Bromodomain-Containing Protein BRD4 Is Hyperphosphorylated in Mitosis




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. BRD4 Is Hyperphosphorylated during Mitosis
2.2. CDK1 Is a Potential Kinase That Mediates BRD4 Hyperphosphorylation during Mitosis
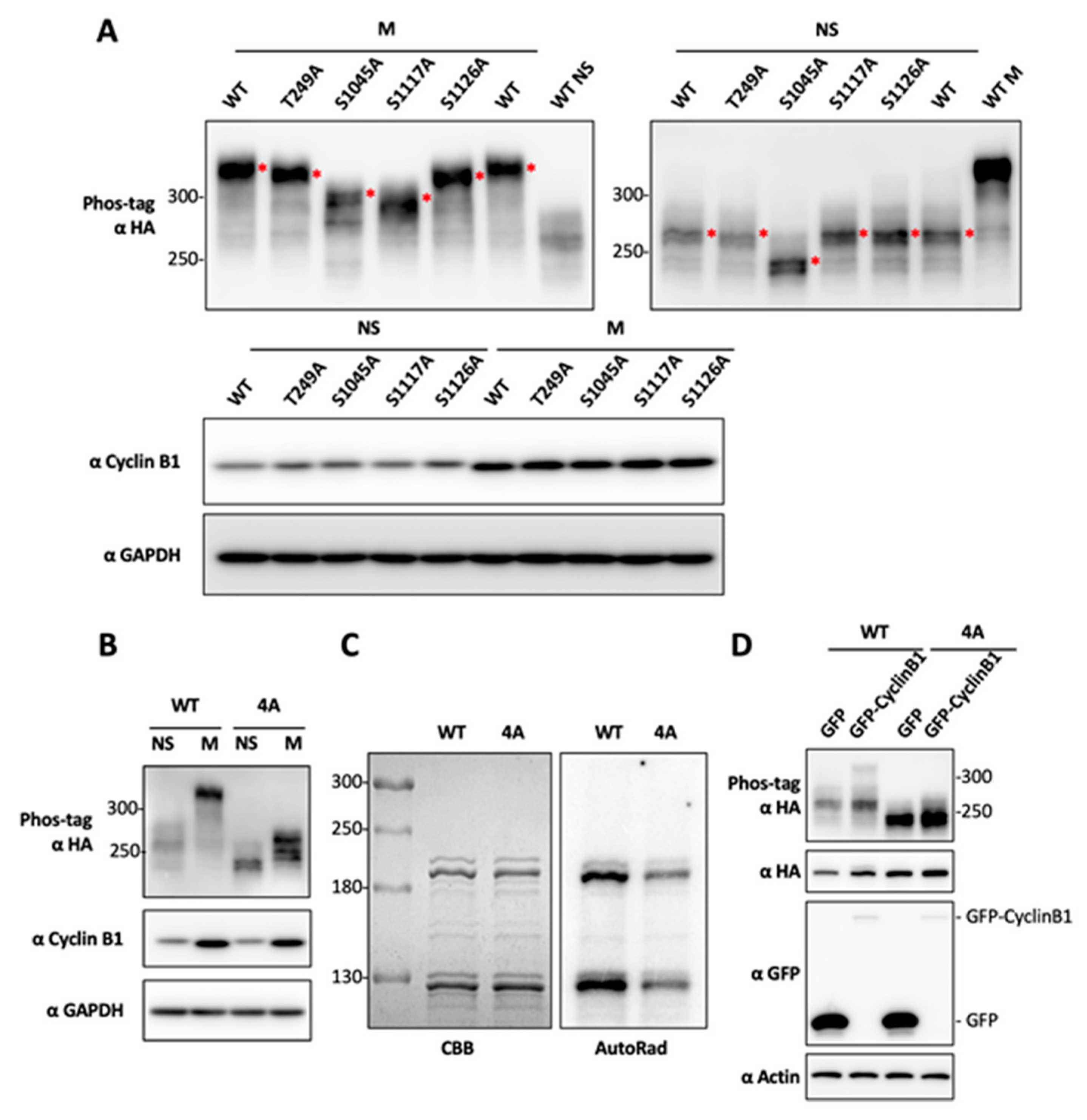
2.3. Determination of BRD4 Mitotic Phosphorylation Sites by Mutagenesis
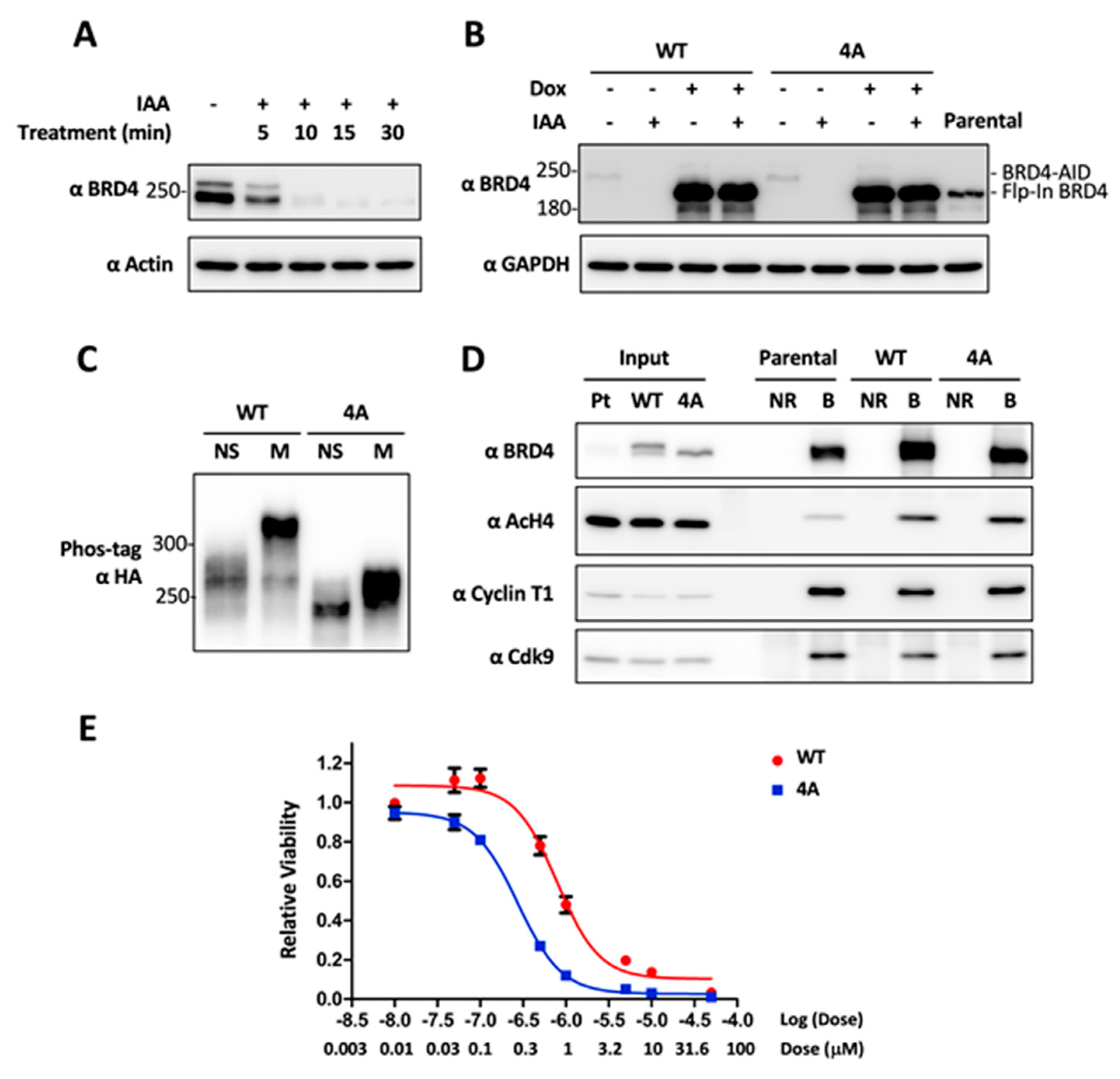
2.4. CDK1-Mediated BRD4 Hyperphosphorylation Contributes to BETi Resistance
2.5. Combinatory Inhibition of CDK1 and BRD4 Chromatin Binding Induces a Synergistic Antitumor Effect
3. Discussion
4. Materials and Methods
4.1. Recombinant Plasmid Constructs
4.2. Cell Culture and Transfection
4.3. Phos-tag Gel Technique
4.4. In Vitro Kinase Assay
4.5. Chemical Inhibitors
4.6. Western Blot Analyses
4.7. Generation of DLD-1 Cells with CRISPRed BRD4-AID and Flp-In BRD4 WT or 4A
4.8. Viability Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The double bromodomain protein brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8758–8763. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Chiang, C.M. The double bromodomain-containing chromatin adaptor brd4 and transcriptional regulation. J. Biol. Chem. 2007, 282, 13141–13145. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The brd4 extraterminal domain confers transcription activation independent of ptefb by recruiting multiple proteins, including nsd3. Mol. Cell Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef] [PubMed]
- Loven, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef]
