Current and Future Treatments in the Fight against Non-Alcoholic Fatty Liver Disease



Abstract
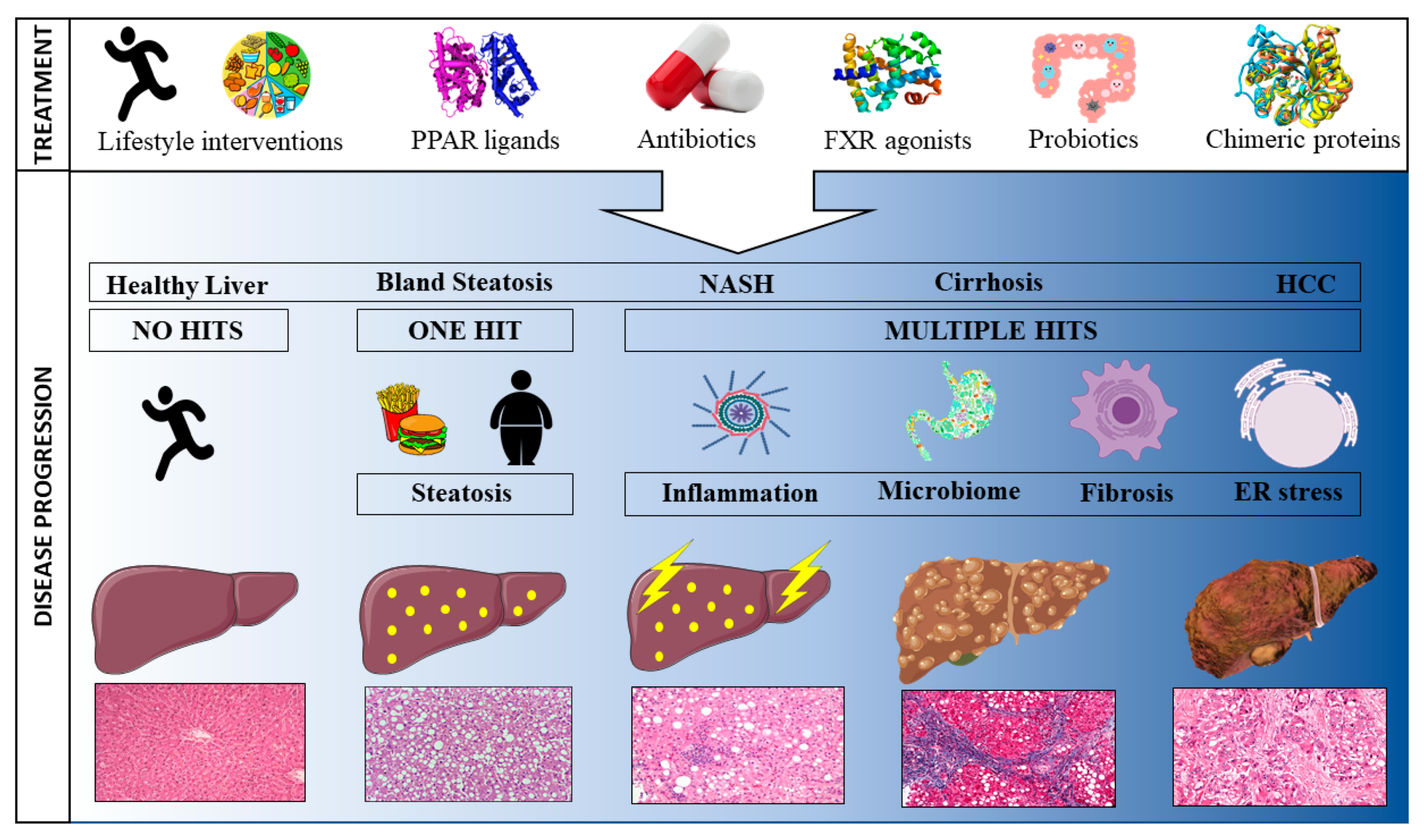
1. Introduction
2. Treatments
2.1. Hallmark Characteristics Driving NAFLD
2.1.1. Steatosis
PPARα Agonists
PPARδ Agonists
PPARγ Agonists
Dual PPAR Agonists
PPAR-pan Agonists
Non-PPAR Treatments
2.1.2. Inflammation
2.1.3. Fibrosis
2.2. Gut Microbiome
2.2.1. Antibiotics
2.2.2. Probiotics
2.2.3. Faecal Microbiota Transplantation (FMT)
2.2.4. Leaky Gut
3. Lean NAFLD
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Reibe, S.; Febbraio, M.A. Relieving ER stress to target NASH-driven hepatocellular carcinoma. Nat. Rev. Endocrinol. 2019, 15, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Febbraio, M.A.; Reibe, S.; Shalapour, S.; Ooi, G.J.; Watt, M.J.; Karin, M. Preclinical Models for Studying NASH-Driven HCC: How Useful Are They? Cell Metab. 2019, 29, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Fitzmaurice, C.; Akinyemiju, T.F.; Al Lami, F.H.; Alam, T.; Alizadeh-Navaei, R.; Allen, C.; Alsharif, U.; Alvis-Guzman, N.; Amini, E.; Anderson, B.O.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2018, 4, 1553–1568. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Younes, R.; Bugianesi, E. NASH in Lean Individuals. Semin. Liver Dis. 2019, 39, 86–95. [Google Scholar] [CrossRef]
- Scaglioni, F.; Ciccia, S.; Marino, M.; Bedogni, G.; Bellentani, S. ASH and NASH. Dig. Dis. 2011, 29, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Minato, T.; Tsutsumi, M.; Tsuchishima, M.; Hayashi, N.; Saito, T.; Matsue, Y.; Toshikuni, N.; Arisawa, T.; George, J. Binge alcohol consumption aggravates oxidative stress and promotes pathogenesis of NASH from obesity-induced simple steatosis. Mol. Med. 2014, 20, 490–502. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Blissett, D.; Blissett, R.; Henry, L.; Stepanova, M.; Younossi, Y.; Racila, A.; Hunt, S.; Beckerman, R. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 2016, 64, 1577–1586. [Google Scholar] [CrossRef]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: Distinct clinicopathologic meanings. Hepatology 2011, 53, 810–820. [Google Scholar] [CrossRef]
- Iqbal, U.; Perumpail, B.J.; Akhtar, D.; Kim, D.; Ahmed, A. The Epidemiology, Risk Profiling and Diagnostic Challenges of Nonalcoholic Fatty Liver Disease. Medicines 2019, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Charlotte, F.; Heurtier, A.; Gombert, S.; Giral, P.; Bruckert, E.; Grimaldi, A.; Capron, F.; Poynard, T. Sampling variability of liver biopsy in nonalcoholic fatty liver disease. Gastroenterology 2005, 128, 1898–1906. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Koo, S.H. Nonalcoholic fatty liver disease: Molecular mechanisms for the hepatic steatosis. Clin. Mol. Hepatol. 2013, 19, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed]
- Pessayre, D. Role of mitochondria in non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2007, 22 (Suppl. 1), S20–S27. [Google Scholar] [CrossRef]
- Alonso, C.; Fernández-Ramos, D.; Varela-Rey, M.; Martínez-Arranz, I.; Navasa, N.; Van Liempd, S.M.; Lavín Trueba, J.L.; Mayo, R.; Ilisso, C.P.; de Juan, V.G.; et al. Metabolomic Identification of Subtypes of Nonalcoholic Steatohepatitis. Gastroenterology 2017, 152, 1449–1461. [Google Scholar] [CrossRef] [PubMed]
- Toshimitsu, K.; Matsuura, B.; Ohkubo, I.; Niiya, T.; Furukawa, S.; Hiasa, Y.; Kawamura, M.; Ebihara, K.; Onji, M. Dietary habits and nutrient intake in non-alcoholic steatohepatitis. Nutrition 2007, 23, 46–52. [Google Scholar] [CrossRef]
- Croci, I.; Coombes, J.S.; Bucher Sandbakk, S.; Keating, S.E.; Nauman, J.; Macdonald, G.A.; Wisloff, U. Non-alcoholic fatty liver disease: Prevalence and all-cause mortality according to sedentary behaviour and cardiorespiratory fitness. The HUNT Study. Prog. Cardiovasc. Dis. 2019, 62, 127–134. [Google Scholar] [CrossRef]
- Houghton, D.; Thoma, C.; Hallsworth, K.; Cassidy, S.; Hardy, T.; Burt, A.D.; Tiniakos, D.; Hollingsworth, K.G.; Taylor, R.; Day, C.P.; et al. Exercise Reduces Liver Lipids and Visceral Adiposity in Patients with Nonalcoholic Steatohepatitis in a Randomized Controlled Trial. Clin. Gastroenterol. Hepatol. 2017, 15, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Hallsworth, K.; Adams, L.A. Lifestyle modification in NAFLD/NASH: Facts and figures. Jhep. Rep. 2019, 1, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Lazo, M.; Solga, S.F.; Horska, A.; Bonekamp, S.; Diehl, A.M.; Brancati, F.L.; Wagenknecht, L.E.; Pi-Sunyer, F.X.; Kahn, S.E.; Clark, J.M. Effect of a 12-month intensive lifestyle intervention on hepatic steatosis in adults with type 2 diabetes. Diabetes Care 2010, 33, 2156–2163. [Google Scholar] [CrossRef] [PubMed]
- Promrat, K.; Kleiner, D.E.; Niemeier, H.M.; Jackvony, E.; Kearns, M.; Wands, J.R.; Fava, J.L.; Wing, R.R. Randomized controlled trial testing the effects of weight loss on nonalcoholic steatohepatitis. Hepatology 2010, 51, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Bozzetto, L.; Prinster, A.; Annuzzi, G.; Costagliola, L.; Mangione, A.; Vitelli, A.; Mazzarella, R.; Longobardo, M.; Mancini, M.; Vigorito, C.; et al. Liver fat is reduced by an isoenergetic MUFA diet in a controlled randomized study in type 2 diabetic patients. Diabetes Care 2012, 35, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Stewart, K.E.; Haller, D.L.; Sargeant, C.; Levenson, J.L.; Puri, P.; Sanyal, A.J. Readiness for behaviour change in non-alcoholic fatty liver disease: Implications for multidisciplinary care models. Liver. Int. 2015, 35, 936–943. [Google Scholar] [CrossRef]
