Acquisition of Cisplatin Resistance Shifts Head and Neck Squamous Cell Carcinoma Metabolism toward Neutralization of Oxidative Stress

 ,
, 
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Background
2. Methods
2.1. Cells
2.2. Drug Effect Studies
2.3. RNA Sequencing and Analysis
2.4. Metabolomic Profiling by Targeted MS
2.5. Metabolite and RNA-Seq Data Integration
2.6. HNSCC Tumors
2.7. Statistical Analysis
3. Results
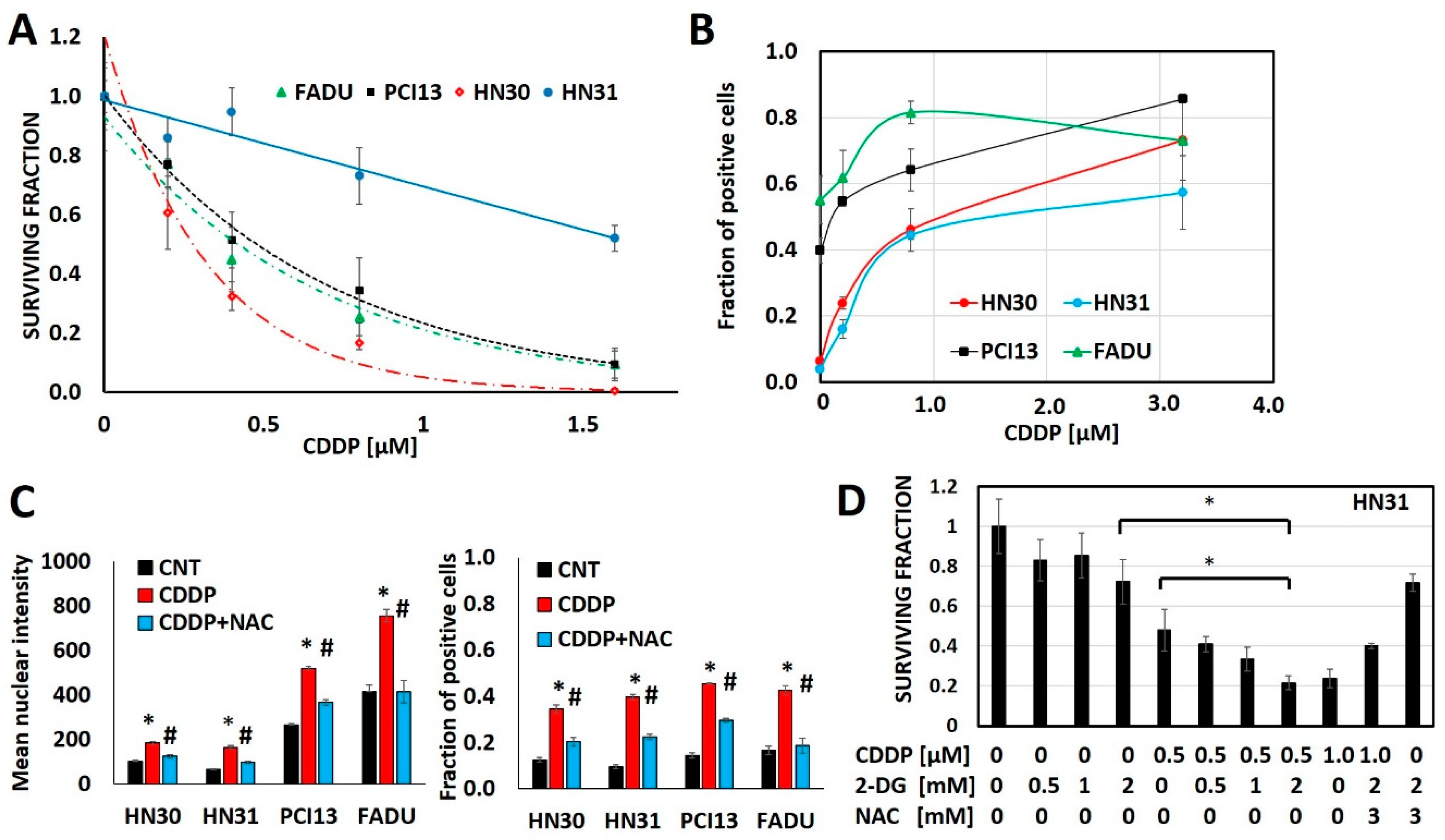
3.1. Cisplatin Anti-Tumor Effects are Linked to Generation of Oxidative Stress
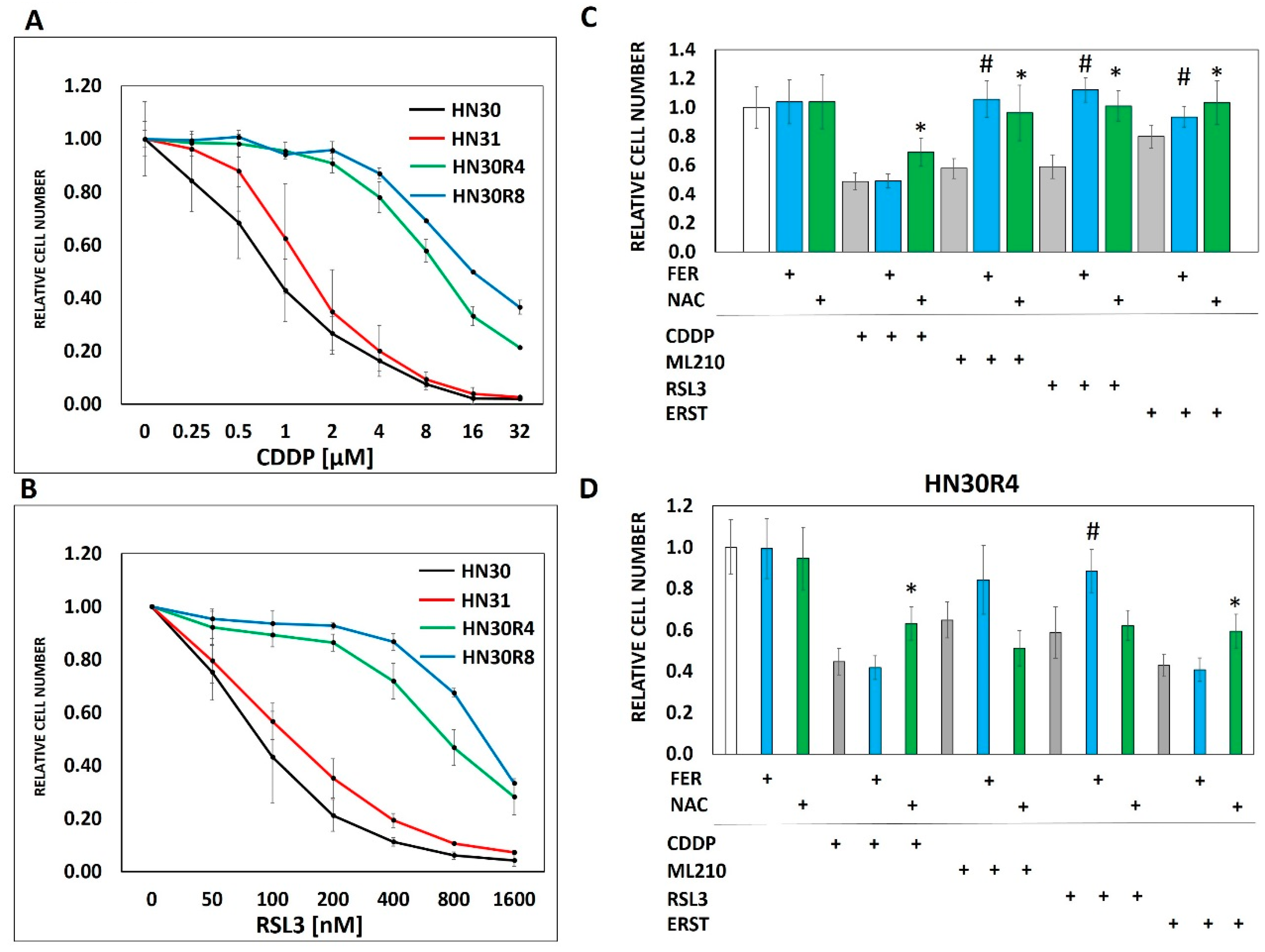
3.2. Ferroptosis Cross-Resistance Indicates Reprogramming of the Oxidative Stress Response
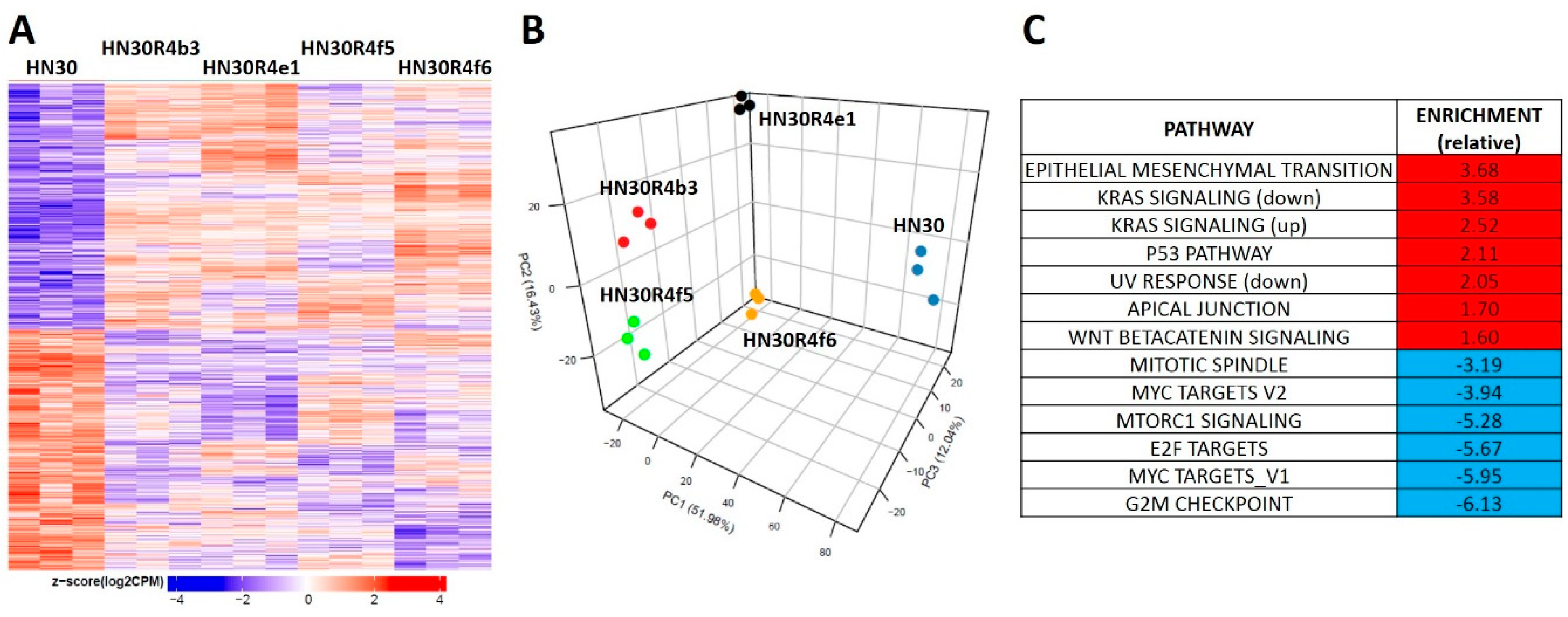
3.3. Acquisition of Cisplatin Resistance in HNSCC is Accompanied by Dysregulation of Stress Response Pathways
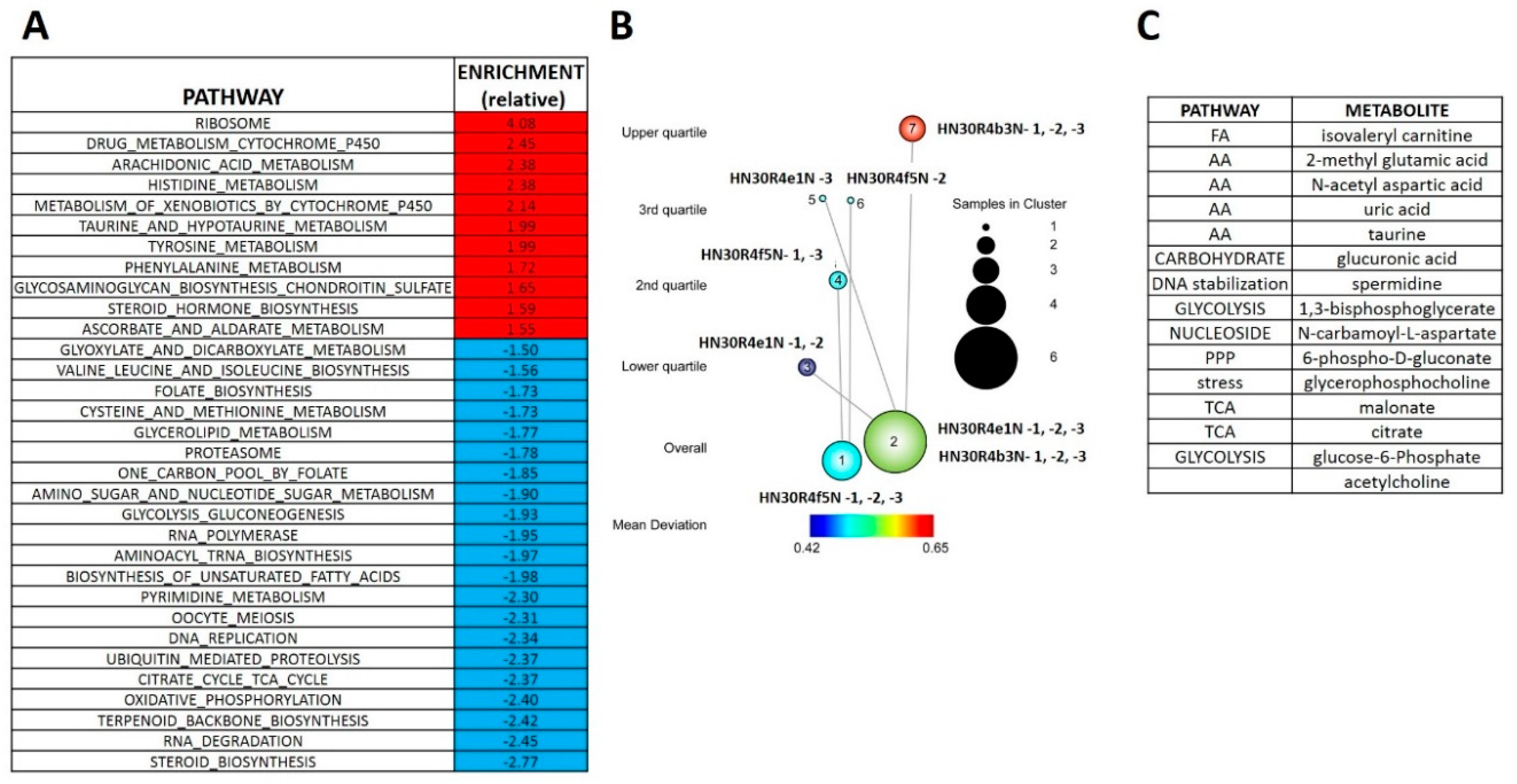
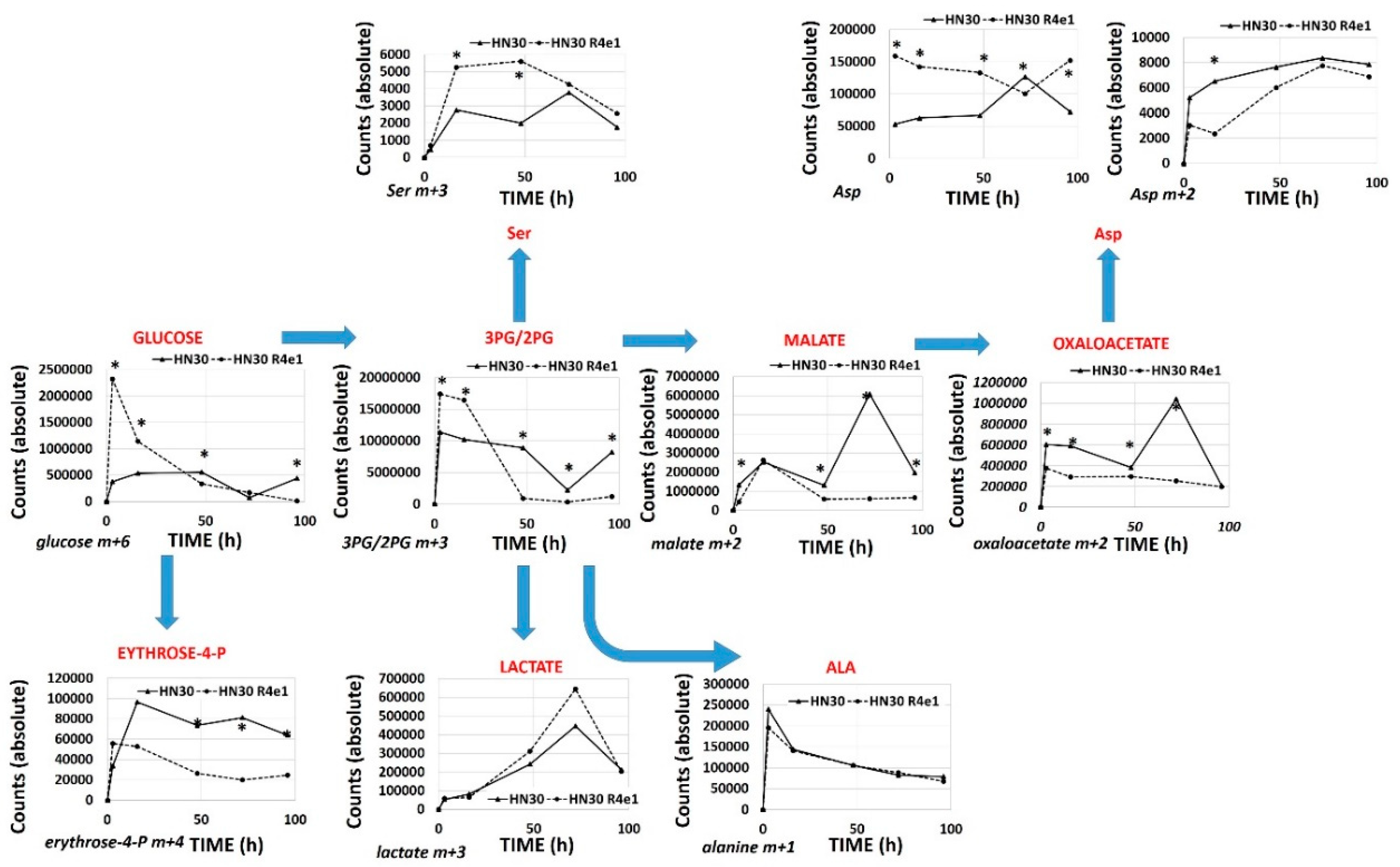
3.4. Acquisition of Cisplatin Resistance Triggers an Enhanced Catabolic Phenotype Supported by Changes in Metabolic Gene Expression
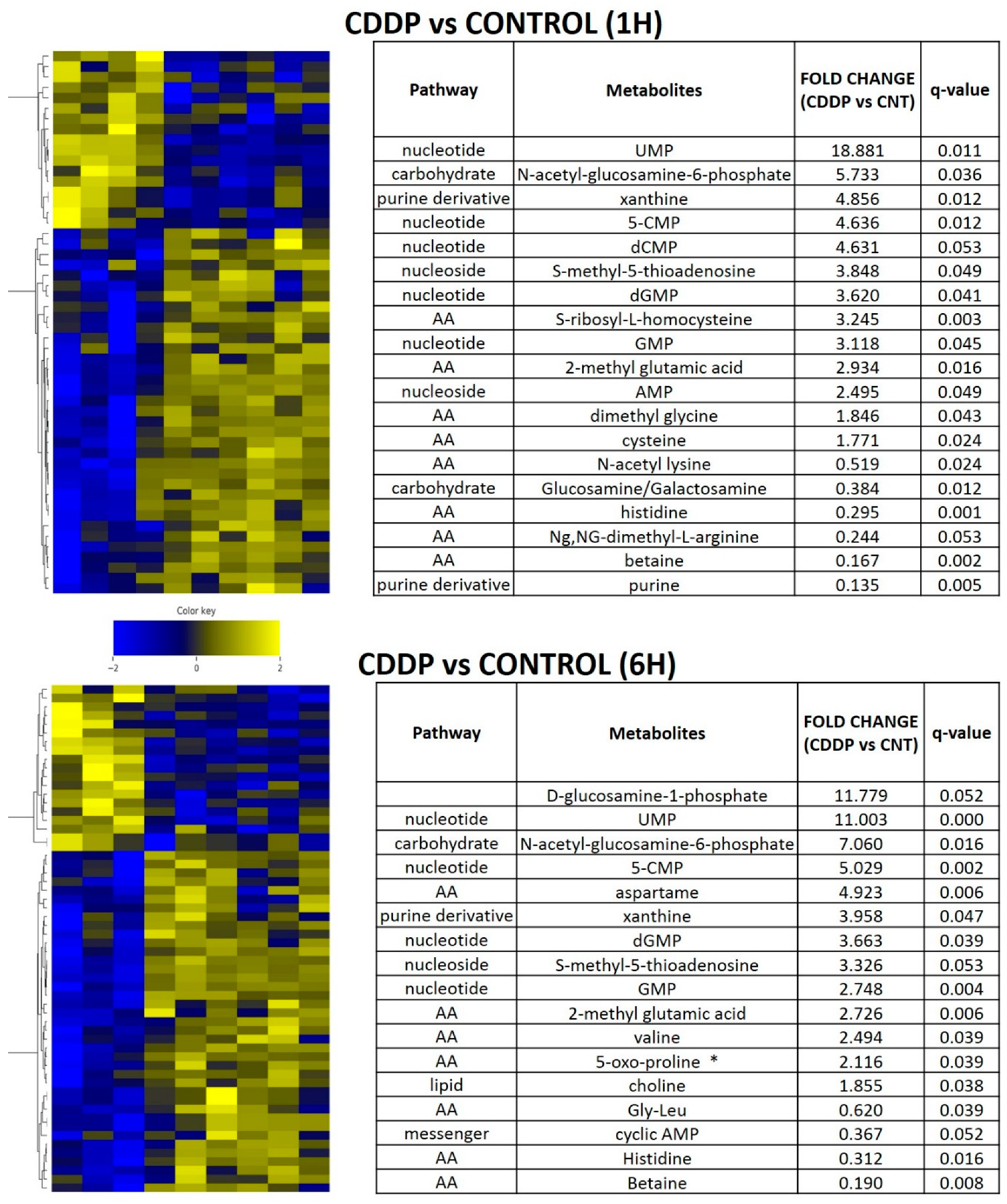
3.5. Acute Cisplatin Exposure Generates Shifts Consistent with a Metabolic Stress Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ahmed, Z.; Deyama, Y.; Yoshimura, Y.; Suzuki, K. Cisplatin sensitivity of oral squamous carcinoma cells is regulated by Na+,K+-ATPase activity rather than copper-transporting P-type ATPases, ATP7A and ATP7B. Cancer Chemother. Pharmacol. 2008, 63, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Bradford, C.R.; Zhu, S.; Ogawa, H.; Ogawa, T.; Ubell, M.; Narayan, A.; Johnson, G.; Wolf, G.T.; Fisher, S.G.; Carey, T.E. P53 mutation correlates with cisplatin sensitivity in head and neck squamous cell carcinoma lines. Head Neck 2003, 25, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Trotti, A.M.; Harris, J.; Eisbruch, A.; Harari, P.M.; Adelstein, D.J.; Jordan, R.C.K.; Zhao, W.; Sturgis, E.M.; Burtness, B.; et al. Radiotherapy plus cetuximab or cisplatin in human papillomavirus-positive oropharyngeal cancer (NRG Oncology RTOG 1016): A randomised, multicentre, non-inferiority trial. Lancet 2019, 393, 40–50. [Google Scholar] [CrossRef]
- Mattson, D.M.; Ahmad, I.; Dayal, D.; Parsons, A.D.; Aykin-Burns, N.; Li, L.; Orcutt, K.P.; Spitz, D.R.; Dornfeld, K.J.; Simons, A.L. Cisplatin combined with zidovudine enhances cytotoxicity and oxidative stress in human head and neck cancer cells via a thiol-dependent mechanism. Free Radic. Biol. Med. 2009, 46, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Robson, H.; Meyer, S.; Shalet, S.; Anderson, E.; Roberts, S.; Eden, O. Platinum agents in the treatment of osteosarcoma: Efficacy of cisplatin vs. carboplatin in human osteosarcoma cell lines. Med. Pediatr. Oncol. 2002, 39, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Sedletska, Y.; Giraud-Panis, M.-J.; Malinge, J.-M. Cisplatin is a DNA-damaging antitumour compound triggering multifactorial biochemical responses in cancer cells: Importance of apoptotic pathways. Curr. Med. Chem. Agents 2005, 5, 251–265. [Google Scholar] [CrossRef]
- Bankson, J.A.; Walker, C.; Ramirez, M.S.; Stefan, W.; Fuentes, D.; Merritt, M.E.; Lee, J.; Sandulache, V.C.; Chen, Y.; Phan, L.; et al. Kinetic Modeling and Constrained Reconstruction of Hyperpolarized [1-13C]-Pyruvate Offers Improved Metabolic Imaging of Tumors. Cancer Res. 2015, 75, 4708–4717. [Google Scholar] [CrossRef]
- Sandulache, V.C.; Chen, Y.; Feng, L.; William, W.N.; Skinner, H.D.; Myers, J.N.; Meyn, R.E.; Li, J.; Mijiti, A.; Bankson, J.A.; et al. Metabolic interrogation as a tool to optimize chemotherapeutic regimens. Oncotarget 2017, 8, 18154–18165. [Google Scholar] [CrossRef]
