BRAF Mutant Melanoma Adjusts to BRAF/MEK Inhibitors via Dependence on Increased Antioxidant SOD2 and Increased Reactive Oxygen Species Levels

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
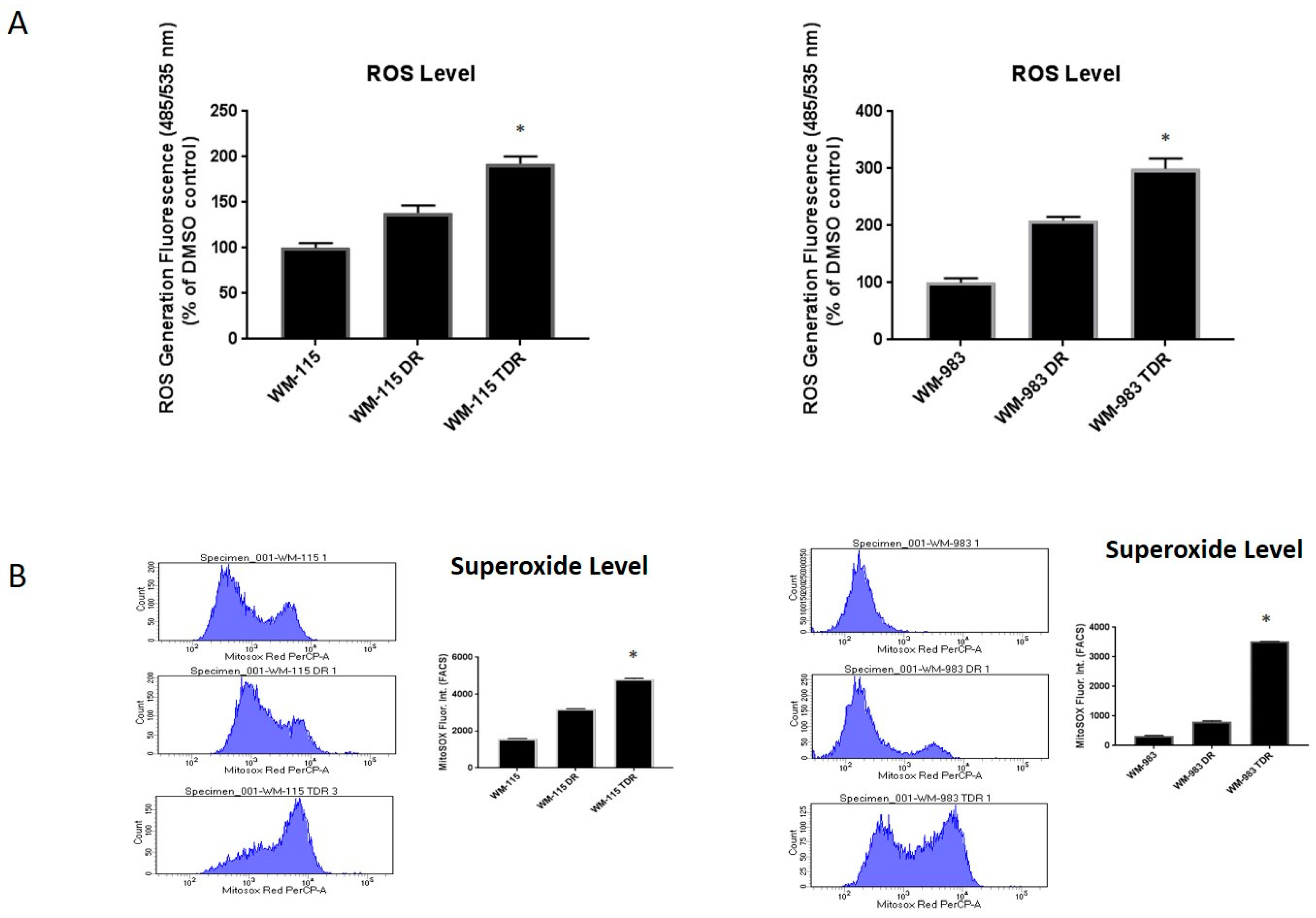
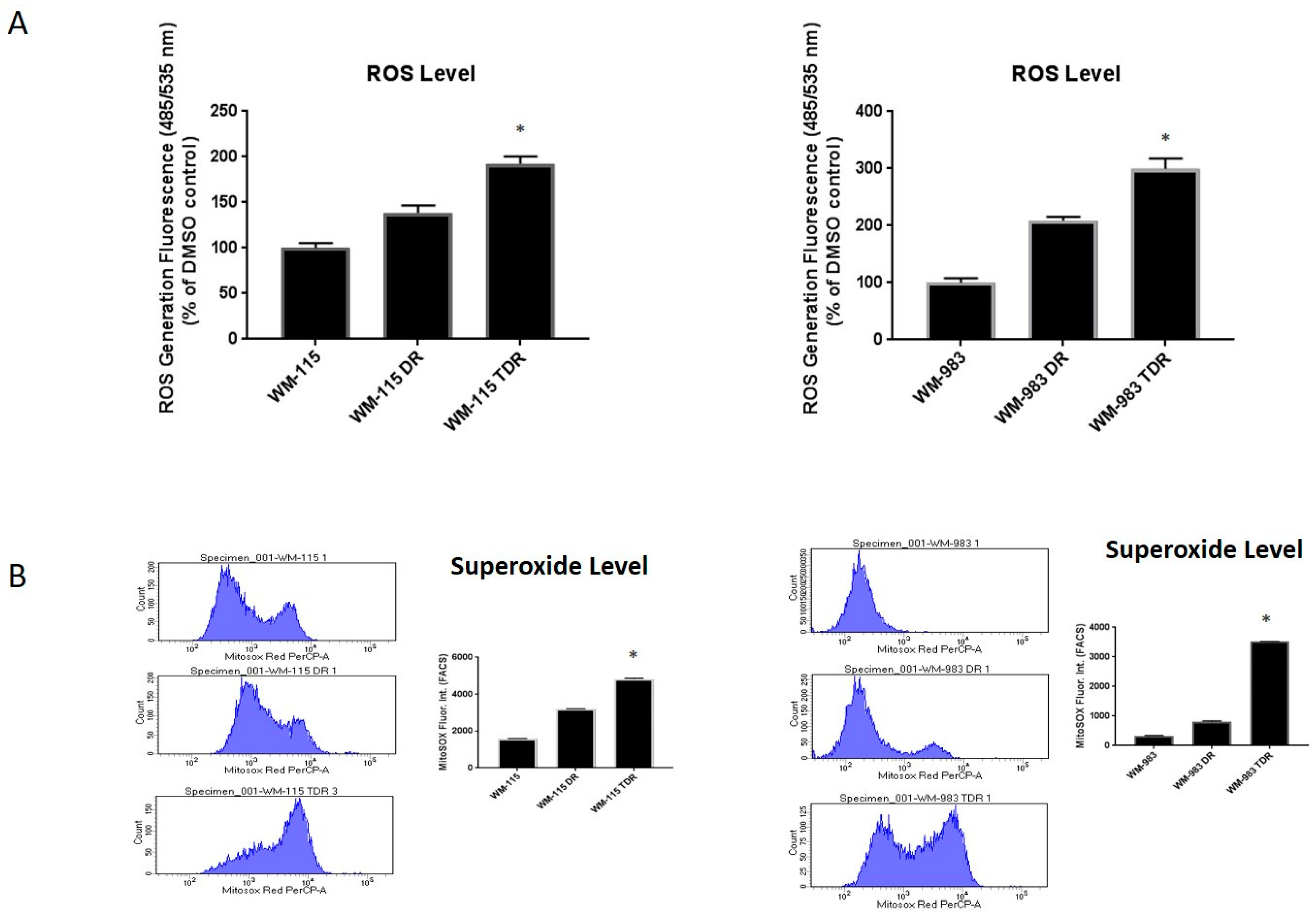
2.1. ROS Levels Are Increased in BRAF Mutant Melanoma in Response to Treatment with BRAF and MEK Inhibitors
2.2. ROS Level Is Upregulated upon Drug Resistance in BRAF Mutant Melanoma Cell Lines
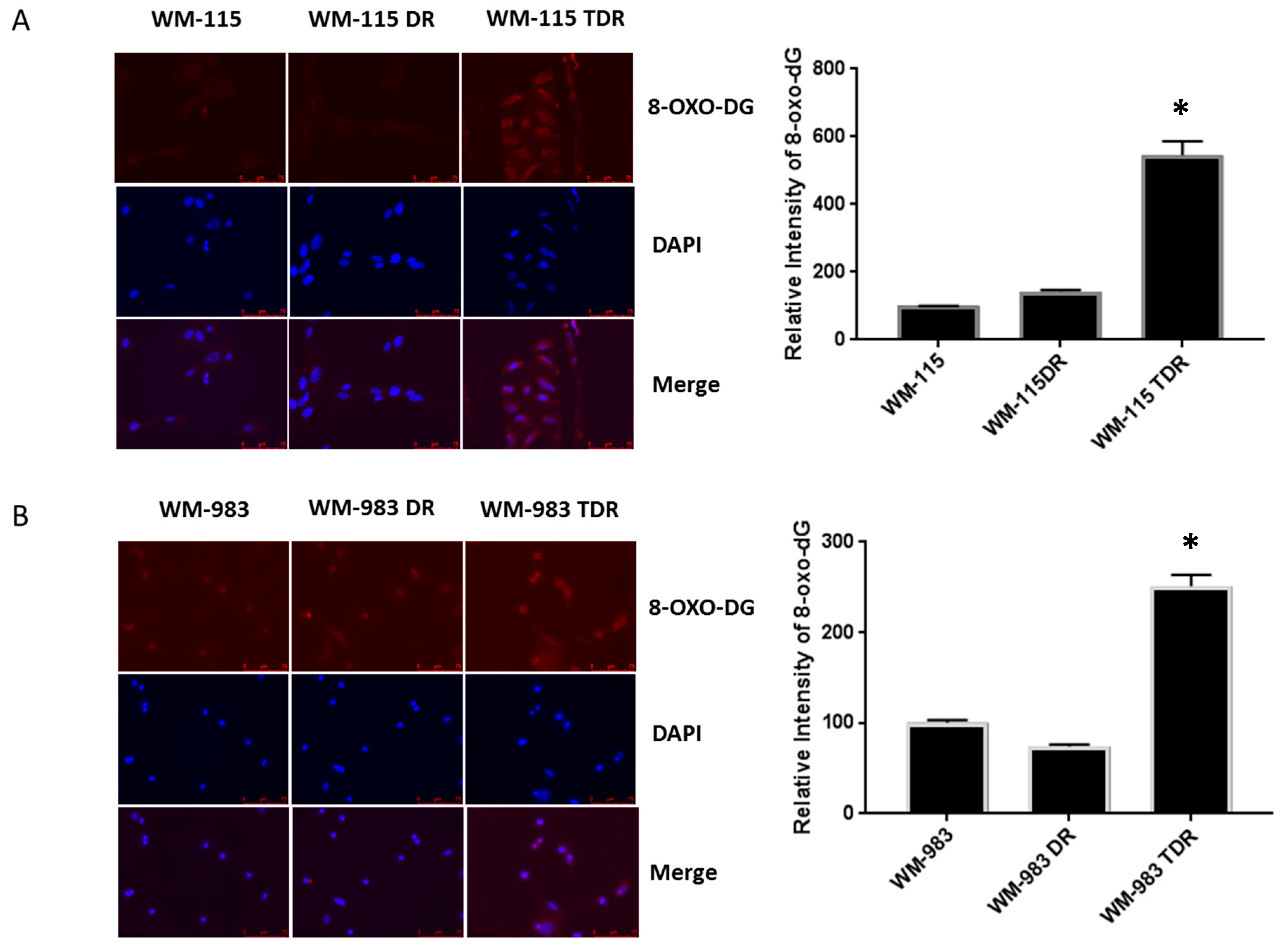
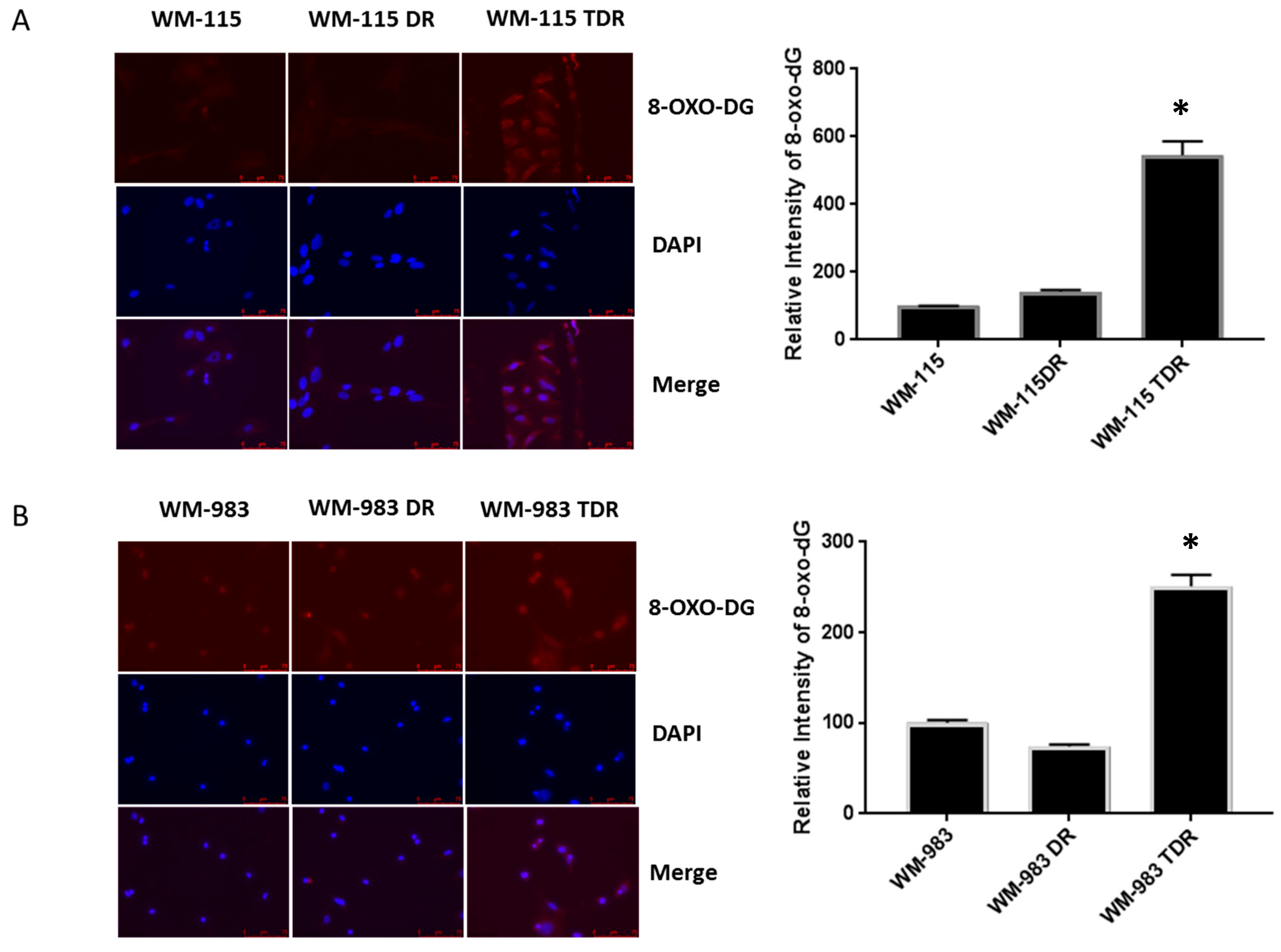
2.3. BRAF Mutant Melanoma Cells Resistant to BRAF and MEK Inhibition Show Increases in DNA Damage
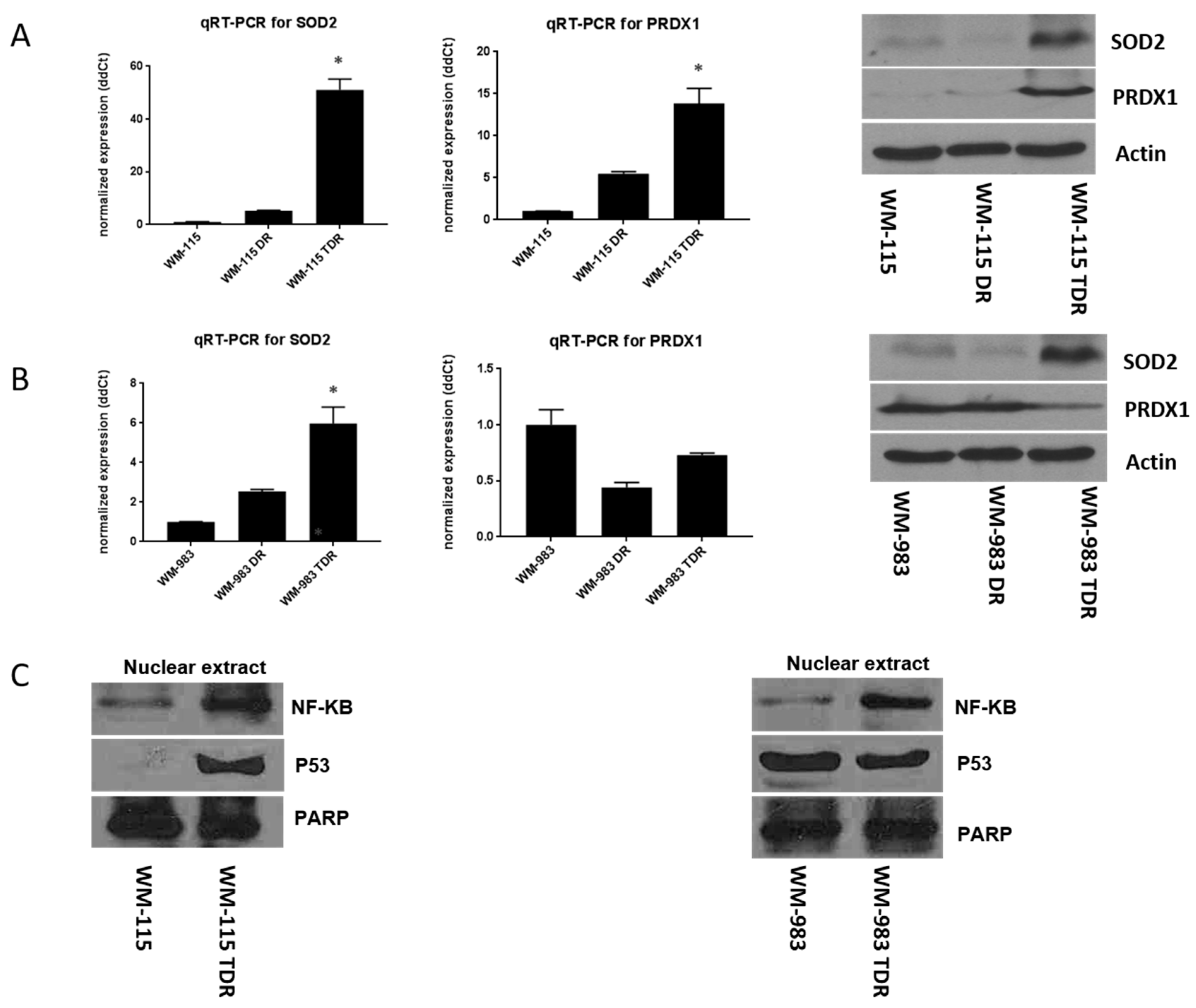
2.4. SOD2 and NF-κB Levels Are Increased in BRAF Pathway Inhibitor-Resistant Melanoma Cells
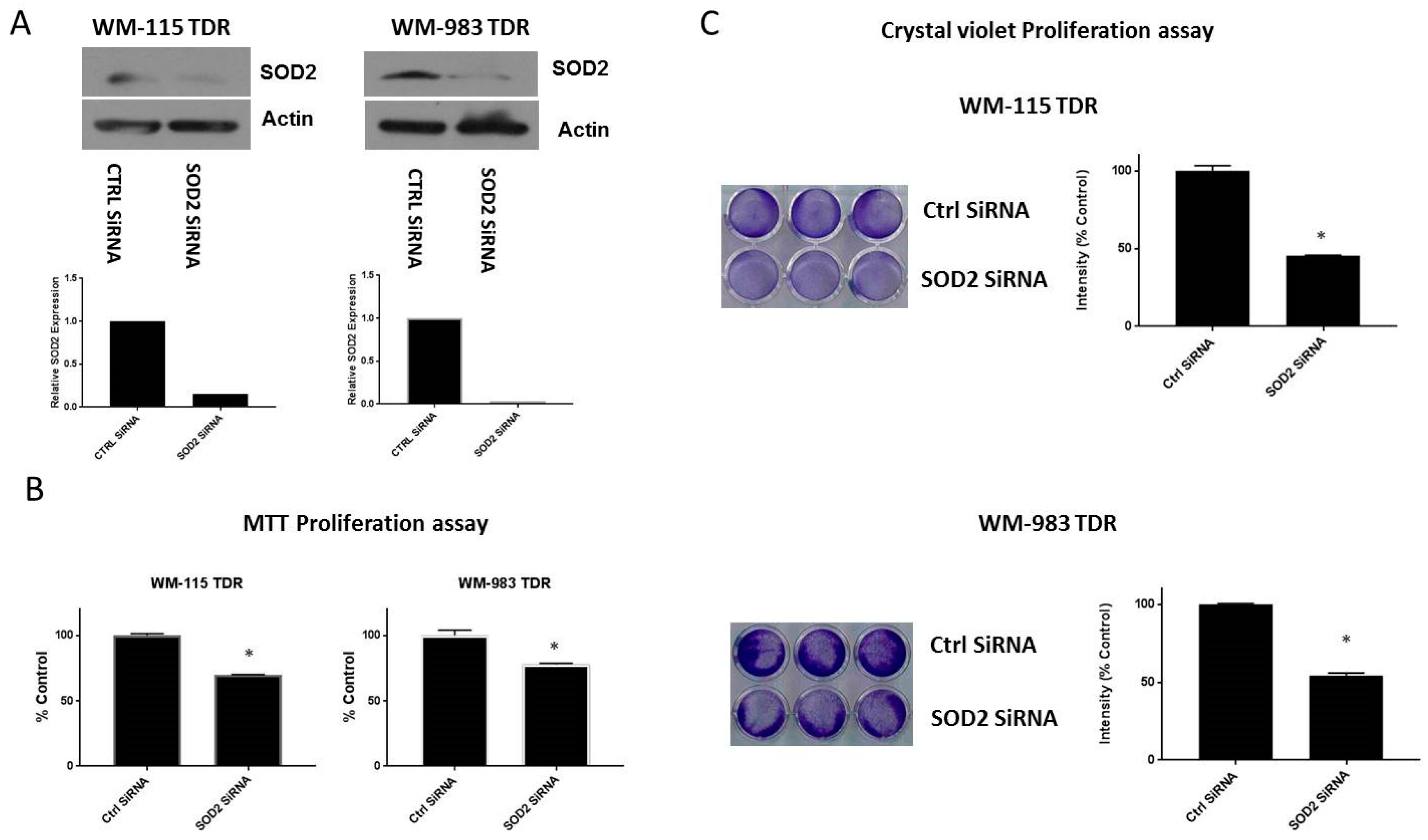
2.5. Knockdown of SOD2 Suppresses the Growth of BRAF Pathway Inhibitor Resistant Melanoma Cells
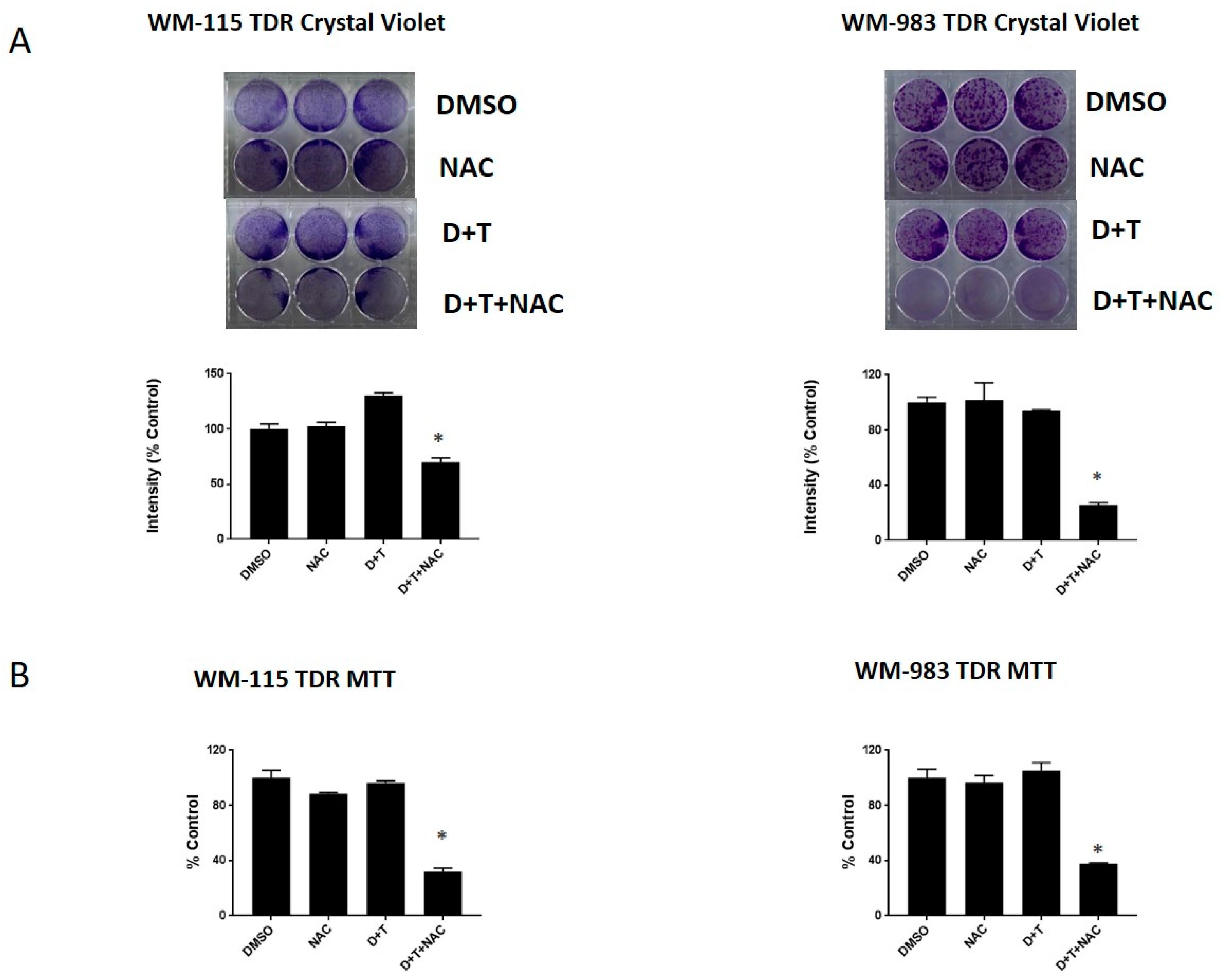
2.6. NAC, a ROS Scavenger, Can Re-Sensitize the BRAF Inhibitor in Melanoma Cells
3. Discussion
4. Material and Methods
4.1. Cell Culture and Inhibitors
4.2. Generation of Trametinib- and Dabrafenib-Resistant (TDR) Cell Lines
4.3. Immunoblotting
4.4. Transient Transfection
4.5. MTT Cell Proliferation Assay
4.6. Crystal Violet Cell Proliferation Assay
4.7. Measurement of Intracellular ROS
4.8. qPCR Analysis of Antioxidant mRNA
4.9. Immunofluorescence
4.10. Generation of Stable Reporter Cell Lines
4.11. In Vitro Luciferase Assays
4.12. In Vivo Xenograft Experiments