- Hussain, S.S.; Bloom, S.R. The regulation of food intake by the gut-brain axis: Implications for obesity. Int. J. Obes. 2013, 37, 625–633. [Google Scholar] [CrossRef]
- Haufe, S.; Haas, V.; Utz, W.; Birkenfeld, A.L.; Jeran, S.; Böhnke, J.; Mähler, A.; Luft, F.C.; Schulz-Menger, J.; Boschmann, M.; et al. Long-lasting improvements in liver fat and metabolism despite body weight regain after dietary weight loss. Diabetes Care 2013, 36, 3786–3792. [Google Scholar] [CrossRef]
- Van der Windt, D.J.; Sud, V.; Zhang, H.; Tsung, A.; Huang, H. The Effects of Physical Exercise on Fatty Liver Disease. Gene Expr. 2018, 18, 89–101. [Google Scholar] [CrossRef]
- Szary, N.; Rector, R.S.; Uptergrove, G.M.; Ridenhour, S.E.; Shukla, S.D.; Thyfault, J.P.; Koch, L.G.; Britton, S.L.; Ibdah, J.A. High Intrinsic Aerobic Capacity Protects against Ethanol-Induced Hepatic Injury and Metabolic Dysfunction: Study Using High Capacity Runner Rat Model. Biomolecules 2015, 5, 3295–3308. [Google Scholar] [CrossRef]
- Jain, M.R.; Giri, S.R.; Trivedi, C.; Bhoi, B.; Rath, A.; Vanage, G.; Vyas, P.; Ranvir, R.; Patel, P.R. Saroglitazar, a novel PPARalpha/gamma agonist with predominant PPARalpha activity, shows lipid-lowering and insulin-sensitizing effects in preclinical models. Pharm. Res. Perspect. 2015, 3, e00136. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, N.S.; Kumar, N.; Duseja, A. Peroxisome Proliferator-Activated Receptors and Their Agonists in Nonalcoholic Fatty Liver Disease. J. Clin. Exp. Hepatol. 2019, 9, 731–739. [Google Scholar] [CrossRef]
- Wagner, M.; Zollner, G.; Trauner, M. Nuclear receptors in liver disease. Hepatology 2011, 53, 1023–1034. [Google Scholar] [CrossRef]
- Basaranoglu, M.; Acbay, O.; Sonsuz, A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J. Hepatol. 1999, 31, 384. [Google Scholar] [CrossRef]
- Fernandez-Miranda, C.; Perez-Carreras, M.; Colina, F.; Lopez-Alonso, G.; Vargas, C.; Solis-Herruzo, J.A. A pilot trial of fenofibrate for the treatment of non-alcoholic fatty liver disease. Dig. Liver Dis. 2008, 40, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Laurin, J.; Lindor, K.D.; Crippin, J.S.; Gossard, A.; Gores, G.J.; Ludwig, J.; Rakela, J.; McGill, D.B. Ursodeoxycholic acid or clofibrate in the treatment of non-alcohol-induced steatohepatitis: A pilot study. Hepatology 1996, 23, 1464–1467. [Google Scholar] [CrossRef]
- Scorletti, E.; Byrne, C.D. Omega-3 fatty acids, hepatic lipid metabolism, and nonalcoholic fatty liver disease. Annu. Rev. Nutr. 2013, 33, 231–248. [Google Scholar] [CrossRef]
- Scorletti, E.; Bhatia, L.; McCormick, K.G.; Clough, G.F.; Nash, K.; Hodson, L.; Moyses, H.E.; Calder, P.C.; Byrne, C.D. Effects of purified eicosapentaenoic and docosahexaenoic acids in nonalcoholic fatty liver disease: Results from the Welcome* study. Hepatology 2014, 60, 1211–1221. [Google Scholar] [CrossRef]
- Tailleux, A.; Wouters, K.; Staels, B. Roles of PPARs in NAFLD: Potential therapeutic targets. Biochim. Biophys. Acta 2012, 1821, 809–818. [Google Scholar] [CrossRef]
- Shearer, B.G.; Patel, H.S.; Billin, A.N.; Way, J.M.; Winegar, D.A.; Lambert, M.H.; Xu, R.X.; Leesnitzer, L.M.; Merrihew, R.V.; Huet, S.; et al. Discovery of a novel class of PPARdelta partial agonists. Bioorg. Med. Chem. Lett. 2008, 18, 5018–5022. [Google Scholar] [CrossRef]
- Choi, Y.J.; Roberts, B.K.; Wang, X.; Geaney, J.C.; Naim, S.; Wojnoonski, K.; Karpf, D.B.; Krauss, R.M. Effects of the PPAR-delta agonist MBX-8025 on atherogenic dyslipidemia. Atherosclerosis 2012, 220, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Haczeyni, F.; Wang, H.; Barn, V.; Mridha, A.R.; Yeh, M.M.; Haigh, W.G.; Ioannou, G.N.; Choi, Y.J.; McWherter, C.A.; Teoh, N.C.; et al. The selective peroxisome proliferator-activated receptor-delta agonist seladelpar reverses nonalcoholic steatohepatitis pathology by abrogating lipotoxicity in diabetic obese mice. Hepatol. Commun. 2017, 1, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Da’adoosh, B.; Marcus, D.; Rayan, A.; King, F.; Che, J.; Goldblum, A. Discovering highly selective and diverse PPAR-delta agonists by ligand based machine learning and structural modeling. Sci. Rep. 2019, 9, 1106. [Google Scholar] [CrossRef] [PubMed]
- Konerman, M.A.; Jones, J.C.; Harrison, S.A. Pharmacotherapy for NASH: Current and emerging. J. Hepatol. 2018, 68, 362–375. [Google Scholar] [CrossRef]
- Nesto, R.W.; Bell, D.; Bonow, R.O.; Fonseca, V.; Grundy, S.M.; Horton, E.S.; Le Winter, M.; Porte, D.; Semenkovich, C.F.; Smith, S.; et al. Thiazolidinedione use, fluid retention, and congestive heart failure: A consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care 2004, 27, 256–263. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients with Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann. Intern. Med. 2016, 165, 305–315. [Google Scholar] [CrossRef]
- Lee, Y.H.; Kim, J.H.; Kim, S.R.; Jin, H.Y.; Rhee, E.J.; Cho, Y.M.; Lee, B.W. Lobeglitazone, a Novel Thiazolidinedione, Improves Non-Alcoholic Fatty Liver Disease in Type 2 Diabetes: Its Efficacy and Predictive Factors Related to Responsiveness. J. Korean Med. Sci. 2017, 32, 60–69. [Google Scholar] [CrossRef]
- Staels, B.; Rubenstrunk, A.; Noel, B.; Rigou, G.; Delataille, P.; Millatt, L.J.; Baron, M.; Lucas, A.; Tailleux, A.; Hum, D.W.; et al. Hepatoprotective effects of the dual peroxisome proliferator-activated receptor alpha/delta agonist, GFT505, in rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2013, 58, 1941–1952. [Google Scholar] [CrossRef]