- Sandulache, V.C.; Chen, Y.; Lee, J.; Rubinstein, A.; Ramirez, M.S.; Skinner, H.D.; Walker, C.; Williams, M.D.; Tailor, R.; Court, L.E.; et al. Evaluation of Hyperpolarized [1-13C]-Pyruvate by Magnetic Resonance to Detect Ionizing Radiation Effects in Real Time. PLoS ONE 2014, 9, e87031. [Google Scholar] [CrossRef]
- Sandulache, V.C.; Chen, Y.; Skinner, H.D.; Lu, T.; Feng, L.; Court, L.E.; Myers, J.N.; Meyn, R.E.; Fuller, C.D.; Bankson, J.A.; et al. Acute Tumor Lactate Perturbations as a Biomarker of Genotoxic Stress: Development of a Biochemical Model. Mol. Cancer Ther. 2015, 14, 2901–2908. [Google Scholar] [CrossRef]
- Yu, W.; Chen, Y.; Dubrulle, J.; Stossi, F.; Putluri, V.; Sreekumar, A.; Putluri, N.; Baluya, D.; Lai, S.Y.; Sandulache, V.C. Cisplatin generates oxidative stress which is accompanied by rapid shifts in central carbon metabolism. Sci. Rep. 2018, 8, 4306. [Google Scholar] [CrossRef] [PubMed]
- Sandulache, V.C.; Ow, T.J.; Pickering, C.R.; Frederick, M.J.; Zhou, G.; Fokt, I.; Davis-Malesevich, M.; Priebe, W.; Myers, J.N. Glucose, not glutamine, is the dominant energy source required for proliferation and survival of head and neck squamous carcinoma cells. Cancer 2011, 117, 2926–2938. [Google Scholar] [CrossRef] [PubMed]
- Sandulache, V.C.; Skinner, H.D.; Ow, T.J.; Zhang, A.; Xia, X.; Luchak, J.M.; Wong, L.-J.C.; Pickering, C.R.; Zhou, G.; Myers, J.N. Individualizing antimetabolic treatment strategies for head and neck squamous cell carcinoma based on TP53 mutational status. Cancer 2011, 118, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Sandulache, V.C.; Skinner, H.D.; Wang, Y.; Chen, Y.; Dodge, C.T.; Ow, T.J.; Bankson, J.A.; Myers, J.N.; Lai, S.Y. Glycolytic inhibition alters anaplastic thyroid carcinoma tumor metabolism and improves response to conventional chemotherapy and radiation. Mol. Cancer Ther. 2012, 11, 1373–1380. [Google Scholar] [CrossRef]