4.13. In Vivo Luciferase Experiments
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Yacoub, N.; Mishra, R.; White, A.; Long, Y.; Alanazi, S.; Garrett, J.T. Current Advances in the Treatment of BRAF-Mutant Melanoma. Cancers 2020, 12, 482. [Google Scholar] [CrossRef] [Green Version]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, X.; Gandhi, V. ROS-activated anticancer prodrugs: A new strategy for tumor-specific damage. Ther. Deliv. 2012, 3, 823–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Mishra, R.; Patel, H.; Abdulsalam, S.; Greis, K.D.; Kadekaro, A.L.; Merino, E.J.; Garrett, J.T. Utilization of Reactive Oxygen Species Targeted Therapy to Prolong the Efficacy of BRAF Inhibitors in Melanoma. J. Cancer 2018, 9, 4665–4676. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Joseph, S.; Khan, A.; Epstein, C.J.; Sobel, R.; Huang, T.T. Enhanced expression of mitochondrial superoxide dismutase leads to prolonged in vivo cell cycle progression and up-regulation of mitochondrial thioredoxin. Free Radic. Biol. Med. 2010, 48, 1501–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, X.; Ratti, B.A.; O’Brien, J.G.; Lautenschlager, S.O.; Gius, D.R.; Bonini, M.G.; Zhu, Y. Manganese superoxide dismutase (SOD2): Is there a center in the universe of mitochondrial redox signaling? J. Bioenerg. Biomembr. 2017, 49, 325–333. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Takaoka, A.; Yanai, H.; Kondo, S.; Duncan, G.; Negishi, H.; Mizutani, T.; Kano, S.; Honda, K.; Ohba, Y.; Mak, T.W.; et al. Integral role of IRF-5 in the gene induction programme activated by Toll-like receptors. Nature 2005, 434, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.P.; Roe, J.H.; Chock, P.B.; Yim, M.B. Transcriptional activation of the human manganese superoxide dismutase gene mediated by tetradecanoylphorbol acetate. J. Biol. Chem. 1999, 274, 37455–37460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storz, P.; Doppler, H.; Toker, A. Protein kinase D mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol. Cell Biol. 2005, 25, 8520–8530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.L.; Ping, D.; Boss, J.M. Tumor necrosis factor alpha and interleukin-1beta regulate the murine manganese superoxide dismutase gene through a complex intronic enhancer involving C/EBP-beta and NF-kappaB. Mol. Cell Biol. 1997, 17, 6970–6981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djavaheri-Mergny, M.; Javelaud, D.; Wietzerbin, J.; Besancon, F. NF-kappaB activation prevents apoptotic oxidative stress via an increase of both thioredoxin and MnSOD levels in TNFalpha-treated Ewing sarcoma cells. FEBS Lett. 2004, 578, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Das, K.C.; Lewis-Molock, Y.; White, C.W. Activation of NF-kappa B and elevation of MnSOD gene expression by thiol reducing agents in lung adenocarcinoma (A549) cells. Am. J. Physiol. 1995, 269, L588–L602. [Google Scholar] [CrossRef]