- Cariou, B.; Hanf, R.; Lambert-Porcheron, S.; Zair, Y.; Sauvinet, V.; Noel, B.; Flet, L.; Vidal, H.; Staels, B.; Laville, M. Dual peroxisome proliferator-activated receptor alpha/delta agonist GFT505 improves hepatic and peripheral insulin sensitivity in abdominally obese subjects. Diabetes Care 2013, 36, 2923–2930. [Google Scholar] [CrossRef]
- Cariou, B.; Zair, Y.; Staels, B.; Bruckert, E. Effects of the new dual PPAR alpha/delta agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care 2011, 34, 2008–2014. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Harrison, S.A.; Francque, S.; Bedossa, P.; Lehert, P.; Serfaty, L.; Romero-Gomez, M.; Boursier, J.; Abdelmalek, M.; Caldwell, S.; et al. Elafibranor, an Agonist of the Peroxisome Proliferator-Activated Receptor-alpha and -delta, Induces Resolution of Nonalcoholic Steatohepatitis Without Fibrosis Worsening. Gastroenterology 2016, 150, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.R.; Giri, S.R.; Bhoi, B.; Trivedi, C.; Rath, A.; Rathod, R.; Ranvir, R.; Kadam, S.; Patel, H.; Swain, P.; et al. Dual PPARalpha/gamma agonist saroglitazar improves liver histopathology and biochemistry in experimental NASH models. Liver. Int. 2018, 38, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Boubia, B.; Poupardin, O.; Barth, M.; Binet, J.; Peralba, P.; Mounier, L.; Jacquier, E.; Gauthier, E.; Lepais, V.; Chatar, M.; et al. Design, Synthesis, and Evaluation of a Novel Series of Indole Sulfonamide Peroxisome Proliferator Activated Receptor (PPAR) α/γ/δ Triple Activators: Discovery of Lanifibranor, a New Antifibrotic Clinical Candidate. J. Med. Chem. 2018, 61, 2246–2265. [Google Scholar] [CrossRef] [PubMed]
- Wettstein, G.; Luccarini, J.M.; Poekes, L.; Faye, P.; Kupkowski, F.; Adarbes, V.; Defrene, E.; Estivalet, C.; Gawronski, X.; Jantzen, I.; et al. The new-generation pan-peroxisome proliferator-activated receptor agonist IVA337 protects the liver from metabolic disorders and fibrosis. Hepatol. Commun. 2017, 1, 524–537. [Google Scholar] [CrossRef]
- Ament, Z.; West, J.A.; Stanley, E.; Ashmore, T.; Roberts, L.D.; Wright, J.; Nicholls, A.W.; Griffin, J.L. PPAR-pan activation induces hepatic oxidative stress and lipidomic remodelling. Free Radic. Biol. Med. 2016, 95, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Chang, F.; Jaber, L.A.; Berlie, H.D.; O’Connell, M.B. Evolution of peroxisome proliferator-activated receptor agonists. Ann. Pharm. 2007, 41, 973–983. [Google Scholar] [CrossRef]
- Feng, L.; Luo, H.; Xu, Z.; Yang, Z.; Du, G.; Zhang, Y.; Yu, L.; Hu, K.; Zhu, W.; Tong, Q.; et al. Bavachinin, as a novel natural pan-PPAR agonist, exhibits unique synergistic effects with synthetic PPAR-gamma and PPAR-alpha agonists on carbohydrate and lipid metabolism in db/db and diet-induced obese mice. Diabetologia 2016, 59, 1276–1286. [Google Scholar] [CrossRef]
- Drucker, D.J.; Habener, J.F.; Holst, J.J. Discovery, characterization, and clinical development of the glucagon-like peptides. J. Clin. Investig. 2017, 127, 4217–4227. [Google Scholar] [CrossRef]
- Bode, B. An overview of the pharmacokinetics, efficacy and safety of liraglutide. Diabetes Res. Clin. Pract. 2012, 97, 27–42. [Google Scholar] [CrossRef]
- Eguchi, Y.; Kitajima, Y.; Hyogo, H.; Takahashi, H.; Kojima, M.; Ono, M.; Araki, N.; Tanaka, K.; Yamaguchi, M.; Matsuda, Y.; et al. Pilot study of liraglutide effects in non-alcoholic steatohepatitis and non-alcoholic fatty liver disease with glucose intolerance in Japanese patients (LEAN-J). Hepatol. Res. 2015, 45, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Newsome, P.; Francque, S.; Harrison, S.; Ratziu, V.; Van Gaal, L.; Calanna, S.; Hansen, M.; Linder, M.; Sanyal, A. Effect of semaglutide on liver enzymes and markers of inflammation in subjects with type 2 diabetes and/or obesity. Aliment. Pharm. 2019, 50, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Flowers, M.T.; Ntambi, J.M. Role of stearoyl-coenzyme A desaturase in regulating lipid metabolism. Curr. Opin. Lipidol. 2008, 19, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Leikin-Frenkel, A.; Gonen, A.; Shaish, A.; Goldiner, I.; Leikin-Gobbi, D.; Konikoff, F.M.; Harats, D.; Gilat, T. Fatty acid bile acid conjugate inhibits hepatic stearoyl coenzyme A desaturase and is non-atherogenic. Arch. Med. Res. 2010, 41, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Iruarrizaga-Lejarreta, M.; Varela-Rey, M.; Fernandez-Ramos, D.; Martinez-Arranz, I.; Delgado, T.C.; Simon, J.; Juan, V.G.; delaCruz-Villar, L.; Azkargorta, M.; Lavin, J.L.; et al. Role of Aramchol in steatohepatitis and fibrosis in mice. Hepatol. Commun. 2017, 1, 911–927. [Google Scholar] [CrossRef] [PubMed]
- Safadi, R.; Konikoff, F.M.; Mahamid, M.; Zelber-Sagi, S.; Halpern, M.; Gilat, T.; Oren, R. The fatty acid-bile acid conjugate Aramchol reduces liver fat content in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2014, 12, 2085–2091. [Google Scholar] [CrossRef] [PubMed]
- Tahrani, A.A.; Barnett, A.H.; Bailey, C.J. SGLT inhibitors in management of diabetes. Lancet Diabetes Endocrinol. 2013, 1, 140–151. [Google Scholar] [CrossRef]
- Jojima, T.; Tomotsune, T.; Iijima, T.; Akimoto, K.; Suzuki, K.; Aso, Y. Empagliflozin (an SGLT2 inhibitor), alone or in combination with linagliptin (a DPP-4 inhibitor), prevents steatohepatitis in a novel mouse model of non-alcoholic steatohepatitis and diabetes. Diabetol. Metab. Syndr. 2016, 8, 45. [Google Scholar] [CrossRef]