- Skinner, H.D.; McCurdy, M.; Echeverria, A.E.; Lin, S.H.; Welsh, J.W.; O’Reilly, M.S.; Hofstetter, W.L.; Ajani, J.A.; Komaki, R.; Cox, J.D.; et al. Metformin use and improved response to therapy in esophageal adenocarcinoma. Acta Oncol. 2012, 52, 1002–1009. [Google Scholar] [CrossRef]
- Woo, S.H.; Sandulache, V.C.; Yang, L.; Skinner, H.D. Evaluating response to metformin/cisplatin combination in cancer cells via metabolic measurement and clonogenic survival. Methods Mol Biol. 2014, 1165, 11–18. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Laurent, K.; Giuliano, S.; Larbret, F.; Ponzio, G.; Gounon, P.; Le Marchand-Brustel, Y.; Giorgetti-Peraldi, S.; Cormont, M.; Bertolotto, C.; et al. Targeting Cancer Cell Metabolism: The Combination of Metformin and 2-Deoxyglucose Induces p53-Dependent Apoptosis in Prostate Cancer Cells. Cancer Res. 2010, 70, 2465–2475. [Google Scholar] [CrossRef]
- Damelin, L.H.; Jivan, R.; Veale, R.B.; Rousseau, A.L.; Mavri-Damelin, D. Metformin induces an intracellular reductive state that protects oesophageal squamous cell carcinoma cells against cisplatin but not copper-bis(thiosemicarbazones). BMC Cancer 2014, 14, 314. [Google Scholar] [CrossRef]
- Zhao, M.; Sano, D.; Pickering, C.R.; Jasser, S.A.; Henderson, Y.C.; Clayman, G.L.; Sturgis, E.M.; Ow, T.J.; Lotan, R.; Carey, T.E.; et al. Assembly and initial characterization of a panel of 85 genomically validated cell lines from diverse head and neck tumor sites. Clin. Cancer Res. 2011, 17, 7248–7264. [Google Scholar] [CrossRef]
- Rage, R.; Mitchen, J.; Wilding, G. DNA fluorometric assay in 96-well tissue culture plates using Hoechst 33258 after cell lysis by freezing in distilled water. Anal. Biochem. 1990, 191, 31–34. [Google Scholar] [CrossRef]