- Mochizuki, K.; Nishiyama, A.; Jang, M.K.; Dey, A.; Ghosh, A.; Tamura, T.; Natsume, H.; Yao, H.; Ozato, K. The bromodomain protein brd4 stimulates g1 gene transcription and promotes progression to s phase. J. Biol. Chem. 2008, 283, 9040–9048. [Google Scholar] [CrossRef]
- Yang, Z.; He, N.; Zhou, Q. Brd4 recruits p-tefb to chromosomes at late mitosis to promote g1 gene expression and cell cycle progression. Mol. Cell Biol. 2008, 28, 967–976. [Google Scholar] [CrossRef]
- Zhao, R.; Nakamura, T.; Fu, Y.; Lazar, Z.; Spector, D.L. Gene bookmarking accelerates the kinetics of post-mitotic transcriptional re-activation. Nat. Cell Biol. 2011, 13, 1295–1304. [Google Scholar] [CrossRef]
- Dey, A.; Nishiyama, A.; Karpova, T.; McNally, J.; Ozato, K. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol. Biol. Cell 2009, 20, 4899–4909. [Google Scholar] [CrossRef]
- You, J.; Li, Q.; Wu, C.; Kim, J.; Ottinger, M.; Howley, P.M. Regulation of aurora b expression by the bromodomain protein brd4. Mol. Cell Biol. 2009, 29, 5094–5103. [Google Scholar] [CrossRef]
- Dey, A.; Ellenberg, J.; Farina, A.; Coleman, A.E.; Maruyama, T.; Sciortino, S.; Lippincott-Schwartz, J.; Ozato, K. A bromodomain protein, mcap, associates with mitotic chromosomes and affects g(2)-to-m transition. Mol. Cell Biol. 2000, 20, 6537–6549. [Google Scholar] [CrossRef]
- Maruyama, T.; Farina, A.; Dey, A.; Cheong, J.; Bermudez, V.P.; Tamura, T.; Sciortino, S.; Shuman, J.; Hurwitz, J.; Ozato, K. A mammalian bromodomain protein, brd4, interacts with replication factor c and inhibits progression to s phase. Mol. Cell Biol. 2002, 22, 6509–6520. [Google Scholar] [CrossRef] [PubMed]