- Honda, Y.; Imajo, K.; Kato, T.; Kessoku, T.; Ogawa, Y.; Tomeno, W.; Kato, S.; Mawatari, H.; Fujita, K.; Yoneda, M.; et al. The Selective SGLT2 Inhibitor Ipragliflozin Has a Therapeutic Effect on Nonalcoholic Steatohepatitis in Mice. PLoS ONE 2016, 11, e0146337. [Google Scholar] [CrossRef]
- Shiba, K.; Tsuchiya, K.; Komiya, C.; Miyachi, Y.; Mori, K.; Shimazu, N.; Yamaguchi, S.; Ogasawara, N.; Katoh, M.; Itoh, M.; et al. Canagliflozin, an SGLT2 inhibitor, attenuates the development of hepatocellular carcinoma in a mouse model of human NASH. Sci. Rep. 2018, 8, 2362. [Google Scholar] [CrossRef]
- Qiang, S.; Nakatsu, Y.; Seno, Y.; Fujishiro, M.; Sakoda, H.; Kushiyama, A.; Mori, K.; Matsunaga, Y.; Yamamotoya, T.; Kamata, H.; et al. Treatment with the SGLT2 inhibitor luseogliflozin improves nonalcoholic steatohepatitis in a rodent model with diabetes mellitus. Diabetol. Metab. Syndr. 2015, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Suzuki, K.; Kato, K.; Jojima, T.; Iijima, T.; Murohisa, T.; Iijima, M.; Takekawa, H.; Usui, I.; Hiraishi, H.; et al. Evaluation of the effects of dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on hepatic steatosis and fibrosis using transient elastography in patients with type 2 diabetes and non-alcoholic fatty liver disease. Diabetes Obes. Metab. 2019, 21, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.W.; Lundkvist, P.; Jansson, P.A.; Johansson, L.; Kvarnstrom, M.; Moris, L.; Miliotis, T.; Forsberg, G.B.; Riserus, U.; Lind, L.; et al. Effects of dapagliflozin and n-3 carboxylic acids on non-alcoholic fatty liver disease in people with type 2 diabetes: A double-blind randomised placebo-controlled study. Diabetologia 2018, 61, 1923–1934. [Google Scholar] [CrossRef]
- Findeisen, M.; Allen, T.L.; Henstridge, D.C.; Kammoun, H.; Brandon, A.E.; Baggio, L.L.; Watt, K.I.; Pal, M.; Cron, L.; Estevez, E.; et al. Treatment of type 2 diabetes with the designer cytokine IC7Fc. Nature 2019, 574, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364. [Google Scholar] [CrossRef]
- Jiao, Y.; Lu, Y.; Li, X.Y. Farnesoid X receptor: A master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharm. Sin. 2015, 36, 44–50. [Google Scholar] [CrossRef]
- D’Hellencourt, C.L.; Diaw, L.; Cornillet, P.; Guenounou, M. Differential regulation of TNF alpha, IL-1 beta, IL-6, IL-8, TNF beta, and IL-10 by pentoxifylline. Int. J. Immunopharmacol. 1996, 18, 739–748. [Google Scholar] [CrossRef]
- Zein, C.O.; Yerian, L.M.; Gogate, P.; Lopez, R.; Kirwan, J.P.; Feldstein, A.E.; McCullough, A.J. Pentoxifylline improves nonalcoholic steatohepatitis: A randomized placebo-controlled trial. Hepatology 2011, 54, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.; Nazmul Hasan, S.; Mustafa, G.; Alam, M.; Kamal, M.; Ahmad, N. Effect of Pentoxifylline on Histological Activity and Fibrosis of Nonalcoholic Steatohepatitis Patients: A One Year Randomized Control Trial. J. Transl. Int. Med. 2017, 5, 155–163. [Google Scholar] [CrossRef]
- Sterling, R.K.; Sanyal, A.J. Pentoxifylline for steatohepatitis: Magic bullet or smoking gun? Hepatology 2011, 54, 1496–1499. [Google Scholar] [CrossRef]
- Vieth, R. What is the optimal vitamin D status for health? Prog. Biophys. Mol. Boil. 2006, 92, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Chiu, K.C.; Chu, A.; Go, V.L.W.; Saad, M.F. Hypovitaminosis D is associated with insulin resistance and beta cell dysfunction. Am. J. Clin. Nutr. 2004, 79, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Wortsman, J.; Matsuoka, L.Y.; Chen, T.; Lu, Z.; Holick, M.F. Decreased bioavailability of vitamin D in obesity. Am. J. Clin. Nutr. 2000, 72, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Ford, E.S.; Ajani, U.A.; McGuire, L.C.; Liu, S. Concentrations of serum vitamin D and the metabolic syndrome among U.S. adults. Diabetes Care 2005, 28, 1228–1230. [Google Scholar] [CrossRef] [PubMed]
- Roth, C.L.; Elfers, C.; Figlewicz, D.P.; Melhorn, S.J.; Morton, G.J.; Hoofnagle, A.; Yeh, M.M.; Nelson, J.E.; Kowdley, K.V. Vitamin D deficiency in obese rats exacerbates nonalcoholic fatty liver disease and increases hepatic resistin and toll-like receptor activation. Hepatology 2012, 55, 1103–1111. [Google Scholar] [CrossRef]
- Nelson, J.E.; Roth, C.L.; Wilson, L.A.; Yates, K.P.; Aouizerat, B.; Morgan-Stevenson, V.; Whalen, E.; Hoofnagle, A.; Mason, M.; Gersuk, V.; et al. Vitamin D Deficiency Is Associated with Increased Risk of Non-alcoholic Steatohepatitis in Adults with Non-alcoholic Fatty Liver Disease: Possible Role for MAPK and NF-κB? Am. J. Gastroenterol. 2016, 111, 852–863. [Google Scholar] [CrossRef]
- Geier, A.; Eichinger, M.; Stirnimann, G.; Semela, D.; Tay, F.; Seifert, B.; Tschopp, O.; Bantel, H.; Jahn, D.; Maggio, E.M.; et al. Treatment of non-alcoholic steatohepatitis patients with vitamin D: A double-blinded, randomized, placebo-controlled pilot study. Scand. J. Gastroenterol. 2018, 53, 1114–1120. [Google Scholar] [CrossRef]
- Yodoshi, T.; Orkin, S.; Arce-Clachar, A.C.; Bramlage, K.; Liu, C.; Fei, L.; El-Khider, F.; Dasarathy, S.; Xanthakos, S.A.; Mouzaki, M. Vitamin D deficiency: Prevalence and association with liver disease severity in pediatric nonalcoholic fatty liver disease. Eur. J. Clin. Nutr. 2019, 74, 427–435. [Google Scholar] [CrossRef]