- Szafran, A.T.; Mancini, M. The myImageAnalysis Project: A Web-Based Application for High-Content Screening. ASSAY Drug Dev. Technol. 2014, 12, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.R.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef]
- Law, C.W.; Alhamdoosh, M.; Su, S.; Dong, X.; Tian, L.; Smyth, G.K.; Ritchie, M.E. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Res 2016, 5. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Jeitziner, R.; Carrière, M.; Rougemont, J.; Oudot, S.; Hess, K.; Brisken, C. Two-Tier Mapper, an unbiased topology-based clustering method for enhanced global gene expression analysis. Bioinformatics 2019, 35, 3339–3347. [Google Scholar] [CrossRef]
- Skinner, H.D.; Sandulache, V.C.; Ow, T.J.; Meyn, R.E.; Yordy, J.S.; Beadle, B.M.; Fitzgerald, A.L.; Giri, U.; Ang, K.K.; Myers, J.N. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin. Cancer Res. 2011, 18, 290–300. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Hirschhorn, T.; Stockwell, B.R. The development of the concept of ferroptosis. Free Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xu, B.; Han, Q.; Zhou, H.; Xia, Y.; Gong, C.; Dai, X.; Li, Z.; Wu, G. Ferroptosis: A Novel Anti-tumor Action for Cisplatin. Cancer Res. Treat. 2018, 50, 445–460. [Google Scholar] [CrossRef] [PubMed]
- Mehanna, H.; Robinson, M.; Hartley, A.; Kong, A.; Foran, B.; Fulton-Lieuw, T.; Dalby, M.; Mistry, P.; Sen, M.; O’Toole, L.; et al. Radiotherapy plus cisplatin or cetuximab in low-risk human papillomavirus-positive oropharyngeal cancer (De-ESCALaTE HPV): An open-label randomised controlled phase 3 trial. Lancet 2019, 393, 51–60. [Google Scholar] [CrossRef]