- Van de Bittner, G.C.; Bertozzi, C.R.; Chang, C.J. Strategy for dual-analyte luciferin imaging: In vivo bioluminescence detection of hydrogen peroxide and caspase activity in a murine model of acute inflammation. J. Am. Chem. Soc. 2013, 135, 1783–1795. [Google Scholar] [CrossRef] [Green Version]
- Patel, H.; Mishra, R.; Joan, G. Abstract 1284: Resistance to BRAF and MEK inhibitors in BRAFV600E mutant melanoma. Cancer Res. 2019, 79, 1284. [Google Scholar] [CrossRef]
- Liou, G.Y.; Storz, P. Detecting reactive oxygen species by immunohistochemistry. Met. Mol. Biol. 2015, 1292, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Floyd, R.A.; West, M.S.; Eneff, K.L.; Hogsett, W.E.; Tingey, D.T. Hydroxyl free radical mediated formation of 8-hydroxyguanine in isolated DNA. Arch. Biochem. Biophys. 1988, 262, 266–272. [Google Scholar] [CrossRef]
- Obrador, E.; Liu-Smith, F.; Dellinger, R.W.; Salvador, R.; Meyskens, F.L.; Estrela, J.M. Oxidative stress and antioxidants in the pathophysiology of malignant melanoma. Biol. Chem. 2019, 400, 589–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittgen, H.G.; van Kempen, L.C. Reactive oxygen species in melanoma and its therapeutic implications. Melanoma Res. 2007, 17, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Venza, M.; Visalli, M.; Beninati, C.; De Gaetano, G.V.; Teti, D.; Venza, I. Cellular Mechanisms of Oxidative Stress and Action in Melanoma. Oxid. Med. Cell Longev. 2015, 2015, 481782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candas, D.; Li, J.J. MnSOD in oxidative stress response-potential regulation via mitochondrial protein influx. Antioxid. Redox. Signal. 2014, 20, 1599–1617. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.M.; Wu, T.C.; Wang, Y.C.; Cheng, Y.W.; Sheu, G.T.; Chen, C.Y.; Lee, H. Activation of NF-kappaB by SOD2 promotes the aggressiveness of lung adenocarcinoma by modulating NKX2-1-mediated IKKbeta expression. Carcinogenesis 2013, 34, 2655–2663. [Google Scholar] [CrossRef] [Green Version]
- Hempel, N.; Ye, H.; Abessi, B.; Mian, B.; Melendez, J.A. Altered redox status accompanies progression to metastatic human bladder cancer. Free Radic. Biol. Med. 2009, 46, 42–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hempel, N.; Carrico, P.M.; Melendez, J.A. Manganese superoxide dismutase (Sod2) and redox-control of signaling events that drive metastasis. Anticancer. Agents Med. Chem. 2011, 11, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Kamarajugadda, S.; Cai, Q.; Chen, H.; Nayak, S.; Zhu, J.; He, M.; Jin, Y.; Zhang, Y.; Ai, L.; Martin, S.S.; et al. Manganese superoxide dismutase promotes anoikis resistance and tumor metastasis. Cell Death Dis. 2013, 4, e504. [Google Scholar] [CrossRef]
- Pani, G.; Colavitti, R.; Bedogni, B.; Fusco, S.; Ferraro, D.; Borrello, S.; Galeotti, T. Mitochondrial superoxide dismutase: A promising target for new anticancer therapies. Curr. Med. Chem. 2004, 11, 1299–1308. [Google Scholar] [CrossRef]
- Xian, D.; Lai, R.; Song, J.; Xiong, X.; Zhong, J. Emerging Perspective: Role of Increased ROS and Redox Imbalance in Skin Carcinogenesis. Oxid. Med. Cell Longev. 2019, 2019, 8127362. [Google Scholar] [CrossRef] [Green Version]