- Recio, C.; Lucy, D.; Purvis, G.S.D.; Iveson, P.; Zeboudj, L.; Iqbal, A.J.; Lin, D.; O’Callaghan, C.; Davison, L.; Griesbach, E.; et al. Activation of the Immune-Metabolic Receptor GPR84 Enhances Inflammation and Phagocytosis in Macrophages. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Puengel, T.; De Vos, S.; Hundertmark, J.; Kohlhepp, M.; Guldiken, N.; Pujuguet, P.; Auberval, M.; Marsais, F.; Shoji, K.F.; Saniere, L.; et al. The Medium-Chain Fatty Acid Receptor GPR84 Mediates Myeloid Cell Infiltration Promoting Steatohepatitis and Fibrosis. J. Clin. Med. 2020, 9, 1140. [Google Scholar] [CrossRef]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Yu, L.-X.; Schwabe, R.F. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Hagström, H.; Nasr, P.; Ekstedt, M.; Hammar, U.; Stal, P.; Hultcrantz, R.; Kechagias, S. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J. Hepatol. 2017, 67, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef] [PubMed]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradère, J.-P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Kohli, R. Bile acid metabolism and signaling: Potential therapeutic target for nonalcoholic fatty liver disease. Clin. Transl. Gastroenterol. 2018, 9, 164. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.M.; Afonso, M.B.; Simão, A.L.; Carvalho, C.C.; Trindade, A.; Duarte, A.; Borralho, P.M.; Machado, M.V.; Cortez-Pinto, H.; Rodrigues, C.M.; et al. miR-21 ablation and obeticholic acid ameliorate nonalcoholic steatohepatitis in mice. Cell Death Dis. 2017, 8, e2825. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Itoh, M.; Suganami, T.; Kanai, S.; Shirakawa, I.; Sakai, T.; Asakawa, M.; Yoneyama, T.; Kai, T.; Ogawa, Y. Obeticholic acid protects against hepatocyte death and liver fibrosis in a murine model of nonalcoholic steatohepatitis. Sci. Rep. 2018, 8, 8157. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2014, 385, 956–965. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef]
- Wójcik, M.; Janus, D.; Dolezal-Oltarzewska, K.; Kalicka-Kasperczyk, A.; Poplawska, K.; Drozdz, D.; Sztefko, K.; Starzyk, J.B. A decrease in fasting FGF19 levels is associated with the development of non-alcoholic fatty liver disease in obese adolescents. J. Pediatr. Endocrinol. Metab. 2012, 25, 1089–1093. [Google Scholar] [CrossRef] [PubMed]
- Nicholes, K.; Guillet, S.; Tomlinson, E.; Hillan, K.; Wright, B.; Frantz, G.D.; Pham, T.A.; Dillard-Telm, L.; Tsai, S.P.; Stephan, J.-P.; et al. A Mouse Model of Hepatocellular Carcinoma. Am. J. Pathol. 2002, 160, 2295–2307. [Google Scholar] [CrossRef]
- Zhou, M.; Yang, H.; Learned, R.M.; Tian, H.; Ling, L. Non-cell-autonomous activation of IL-6/STAT3 signaling mediates FGF19-driven hepatocarcinogenesis. Nat. Commun. 2017, 8, 15433. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Rinella, M.E.; Abdelmalek, M.F.; Trotter, J.F.; Paredes, A.H.; Arnold, H.L.; Kugelmas, M.; Bashir, M.R.; Jaros, M.J.; Ling, L.; et al. NGM282 for treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2018, 391, 1174–1185. [Google Scholar] [CrossRef]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-Mediated Bile Acid Sensing Controls Glucose Homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Finn, P.D.; Rodriguez, D.; Kohler, J.; Jiang, Z.; Wan, S.; Blanco, E.; King, A.J.; Chen, T.; Bell, N.; Dragoli, D.; et al. Intestinal TGR5 agonism improves hepatic steatosis and insulin sensitivity in Western diet-fed mice. Am. J. Physiol. Liver Physiol. 2019, 316, G412–G424. [Google Scholar] [CrossRef]
- Mcmahan, R.H.; Wang, X.X.; Cheng, L.L.; Krisko, T.; Smith, M.; El Kasmi, K.; Pruzanski, M.; Adorini, L.; Golden-Mason, L.; Levi, M.; et al. Bile Acid Receptor Activation Modulates Hepatic Monocyte Activity and Improves Nonalcoholic Fatty Liver Disease. J. Boil. Chem. 2013, 288, 11761–11770. [Google Scholar] [CrossRef]
- Roth, J.D.; Feigh, M.; Veidal, S.S.; Fensholdt, L.K.; Rigbolt, K.T.; Hansen, H.; Chen, L.C.; Petitjean, M.; Friley, W.; Vrang, N.; et al. INT-767 improves histopathological features in a diet-inducedob/obmouse model of biopsy-confirmed non-alcoholic steatohepatitis. World J. Gastroenterol. 2018, 24, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Tabas, I.; Pajvani, U.B. Mechanisms of Fibrosis Development in Nonalcoholic Steatohepatitis. Gastroenterology 2020, 158, 1913–1928. [Google Scholar] [CrossRef]
- Sato, Y.; Murase, K.; Kato, J.; Kobune, M.; Sato, T.; Kawano, Y.; Takimoto, R.; Takada, K.; Miyanishi, K.; Matsunaga, T.; et al. Resolution of liver cirrhosis using vitamin A–coupled liposomes to deliver siRNA against a collagen-specific chaperone. Nat. Biotechnol. 2008, 26, 431–442. [Google Scholar] [CrossRef]
- Loomba, R.; Lawitz, E.; Mantry, P.S.; Jayakumar, S.; Caldwell, S.H.; Arnold, H.; Diehl, A.M.; Djedjos, C.S.; Han, L.; Myers, R.P.; et al. The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: A randomized, phase 2 trial. Hepatology 2017, 67, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Wong, V.W.-S.; Okanoue, T.; Bzowej, N.; Vuppalanchi, R.; Younes, Z.; Kohli, A.; Sarin, S.; Caldwell, S.H.; Alkhouri, N.; et al. Selonsertib for Patients with Bridging Fibrosis or Compensated Cirrhosis Due to NASH: Results from Randomized Ph III STELLAR Trials. J. Hepatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015, 31, 69–75. [Google Scholar] [CrossRef]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Jang, C.; Liu, J.; Uehara, K.; Gilbert, M.; Izzo, L.; Zeng, X.; Trefely, S.; Fernandez, S.; Carrer, A.; et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 2020, 579, 586–591. [Google Scholar] [CrossRef]