- Drayton, R.M.; Catto, J.W. Molecular mechanisms of cisplatin resistance in bladder cancer. Expert Rev. Anticancer. Ther. 2012, 12, 271–281. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2011, 31, 1869–1883. [Google Scholar] [CrossRef]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol. Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef]
- Piulats, J.M.; Jiménez, L.; Del Muro, X.G.; Villanueva, A.; Viñals, F.; Germà-Lluch, J.R. Molecular mechanisms behind the resistance of cisplatin in germ cell tumours. Clin. Transl. Oncol. 2009, 11, 780–786. [Google Scholar] [CrossRef]
- Siddik, Z.H. Biochemical and molecular mechanisms of cisplatin resistance. Cancer Treat Res. 2002, 112, 263–284. [Google Scholar] [CrossRef]
- Zhu, H.; Luo, H.; Zhang, W.; Shen, Z.; Hu, X.; Zhu, X. Molecular mechanisms of cisplatin resistance in cervical cancer. Drug Des. Dev. Ther. 2016, 10, 1885–1895. [Google Scholar] [CrossRef]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Zogg, C. Phosphoglycerate Dehydrogenase: Potential Therapeutic Target and Putative Metabolic Oncogene. J. Oncol. 2014, 2014, 524101. [Google Scholar] [CrossRef]
- Ichihara, A.; Greenberg, D.M. Pathway of serine formation from carbohydrate in rat liver. Proc. Natl. Acad. Sci. USA 1955, 41, 605–609. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Cantley, L.C. Cancer’s Fuel Choice: New Flavors for a Picky Eater. Mol. Cell 2015, 60, 514–523. [Google Scholar] [CrossRef]
- DeNicola, G.; Chen, P.-H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non–small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, H.; Birsoy, K.; Sethumadhavan, S.; Woo, H.-K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef]
- Lenis, Y.Y.; Elmetwally, M.; Maldonado-Estrada, J.G.; Bazer, F.W. Physiological importance of polyamines. Zygote 2017, 25, 244–255. [Google Scholar] [CrossRef]
- Casero, R.A.; Marton, L.J. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 2007, 6, 373–390. [Google Scholar] [CrossRef]
- Casero, R.A., Jr.; Stewart, T.M.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.K.; Dwarakanath, B.S.; Varshney, R. Radiosensitization by 2-deoxy-D-glucose and 6-aminonicotinamide involves activation of redox sensitive ASK1-JNK/p38MAPK signaling in head and neck cancer cells. Free Radic. Biol. Med. 2012, 53, 1500–1513. [Google Scholar] [CrossRef]
- Simons, A.L.; Ahmad, I.; Mattson, D.M.; Dornfeld, K.J.; Spitz, D.R. 2-Deoxy-D-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res. 2007, 67, 3364–3370. [Google Scholar] [CrossRef] [PubMed]
- Simons, A.L.; Fath, M.A.; Mattson, D.M.; Smith, B.J.; Walsh, S.; Graham, M.M.; Hichwa, R.D.; Buatti, J.M.; Dornfeld, K.; Spitz, D.R. Enhanced Response of Human Head and Neck Cancer Xenograft Tumors to Cisplatin Combined With 2-Deoxy-d-Glucose Correlates With Increased 18F-FDG Uptake as Determined by PET Imaging. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 1222–1230. [Google Scholar] [CrossRef] [PubMed]
- Simons, A.L.; Parsons, A.D.; Foster, K.A.; Orcutt, K.P.; Fath, M.A.; Spitz, D.R. Inhibition of Glutathione and Thioredoxin Metabolism Enhances Sensitivity to Perifosine in Head and Neck Cancer Cells. J. Oncol. 2009, 2009, 519563. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Wang, J.; Zhao, M.; Xie, T.-X.; Tanaka, N.; Sano, D.; Patel, A.A.; Ward, A.M.; Sandulache, V.C.; Jasser, S.A.; et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol. Cell 2014, 54, 960–974. [Google Scholar] [CrossRef]
- Feng, R.N.; Niu, Y.C.; Sun, X.W.; Li, Q.; Zhao, C.; Wang, C.; Guo, F.C.; Sun, C.H.; Li, Y. Histidine supplementation improves insulin resistance through suppressed inflammation in obese women with the metabolic syndrome: A randomised controlled trial. Diabetologia 2013, 56, 985–994. [Google Scholar] [CrossRef]
- Watanabe, M.; Suliman, M.E.; Qureshi, A.R.; Garcia-Lopez, E.; Bárány, P.; Heimbürger, O.; Stenvinkel, P.; Lindholm, B. Consequences of low plasma histidine in chronic kidney disease patients: Associations with inflammation, oxidative stress, and mortality. Am. J. Clin. Nutr. 2008, 87, 1860–1866. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, W.; Chen, Y.; Putluri, N.; Coarfa, C.; Robertson, M.J.; Putluri, V.; Stossi, F.; Dubrulle, J.; Mancini, M.A.; Pang, J.C.; et al. Acquisition of Cisplatin Resistance Shifts Head and Neck Squamous Cell Carcinoma Metabolism toward Neutralization of Oxidative Stress. Cancers 2020, 12, 1670. https://doi.org/10.3390/cancers12061670
Yu W, Chen Y, Putluri N, Coarfa C, Robertson MJ, Putluri V, Stossi F, Dubrulle J, Mancini MA, Pang JC, et al. Acquisition of Cisplatin Resistance Shifts Head and Neck Squamous Cell Carcinoma Metabolism toward Neutralization of Oxidative Stress. Cancers. 2020; 12(6):1670. https://doi.org/10.3390/cancers12061670
Chicago/Turabian StyleYu, Wangjie, Yunyun Chen, Nagireddy Putluri, Cristian Coarfa, Matthew J. Robertson, Vasanta Putluri, Fabio Stossi, Julien Dubrulle, Michael A. Mancini, Jonathan C. Pang, and et al. 2020. "Acquisition of Cisplatin Resistance Shifts Head and Neck Squamous Cell Carcinoma Metabolism toward Neutralization of Oxidative Stress" Cancers 12, no. 6: 1670. https://doi.org/10.3390/cancers12061670
APA StyleYu, W., Chen, Y., Putluri, N., Coarfa, C., Robertson, M. J., Putluri, V., Stossi, F., Dubrulle, J., Mancini, M. A., Pang, J. C., Nguyen, T., Baluya, D., Myers, J. N., Lai, S. Y., & Sandulache, V. C. (2020). Acquisition of Cisplatin Resistance Shifts Head and Neck Squamous Cell Carcinoma Metabolism toward Neutralization of Oxidative Stress. Cancers, 12(6), 1670. https://doi.org/10.3390/cancers12061670