- Liu-Smith, F.; Dellinger, R.; Meyskens, F.L., Jr. Updates of reactive oxygen species in melanoma etiology and progression. Arch. Biochem. Biophys. 2014, 563, 51–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Yin, C.; Li, X.X.; Yang, X.Z.; Yang, Y.; Zhang, M.Y.; Wang, H.Y.; Zheng, X.F. Reduced SOD2 expression is associated with mortality of hepatocellular carcinoma patients in a mutant p53-dependent manner. Aging (Albany NY) 2016, 8, 1184–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Kairisalo, M.; Korhonen, L.; Blomgren, K.; Lindholm, D. X-linked inhibitor of apoptosis protein increases mitochondrial antioxidants through NF-kappaB activation. Biochem. Biophys. Res. Commun. 2007, 364, 138–144. [Google Scholar] [CrossRef]
- Kiningham, K.K.; St Clair, D.K. Overexpression of manganese superoxide dismutase selectively modulates the activity of Jun-associated transcription factors in fibrosarcoma cells. Cancer Res. 1997, 57, 5265–5271. [Google Scholar]
- Wan, X.S.; Devalaraja, M.N.; St Clair, D.K. Molecular structure and organization of the human manganese superoxide dismutase gene. DNA Cell Biol. 1994, 13, 1127–1136. [Google Scholar] [CrossRef]
- Xu, Y.; Kiningham, K.K.; Devalaraja, M.N.; Yeh, C.C.; Majima, H.; Kasarskis, E.J.; St Clair, D.K. An intronic NF-kappaB element is essential for induction of the human manganese superoxide dismutase gene by tumor necrosis factor-alpha and interleukin-1beta. DNA Cell Biol. 1999, 18, 709–722. [Google Scholar] [CrossRef]
- Das, K.C.; Lewis-Molock, Y.; White, C.W. Thiol modulation of TNF alpha and IL-1 induced MnSOD gene expression and activation of NF-kappa B. Mol. Cell Biochem. 1995, 148, 45–57. [Google Scholar] [CrossRef]
- Grdina, D.J.; Murley, J.S.; Kataoka, Y.; Calvin, D.P. Differential activation of nuclear transcription factor kappaB, gene expression, and proteins by amifostine’s free thiol in human microvascular endothelial and glioma cells. Semin. Radiat. Oncol. 2002, 12, 103–111. [Google Scholar] [CrossRef]
- Fan, M.; Ahmed, K.M.; Coleman, M.C.; Spitz, D.R.; Li, J.J. Nuclear factor-kappaB and manganese superoxide dismutase mediate adaptive radioresistance in low-dose irradiated mouse skin epithelial cells. Cancer Res. 2007, 67, 3220–3228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nantajit, D.; Fan, M.; Duru, N.; Wen, Y.; Reed, J.C.; Li, J.J. Cyclin B1/Cdk1 phosphorylation of mitochondrial p53 induces anti-apoptotic response. PLoS ONE 2010, 5, e12341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Liu, J.; Robbins, D.; Morris, K.; Sit, A.; Liu, Y.Y.; Zhao, Y. Mutant p53 exhibits trivial effects on mitochondrial functions which can be reactivated by ellipticine in lymphoma cells. Apoptosis 2011, 16, 301–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra215. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [Green Version]
- Herlyn, M.; Balaban, G.; Bennicelli, J.; Guerry, D.; Halaban, R.; Herlyn, D.; Elder, D.E.; Maul, G.G.; Steplewski, Z.; Nowell, P.C.; et al. Primary melanoma cells of the vertical growth phase: Similarities to metastatic cells. J. Natl. Cancer Inst. 1985, 74, 283–289. [Google Scholar]
- Hsu, M.-Y.; Elder, D.E.; Herlyn, M. Melanoma: The Wistar Melanoma (WM) Cell Lines. In Human Cell Culture: Cancer Cell Lines Part 1; Masters, J.R.W., Palsson, B., Eds.; Springer: Dordrecht, The Netherlands, 1999; pp. 259–274. [Google Scholar]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, L.; Mishra, R.; Patel, H.; Alanazi, S.; Wei, X.; Ma, Z.; Garrett, J.T. BRAF Mutant Melanoma Adjusts to BRAF/MEK Inhibitors via Dependence on Increased Antioxidant SOD2 and Increased Reactive Oxygen Species Levels. Cancers 2020, 12, 1661. https://doi.org/10.3390/cancers12061661
Yuan L, Mishra R, Patel H, Alanazi S, Wei X, Ma Z, Garrett JT. BRAF Mutant Melanoma Adjusts to BRAF/MEK Inhibitors via Dependence on Increased Antioxidant SOD2 and Increased Reactive Oxygen Species Levels. Cancers. 2020; 12(6):1661. https://doi.org/10.3390/cancers12061661
Chicago/Turabian StyleYuan, Long, Rosalin Mishra, Hima Patel, Samar Alanazi, Xin Wei, Zhijun Ma, and Joan T. Garrett. 2020. "BRAF Mutant Melanoma Adjusts to BRAF/MEK Inhibitors via Dependence on Increased Antioxidant SOD2 and Increased Reactive Oxygen Species Levels" Cancers 12, no. 6: 1661. https://doi.org/10.3390/cancers12061661
APA StyleYuan, L., Mishra, R., Patel, H., Alanazi, S., Wei, X., Ma, Z., & Garrett, J. T. (2020). BRAF Mutant Melanoma Adjusts to BRAF/MEK Inhibitors via Dependence on Increased Antioxidant SOD2 and Increased Reactive Oxygen Species Levels. Cancers, 12(6), 1661. https://doi.org/10.3390/cancers12061661