- Todoric, J.; Di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; Shalapour, S.; Taniguchi, K.; et al. Fructose-stimulated de novo lipogenesis depends on inflammation. Nat. Med. 2020. Accepted. [Google Scholar]
- Henao-Mejia, J.; Elinav, E.; Jin, C.-C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef]
- Ye, J.; Lv, L.; Wu, W.; Li, Y.; Shi, D.; Fang, D.; Guo, F.; Jiang, H.; Yan, R.; Ye, W.; et al. Butyrate Protects Mice Against Methionine–Choline-Deficient Diet-Induced Non-alcoholic Steatohepatitis by Improving Gut Barrier Function, Attenuating Inflammation and Reducing Endotoxin Levels. Front. Microbiol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Sohn, W.; Jun, D.W.; Lee, K.N.; Lee, H.L.; Lee, O.Y.; Choi, H.S.; Yoon, B.C. Lactobacillus paracasei Induces M2-Dominant Kupffer Cell Polarization in a Mouse Model of Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2015, 60, 3340–3350. [Google Scholar] [CrossRef]
- Ginès, P.; Rimola, A.; Planas, R.; Vargas, V.; Marco, F.; Almela, M.; Forné, M.; Miranda, M.L.; Llach, J.; Salmerón, J.M.; et al. Norfloxacin prevents spontaneous bacterial peritonitis recurrence in cirrhosis: Results of a double-blind, placebo-controlled trial. Hepatology 1990, 12, 716–724. [Google Scholar] [CrossRef]
- Dapito, D.H.; Mencin, A.; Gwak, G.-Y.; Pradère, J.-P.; Jang, M.-K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef]
- Sharma, B.C.; Sharma, P.; Lunia, M.K.; Srivastava, S.; Goyal, R.; Sarin, S.K. A Randomized, Double-Blind, Controlled Trial Comparing Rifaximin Plus Lactulose with Lactulose Alone in Treatment of Overt Hepatic Encephalopathy. Am. J. Gastroenterol. 2013, 108, 1458–1463. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Razik, A.; Mousa, N.; Shabana, W.; Refaey, M.; Elzehery, R.; Elhelaly, R.; Zalata, K.; Abdelsalam, M.; Eldeeb, A.A.; Awad, M.; et al. Rifaximin in nonalcoholic fatty liver disease. Eur. J. Gastroenterol. Hepatol. 2018, 30, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Cobbold, J.; Atkinson, S.; Marchesi, J.R.; Smith, A.; Wai, S.N.; Stove, J.; Shojaee-Moradie, F.; Jackson, N.; Umpleby, A.M.; Fitzpatrick, J.; et al. Rifaximin in non-alcoholic steatohepatitis: An open-label pilot study. Hepatol. Res. 2017, 48, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef]
- Salminen, S.; Bouley, C.; Boutron, M.-C.; Cummings, J.H.; Franck, A.; Gibson, G.R.; Isolauri, E.; Moreau, M.-C.; Roberfroid, M.; Rowland, I. Functional food science and gastrointestinal physiology and function. Br. J. Nutr. 1998, 80, S147–S171. [Google Scholar] [CrossRef] [PubMed]
- Kirpich, I.A.; McClain, C.J. Probiotics in the treatment of the liver diseases. J. Am. Coll. Nutr. 2012, 31, 14–23. [Google Scholar] [CrossRef]
- Zhang, H.-L.; Yu, L.-X.; Yang, W.; Tang, L.; Lin, Y.; Wu, H.; Zhai, B.; Tan, Y.-X.; Shan, L.; Liu, Q.; et al. Profound impact of gut homeostasis on chemically-induced pro-tumorigenic inflammation and hepatocarcinogenesis in rats. J. Hepatol. 2012, 57, 803–812. [Google Scholar] [CrossRef]
- Mencarelli, A.; Cipriani, S.; Renga, B.; Bruno, A.; D’Amore, C.; Distrutti, E.; Fiorucci, S. VSL#3 Resets Insulin Signaling and Protects against NASH and Atherosclerosis in a Model of Genetic Dyslipidemia and Intestinal Inflammation. PLoS ONE 2012, 7, e45425. [Google Scholar] [CrossRef]
- Li, Z.; Lin, H.; Huang, J.; DeSimone, C.; Diehl, A.M.; Yang, S.; Watkins, P.A.; Moser, A.B.; Song, X. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003, 37, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Jena, P.K.; Sheng, L.; Li, Y.; Wan, Y.-J.Y. Probiotics VSL#3 are effective in reversing non-alcoholic steatohepatitis in a mouse model. Hepatobiliary Surg. Nutr. 2020, 9, 170–182. [Google Scholar]
- Esposito, E.; Iacono, A.; Bianco, G.; Autore, G.; Cuzzocrea, S.; Vajro, P.; Canani, R.B.; Calignano, A.; Raso, G.M.; Meli, R. Probiotics Reduce the Inflammatory Response Induced by a High-Fat Diet in the Liver of Young Rats. J. Nutr. 2009, 139, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Alisi, A.; Bedogni, G.; Baviera, G.; Giorgio, V.; Porro, E.; Paris, C.; Giammaria, P.; Reali, L.; Anania, F.; Nobili, V. Randomised clinical trial: The beneficial effects of VSL#3 in obese children with non-alcoholic steatohepatitis. Aliment. Pharm. 2014, 39, 1276–1285. [Google Scholar] [CrossRef]
- Cammarota, G.; Ianiro, G.; Tilg, H.; Rajilic-Stojanovic, M.; Kump, P.; Satokari, R.; Sokol, H.; Arkkila, P.; Pintus, C.; Hart, A.; et al. European consensus conference on faecal microbiota transplantation in clinical practice. Gut 2017, 66, 569–580. [Google Scholar] [CrossRef]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga–Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of Intestinal Microbiota From Lean Donors Increases Insulin Sensitivity in Individuals With Metabolic Syndrome. Gastroenterology 2012, 143, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Bakken, J.S.; Borody, T.J.; Brandt, L.J.; Brill, J.V.; Demarco, D.C.; Franzos, M.A.; Kelly, C.; Khoruts, A.; Louie, T.; Martinelli, L.P.; et al. Treating Clostridium difficile Infection with Fecal Microbiota Transplantation. Clin. Gastroenterol. Hepatol. 2011, 9, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Hoyles, L.; Fernandez-Real, J.-M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H.; et al. Publisher Correction: Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1628. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, M.; Wang, W.; Cao, X.; Piao, M.; Khan, S.; Yan, F.; Cao, H.; Wang, B. Systematic Review: Adverse Events of Fecal Microbiota Transplantation. PLoS ONE 2016, 11, e0161174. [Google Scholar] [CrossRef]