- Houzelstein, D.; Bullock, S.L.; Lynch, D.E.; Grigorieva, E.F.; Wilson, V.A.; Beddington, R.S. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein brd4. Mol. Cell Biol. 2002, 22, 3794–3802. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Dey, A.; Miyazaki, J.; Ozato, K. Brd4 is required for recovery from antimicrotubule drug-induced mitotic arrest: Preservation of acetylated chromatin. Mol. Biol. Cell 2006, 17, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. Rnai screen identifies brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef]
- Rodriguez, R.M.; Huidobro, C.; Urdinguio, R.G.; Mangas, C.; Soldevilla, B.; Dominguez, G.; Bonilla, F.; Fernandez, A.F.; Fraga, M.F. Aberrant epigenetic regulation of bromodomain brd4 in human colon cancer. J. Mol. Med. (Berl.) 2012, 90, 587–595. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. Bet bromodomain inhibition as a therapeutic strategy to target c-myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J., 3rd. Targeting myc dependence in cancer by inhibiting bet bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef]
- Crawford, N.P.; Alsarraj, J.; Lukes, L.; Walker, R.C.; Officewala, J.S.; Yang, H.H.; Lee, M.P.; Ozato, K.; Hunter, K.W. Bromodomain 4 activation predicts breast cancer survival. Proc. Natl. Acad. Sci. USA 2008, 105, 6380–6385. [Google Scholar] [CrossRef]
- Venkataraman, S.; Alimova, I.; Balakrishnan, I.; Harris, P.; Birks, D.K.; Griesinger, A.; Amani, V.; Cristiano, B.; Remke, M.; Taylor, M.D.; et al. Inhibition of brd4 attenuates tumor cell self-renewal and suppresses stem cell signaling in myc driven medulloblastoma. