- Krishna Rao, S.V.; Mejia, G.; Roberts-Thomson, K.; Logan, R. Epidemiology of oral cancer in Asia in the past decade—An update (2000–2012). Asian Pac. J. Cancer Prev. 2013, 14, 5567–5577. [Google Scholar] [PubMed]
- Chiang, C.J.; Chen, Y.C.; Chen, C.J.; You, S.L.; Lai, M.S.; Taiwan Cancer Registry Task Force. Cancer trends in Taiwan. Jpn. J. Clin. Oncol. 2010, 40, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.W.; Lee, C.C.; Liu, H.; Wu, C.S.; Pickering, C.R.; Huang, P.J.; Wang, J.; Chang, I.Y.; Yeh, Y.M.; Chen, C.D.; et al. APOBEC3A is an oral cancer prognostic biomarker in Taiwanese carriers of an APOBEC deletion polymorphism. Nat. Commun. 2017, 8, 465. [Google Scholar] [CrossRef]
- Su, S.C.; Lin, C.W.; Liu, Y.F.; Fan, W.L.; Chen, M.K.; Yu, C.P.; Yang, W.E.; Su, C.W.; Chuang, C.Y.; Li, W.H.; et al. Exome Sequencing of Oral Squamous Cell Carcinoma Reveals Molecular Subgroups and Novel Therapeutic Opportunities. Theranostics 2017, 7, 1088–1099. [Google Scholar] [CrossRef]
- Chen, S.J.; Liu, H.; Liao, C.T.; Huang, P.J.; Huang, Y.; Hsu, A.; Tang, P.; Chang, Y.S.; Chen, H.C.; Yen, T.C. Ultra-deep targeted sequencing of advanced oral squamous cell carcinoma identifies a mutation-based prognostic gene signature. Oncotarget 2015, 6, 18066–18080. [Google Scholar] [CrossRef]
- Hashibe, M.; Brennan, P.; Chuang, S.C.; Boccia, S.; Castellsague, X.; Chen, C.; Curado, M.P.; Dal Maso, L.; Daudt, A.W.; Fabianova, E.; et al. Interaction between tobacco and alcohol use and the risk of head and neck cancer: Pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 541–550. [Google Scholar] [CrossRef]
- Sharan, R.N.; Mehrotra, R.; Choudhury, Y.; Asotra, K. Association of betel nut with carcinogenesis: Revisit with a clinical perspective. PLoS ONE 2012, 7, e42759. [Google Scholar] [CrossRef]
- Omura, K. Current status of oral cancer treatment strategies: Surgical treatments for oral squamous cell carcinoma. Int. J. Clin. Oncol. 2014, 19, 423–430. [Google Scholar] [CrossRef] [PubMed]
- De Paz, D.; Kao, H.K.; Huang, Y.; Chang, K.P. Prognostic Stratification of Patients with Advanced Oral Cavity Squamous Cell Carcinoma. Curr. Oncol. Rep. 2017, 19, 65. [Google Scholar] [CrossRef] [PubMed]
- Yanamoto, S.; Umeda, M.; Kioi, M.; Kirita, T.; Yamashita, T.; Hiratsuka, H.; Yokoo, S.; Tanzawa, H.; Uzawa, N.; Shibahara, T.; et al. Multicenter retrospective study of cetuximab plus platinum-based chemotherapy for recurrent or metastatic oral squamous cell carcinoma. Cancer Chemother. Pharmacol. 2018, 81, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Alsahafi, E.; Begg, K.; Amelio, I.; Raulf, N.; Lucarelli, P.; Sauter, T.; Tavassoli, M. Clinical update on head and neck cancer: Molecular biology and ongoing challenges. Cell Death Dis. 2019, 10, 540. [Google Scholar] [CrossRef]
- Karamboulas, C.; Bruce, J.P.; Hope, A.J.; Meens, J.; Huang, S.H.; Erdmann, N.; Hyatt, E.; Pereira, K.; Goldstein, D.P.; Weinreb, I.; et al. Patient-Derived Xenografts for Prognostication and Personalized Treatment for Head and Neck Squamous Cell Carcinoma. Cell Rep. 2018, 25, 1318–1331 e1314. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Nemunaitis, J. Head and neck cancer: Response to p53-based therapeutics. Head Neck 2011, 33, 131–134. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef]
- Santarelli, A.; Mascitti, M.; Lo Russo, L.; Sartini, D.; Troiano, G.; Emanuelli, M.; Lo Muzio, L. Survivin-Based Treatment Strategies for Squamous Cell Carcinoma. Int. J. Mol. Sci. 2018, 19, 971. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-derived tumour xenografts as models for oncology drug development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef]
- Sereti, E.; Karagianellou, T.; Kotsoni, I.; Magouliotis, D.; Kamposioras, K.; Ulukaya, E.; Sakellaridis, N.; Zacharoulis, D.; Dimas, K. Patient Derived Xenografts (PDX) for personalized treatment of pancreatic cancer: Emerging allies in the war on a devastating cancer? J. Proteom. 2018, 188, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hunter, T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189. [Google Scholar] [CrossRef]
- Manic, G.; Obrist, F.; Sistigu, A.; Vitale, I. Trial Watch: Targeting ATM-CHK2 and ATR-CHK1 pathways for anticancer therapy. Mol. Cell Oncol. 2015, 2, e1012976. [Google Scholar] [CrossRef]
- Bartek, J.; Lukas, J. DNA repair: Damage alert. Nature 2003, 421, 486–488. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Albiges, L.; Goubar, A.; Scott, V.; Vicier, C.; Lefebvre, C.; Alsafadi, S.; Commo, F.; Saghatchian, M.; Lazar, V.; Dessen, P.; et al. Chk1 as a new therapeutic target in triple-negative breast cancer. Breast 2014, 23, 250–258. [Google Scholar] [CrossRef]
- Cole, K.A.; Huggins, J.; Laquaglia, M.; Hulderman, C.E.; Russell, M.R.; Bosse, K.; Diskin, S.J.; Attiyeh, E.F.; Sennett, R.; Norris, G.; et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc. Natl. Acad. Sci. USA 2011, 108, 3336–3341. [Google Scholar] [CrossRef]
- Alcaraz-Sanabria, A.; Nieto-Jimenez, C.; Corrales-Sanchez, V.; Serrano-Oviedo, L.; Andres-Pretel, F.; Montero, J.C.; Burgos, M.; Llopis, J.; Galan-Moya, E.M.; Pandiella, A.; et al. Synthetic Lethality Interaction Between Aurora Kinases and CHEK1 Inhibitors in Ovarian Cancer. Mol. Cancer Ther. 2017, 16, 2552–2562. [Google Scholar] [CrossRef]
- Sriuranpong, V.; Mutirangura, A.; Gillespie, J.W.; Patel, V.; Amornphimoltham, P.; Molinolo, A.A.; Kerekhanjanarong, V.; Supanakorn, S.; Supiyaphun, P.; Rangdaeng, S.; et al. Global gene expression profile of nasopharyngeal carcinoma by laser capture microdissection and complementary DNA microarrays. Clin. Cancer Res. 2004, 10, 4944–4958. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Wang, J.; Huo, L.; Xie, Y.; Xie, W.; Xu, G.; Wang, M. Serine/Threonine Kinase CHEK1-Dependent Transcriptional Regulation of RAD54L Promotes Proliferation and Radio Resistance in Glioblastoma. Transl. Oncol. 2018, 11, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Frederick, M.J.; VanMeter, A.J.; Gadhikar, M.A.; Henderson, Y.C.; Yao, H.; Pickering, C.C.; Williams, M.D.; El-Naggar, A.K.; Sandulache, V.; Tarco, E.; et al. Phosphoproteomic analysis of signaling pathways in head and neck squamous cell carcinoma patient samples. Am. J. Pathol. 2011, 178, 548–571. [Google Scholar] [CrossRef]
- Parikh, R.A.; Appleman, L.J.; Bauman, J.E.; Sankunny, M.; Lewis, D.W.; Vlad, A.; Gollin, S.M. Upregulation of the ATR-CHEK1 pathway in oral squamous cell carcinomas. Genes Chromosomes Cancer 2014, 53, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, A.; Takahashi, H.; Patel, A.A.; Osman, A.A.; Myers, J.N. Targeting the DNA Damage Response in OSCC with TP53 Mutations. J. Dent Res. 2018, 97, 635–644. [Google Scholar] [CrossRef]
- Neskey, D.M.; Osman, A.A.; Ow, T.J.; Katsonis, P.; McDonald, T.; Hicks, S.C.; Hsu, T.K.; Pickering, C.R.; Ward, A.; Patel, A.; et al. Evolutionary Action Score of TP53 Identifies High-Risk Mutations Associated with Decreased Survival and Increased Distant Metastases in Head and Neck Cancer. Cancer Res. 2015, 75, 1527–1536. [Google Scholar] [CrossRef]
- Gadhikar, M.A.; Sciuto, M.R.; Alves, M.V.; Pickering, C.R.; Osman, A.A.; Neskey, D.M.; Zhao, M.; Fitzgerald, A.L.; Myers, J.N.; Frederick, M.J. Chk1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53. Mol. Cancer Ther. 2013, 12, 1860–1873. [Google Scholar] [CrossRef]
- Gadhikar, M.A.; Zhang, J.; Shen, L.; Rao, X.; Wang, J.; Zhao, M.; Kalu, N.N.; Johnson, F.M.; Byers, L.A.; Heymach, J.; et al. CDKN2A/p16 Deletion in Head and Neck Cancer Cells Is Associated with CDK2 Activation, Replication Stress, and Vulnerability to CHK1 Inhibition. Cancer Res. 2018, 78, 781–797. [Google Scholar] [CrossRef]
- Garcia-Escudero, R.; Segrelles, C.; Duenas, M.; Pombo, M.; Ballestin, C.; Alonso-Riano, M.; Nenclares, P.; Alvarez-Rodriguez, R.; Sanchez-Aniceto, G.; Ruiz-Alonso, A.; et al. Overexpression of PIK3CA in head and neck squamous cell carcinoma is associated with poor outcome and activation of the YAP pathway. Oral. Oncol. 2018, 79, 55–63. [Google Scholar] [CrossRef]
- Juric, D.; Rodon, J.; Tabernero, J.; Janku, F.; Burris, H.A.; Schellens, J.H.M.; Middleton, M.R.; Berlin, J.; Schuler, M.; Gil-Martin, M.; et al. Phosphatidylinositol 3-Kinase alpha-Selective Inhibition With Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results From the First-in-Human Study. J. Clin. Oncol. 2018, 36, 1291–1299. [Google Scholar] [CrossRef]
- Markham, A. Alpelisib: First Global Approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Blasina, A.; Hallin, J.; Chen, E.; Arango, M.E.; Kraynov, E.; Register, J.; Grant, S.; Ninkovic, S.; Chen, P.; Nichols, T.; et al. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol. Cancer Ther. 2008, 7, 2394–2404. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A. Novel inhibitors of the PI3K family. Expert Opin. Investig. Drugs 2009, 18, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.M.; Tsai, W.S.; Chiang, S.F.; Lai, Y.H.; Ma, C.P.; Wang, J.H.; Lin, J.; Lu, P.S.; Yang, C.Y.; Tan, B.C.; et al. Comprehensive transcriptome profiling of Taiwanese colorectal cancer implicates an ethnic basis for pathogenesis. Sci. Rep. 2020, 10, 4526. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.M.; Paull, E.O.; Graham, N.A.; Lee, J.K.; Smith, B.A.; Titz, B.; Stoyanova, T.; Faltermeier, C.M.; Uzunangelov, V.; Carlin, D.E.; et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell 2016, 166, 1041–1054. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Patients | ||
|---|---|---|---|
| #1 | #2 | #3 | |
| Age (years) | 47 | 60 | 66 |
| Sex | M | M | M |
| Tumor classification | 4A | 2 | 4A |
| Node classification | 2B | 2B | 0 |
| Overall TNM stage | IV | IV | IV |
| Differentiation a | Moderate | Moderate | Moderate |
| Site | Tongue | Buccal mucosa | Other |
| Alcohol drinking | Y | Y | Y |
| Betel quid chewing | Y | Y | Y |
| Cigarette smoking | N | Y | Y |
| Patient Characteristics | Number of Patients | No. of Patients (%) | p Value | |
|---|---|---|---|---|
| CHEK1 Low Expression | CHEK1 High Expression | |||
| Sex | ||||
| Male | 114 | 57 (50.0) | 57 (50.0) | 1.000 |
| Female | 12 | 6 (50.0) | 6 (50.0) | |
| Age a (year) | 51.8 ± 11.3 | 52.8 ± 1.2 | 0.336 | |
| Tumor classification | ||||
| T1–T2 | 63 | 33 (52.4) | 30 (47.6) | 0.593 |
| T3–T4 | 63 | 30 (47.6) | 33 (52.4) | |
| Node classification | ||||
| N = 0 | 64 | 38 (59.4) | 26 (40.6) | 0.032 b |
| N > 0 | 62 | 25 (40.3) | 37 (59.7) | |
| Overall TNM stage | ||||
| I–II | 38 | 23 (60.5) | 15 (39.5) | 0.120 |
| III–IV | 88 | 40 (45.5) | 48 (54.5) | |
| Extranodal extension | ||||
| No | 94 | 49 (52.1) | 45 (47.9) | 0.413 |
| Yes | 32 | 14 (43.8) | 18 (56.2) | |
| Perineural invasion | ||||
| No | 68 | 36 (52.9) | 32 (47.1) | 0.475 |
| Yes | 58 | 27 (46.6) | 31 (53.4) | |
| Differentiation c | ||||
| Well + Mod | 110 | 57 (51.8) | 53 (48.2) | 0.285 |
| Poor | 16 | 6 (37.5) | 10 (62.5) | |
| Tumor depth | ||||
| mm | 14.3 ± 9.5 | 13.5 ± 10.3 | 0.475 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.-Y.; Liu, C.-R.; Chang, I.Y.-F.; OuYang, C.-N.; Hsieh, C.-H.; Huang, Y.-L.; Wang, C.-I.; Jan, F.-W.; Wang, W.-L.; Tsai, T.-L.; et al. Cotargeting CHK1 and PI3K Synergistically Suppresses Tumor Growth of Oral Cavity Squamous Cell Carcinoma in Patient-Derived Xenografts. Cancers 2020, 12, 1726. https://doi.org/10.3390/cancers12071726
Yang C-Y, Liu C-R, Chang IY-F, OuYang C-N, Hsieh C-H, Huang Y-L, Wang C-I, Jan F-W, Wang W-L, Tsai T-L, et al. Cotargeting CHK1 and PI3K Synergistically Suppresses Tumor Growth of Oral Cavity Squamous Cell Carcinoma in Patient-Derived Xenografts. Cancers. 2020; 12(7):1726. https://doi.org/10.3390/cancers12071726
Chicago/Turabian StyleYang, Chia-Yu, Chiao-Rou Liu, Ian Yi-Feng Chang, Chun-Nan OuYang, Chia-Hsun Hsieh, Yen-Lin Huang, Chun-I Wang, Fei-Wen Jan, Wan-Ling Wang, Ting-Lin Tsai, and et al. 2020. "Cotargeting CHK1 and PI3K Synergistically Suppresses Tumor Growth of Oral Cavity Squamous Cell Carcinoma in Patient-Derived Xenografts" Cancers 12, no. 7: 1726. https://doi.org/10.3390/cancers12071726
APA StyleYang, C.-Y., Liu, C.-R., Chang, I. Y.-F., OuYang, C.-N., Hsieh, C.-H., Huang, Y.-L., Wang, C.-I., Jan, F.-W., Wang, W.-L., Tsai, T.-L., Liu, H., Tseng, C.-P., Chang, Y.-S., Wu, C.-C., & Chang, K.-P. (2020). Cotargeting CHK1 and PI3K Synergistically Suppresses Tumor Growth of Oral Cavity Squamous Cell Carcinoma in Patient-Derived Xenografts. Cancers, 12(7), 1726. https://doi.org/10.3390/cancers12071726