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G.; et al. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830–834. [Google Scholar] [CrossRef]
- Verbeke, L.; Farré, R.; Verbinnen, B.; Covens, K.; Vanuytsel, T.; Verhaegen, J.; Komuta, M.; Roskams, T.; Chatterjee, S.; Annaert, P.; et al. The FXR Agonist Obeticholic Acid Prevents Gut Barrier Dysfunction and Bacterial Translocation in Cholestatic Rats. Am. J. Pathol. 2015, 185, 409–419. [Google Scholar] [CrossRef]
- Ubeda, M.; Lario, M.; Muñoz, L.; Borrero, M.-J.; Serrano, E.M.R.; Díaz, A.M.S.; Del Campo, R.; Lledó, L.; Pastor, Ó.; García-Bermejo, L. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J. Hepatol. 2016, 64, 1049–1057. [Google Scholar] [CrossRef]
- Schneider, K.M.; Bieghs, V.; Heymann, F.; Hu, W.; Dreymueller, D.; Liao, L.; Frissen, M.; Ludwig, A.; Gassler, N.; Pabst, O.; et al. CX3CR1 is a gatekeeper for intestinal barrier integrity in mice: Limiting steatohepatitis by maintaining intestinal homeostasis. Hepatology 2015, 62, 1405–1416. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2017, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Mohan, S. Non-alcoholic Fatty Liver Disease in Lean Subjects: Characteristics and Implications. J. Clin. Transl. Hepatol. 2017, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Cortez-Pinto, H. No need for a large belly to have NASH. J. Hepatol. 2011, 54, 1090–1093. [Google Scholar] [CrossRef]
- Wattacheril, J.; Sanyal, A.J. Lean NAFLD: An Underrecognized Outlier. Curr. Hepatol. Rep. 2016, 15, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Abid, A.; Taha, O.; Nseir, W.; Farah, R.; Grosovski, M.; Assy, N. Soft drink consumption is associated with fatty liver disease independent of metabolic syndrome. J. Hepatol. 2009, 51, 918–924. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; De Michieli, F.; Cassader, M.; Rizzetto, M.; Durazzo, M.; Fagà, E.; Silli, B.; Pagano, G. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology 2003, 37, 909–916. [Google Scholar] [CrossRef]
- Ryu, S.; Chang, Y.; Jung, H.; Yun, K.E.; Kwon, M.-J.; Choi, Y.; Kim, C.-W.; Cho, J.; Suh, B.-S.; Cho, Y.K.; et al. Relationship of sitting time and physical activity with non-alcoholic fatty liver disease. J. Hepatol. 2015, 63, 1229–1237. [Google Scholar] [CrossRef]
- Chen, F.; Esmaili, S.; Rogers, G.B.; Bugianesi, E.; Petta, S.; Marchesini, G.; Bayoumi, A.; Metwally, M.; Azardaryany, M.K.; Coulter, S.; et al. Lean NAFLD: A Distinct Entity Shaped by Differential Metabolic Adaptation. Hepatology 2020, 71, 1213–1227. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Wei, J.L.; Leung, J.C.-F.; Loong, T.C.-W.; Wong, G.L.-H.; Yeung, D.K.-W.; Chan, R.S.M.; Chan, H.L.-Y.; Chim, A.M.-L.; Woo, J.; Chu, W.C.W.; et al. Prevalence and Severity of Nonalcoholic Fatty Liver Disease in Non-Obese Patients: A Population Study Using Proton-Magnetic Resonance Spectroscopy. Am. J. Gastroenterol. 2015, 110, 1306–1314. [Google Scholar] [CrossRef]
- Huang, Y.; Cohen, J.C.; Hobbs, H.H. Expression and characterization of a PNPLA3 protein isoform (I148M) associated with nonalcoholic fatty liver disease. J. Boil. Chem. 2011, 286, 37085–37093. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Marsh, J.; Ayonrinde, O.T.; Olynyk, J.K.; Ang, W.Q.; Beilin, L.J.; Mori, T.A.; Palmer, L.J.; Oddy, W.; Lye, S.J.; et al. Cholesteryl ester transfer protein gene polymorphisms increase the risk of fatty liver in females independent of adiposity. J. Gastroenterol. Hepatol. 2012, 27, 1520–1527. [Google Scholar] [CrossRef] [PubMed]
- Nakatsuka, A.; Matsuyama, M.; Yamaguchi, S.; Katayama, A.; Eguchi, J.; Murakami, K.; Teshigawara, S.; Ogawa, D.; Wada, N.; Yasunaka, T.; et al. Insufficiency of phosphatidylethanolamine N-methyltransferase is risk for lean non-alcoholic steatohepatitis. Sci. Rep. 2016, 6, 21721. [Google Scholar] [CrossRef] [PubMed]
- Pirola, C.J.; Garaycoechea, M.; Flichman, D.; Arrese, M.; Martino, J.S.; Gazzi, C.; Castaño, G.O.; Sookoian, S. Splice variant rs72613567 prevents worst histologic outcomes in patients with nonalcoholic fatty liver disease. J. Lipid Res. 2018, 60, 176–185. [Google Scholar] [CrossRef]
- DeFronzo, R.; Tripathy, D.; Schwenke, D.C.; Banerji, M.; Bray, G.A.; Buchanan, T.A.; Clement, S.C.; Henry, R.R.; Hodis, H.N.; Kitabchi, A.E.; et al. Pioglitazone for Diabetes Prevention in Impaired Glucose Tolerance. N. Engl. J. Med. 2011, 364, 1104–1115. [Google Scholar] [CrossRef]


{kind=link}
| Target | Drug | Trial ID | BRCT | Cohort Medical Conditions | Ref. | Outcome |
|---|---|---|---|---|---|---|
| PPARα | Gemfibrozil | (Turkey) | Y | NASH | [34] | Decreased serum liver enzymes 1 and triglyceride |
| Fenofibrate | (Spain) | N | NAFLD | [35] | Improved metabolic syndrome, decreased serum liver enzymes and triglycerides | |
| Clofibrate | (US – Pre-1997) | N | NASH | [36] | No improvement | |
| Omega-3 PUFA (Omacor) | Welcome – Phase IV, NCT00760513 | Y | NAFLD | [38] | Decreased liver fat percentage | |
| Omega-3 PUFA (Omacor) | Phase III, NCT01277237 | Y | NAFLD | - | TBD | |
| PPARδ | Seladelpar (MBX-8025)± Atorvastatin | Phase II, NCT00701883 | Y | Hyperlipidaemia | [41] | Decreased liver enzymes and improved serum lipid profile |
| Seladelpar (MBX-8025) | Phase II, NCT03551522 | Y | NASH | - | Trial Suspended – unexplained histological findings | |
| PPARγ | Pioglitazone± vitamin E | PIVENS-Phase III, NCT00063622 | Y | NAFLD, NASH | [46] | Reduced liver steatosis, lobular inflammation and serum ALT/AST |
| Pioglitazone | UTHSCSA NASH-Phase IV, NCT00994682 | Y | Type 2 Diabetes, NAFLD, NASH | [47] | Significant decrease in NAS score by ≥ 2 points in 58% participants; resolution of NASH in 51%. | |
| Lobeglitazone | ELLEGANCE - Phase IV, NCT02285205 | N | Type 2 Diabetes, NAFLD | [48] | Improved liver and serum lipid profiles | |
| PPARα/δ | Elafibranor (GFT505) | Phase IIa, NCT01271777 | Y | Insulin resistance + abdominal obesity | [50] | Improved plasma lipids and hepatic insulin resistance, reduced liver inflammation and ALT |
| Elafibranor (GFT505) | Phase II, NCT01271751 | Y | Athero-genic dyslipidaemia + abdominal obesity | [50] | Decreased serum lipids and liver GGT | |
| Elafibranor (GFT505) | Phase II, NCT01275469 | Y | Impaired glucose tolerance + abdominal obesity | [51] | Improved insulin sensitivity (HOMA-IR), fasting blood glucose and decrease in liver GGT | |
| Elafibranor (GFT505) | Phase IIb, NCT01694849 | Y | NASH | [52] | No significant difference between placebo and elafibranor groups for primary outcome (resolution of NASH) 2 | |
| Elafibranor (GFT505) | RESOLVE-IT – Phase III, NCT02704403 | Y | NASH | - | Ongoing (recruiting) | |
| PPARα/γ | Saroglitazar | EVIDENCES VI – Phase II, NCT03863574 | Y | NASH | - | Ongoing (recruiting) |
| PPAR-pan | lanifibranor (IVA337) | NATIVE – Phase IIb, NCT03459079 | Y | NAFLD, Type 2 Diabetes | - | Ongoing (recruiting) |
| Non-PPAR | Oral insulin (ORMD-0801) | Phase II, NCT02653300 | N | NASH, Type 2 diabetes | - | Ongoing (recruitment) |
| Liraglutide (GLP1 agonist) | LEAN-J | N | NASH | [61] | Decreased liver and visceral fat, liver enzymes and FPG | |
| Semaglutide (GLP1 agonist) | Phase II, NCT02453711 | Y | Obesity, metabolic disorder | [62] | Decreased ALT and hsCRP, significant weight loss at all doses | |
| Semaglutide (GLP1 agonist) | SUSTAIN 6 – Phase III, NCT01720446 | Y | Diabetes, Type 2 diabetes | [62] | Decreased ALT and hsCRP, decreased cardiovascular events (death, infarction or stroke) | |
| Armachol (SCD1 inhibition) | Aramchol003 - Phase II, NCT01094158 | Y | NAFLD, NASH, Metabolic syndrome | [66] | Decrease in liver fat percentage at mid-dose | |
| Armachol (SCD1 inhibition) | ARMOR - Phase III/IV, NCT04104321 | Y | NASH | - | Ongoing (recruiting) | |
| Dapagliflozin (SGLT2 inhibitor) | Dokkyo Medical University (Japan) - UMIN000022155 | Y | NAFLD | [72] | Decreased liver fibrosis, visceral fat mass and liver enzymes | |
| Dapagliflozin (SGLT2 inhibitor) + omega-3 carboxylic acid | EFFECTII – Phase II, NCT02279407 | Y | NAFLD, Type 2 diabetes | [73] | Significant reduction in liver fat and liver enzymes | |
| Dapagliflozin (SGLT2 inhibitor) | DEAN – Phase III, NCT03723252 | Y | NASH | - | Ongoing (recruitment) | |
| Pentoxifylline (TNFα inhibitor) | Phase II, NCT00590161 | Y | NASH | [78] | Improved liver steatosis, fibrosis and lobular inflammation | |
| Pentoxifylline (TNFα inhibitor) | (Sri Lanka) -SLCTR/2014/016 | N | NASH | [79] | Lifestyle intervention+pentoxifyllin improved NAS | |
| Pentoxifylline (TNFα inhibitor) | Phase II/III, NCT00267670 | Y | NASH | - | No difference between placebo and pentoxifylline groups | |
| Vitamin D3 | Phase II, NCT01571063 | Y | NASH | [87] | Decreased serum ALT | |
| Obeticholic acid (FXR1 ligand) | FLINT – Phase IIb, NCT01265498 | Y | NASH, NAFLD | [98] | Improved liver NAS in 45% of patients, elevated pruritis | |
| Obeticholic acid (FXR1 ligand) | REGENERATE – Phase III, NCT02548351 | Y | NASH | [100] | Improved fibrosis in 23% of patients, elevated pruritis | |
| NGM282 (FGF19 signalling) | Phase II, NCT02443116 | Y | NASH | [104] | Reduction in liver fat content | |
| ND-L02-s0201 (Vitamin A-coupled siRNA to HSP47 – hepatic stellate cell fibrosis target) | METAVIR F3-4 - Phase Ib/II, NCT02227459 | Y | Hepatic fibrosis | - | TBD | |
| Selonsertib (inhibitor of ASK1) | Multi-center Phase 2 | N | NASH | [111] | Improved liver fibrosis | |
| Selonsertib (inhibitor of ASK1) | STELLAR 3 and 4 - Phase 3, NCT03053050 and NCT03053063 | Y | NASH | [112] | No improvement in fibrosis, trial terminated | |
| Rifaximin (antibiotic) | Phase I, NCT02884037 | Y | NAFLD, NASH | [123] | Reduction in proinflammatory cytokines, liver enzymes and NAFLD-liver fat score; improved insulin sensitivity (HOMA-IR) | |
| Rifaximin (antibiotic) | Phase II, EudraCT 2010–021515-17 | N | NASH | [124] | Trial prematurely ended - no improvement | |
| VSL#3 (Probiotic) | VAIIO – Phase II, NCT01650025 | Y | Obesity | [133] | Significant improvement in NAFLD |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smeuninx, B.; Boslem, E.; Febbraio, M.A. Current and Future Treatments in the Fight against Non-Alcoholic Fatty Liver Disease. Cancers 2020, 12, 1714. https://doi.org/10.3390/cancers12071714
Smeuninx B, Boslem E, Febbraio MA. Current and Future Treatments in the Fight against Non-Alcoholic Fatty Liver Disease. Cancers. 2020; 12(7):1714. https://doi.org/10.3390/cancers12071714
Chicago/Turabian StyleSmeuninx, Benoit, Ebru Boslem, and Mark A. Febbraio. 2020. "Current and Future Treatments in the Fight against Non-Alcoholic Fatty Liver Disease" Cancers 12, no. 7: 1714. https://doi.org/10.3390/cancers12071714
APA StyleSmeuninx, B., Boslem, E., & Febbraio, M. A. (2020). Current and Future Treatments in the Fight against Non-Alcoholic Fatty Liver Disease. Cancers, 12(7), 1714. https://doi.org/10.3390/cancers12071714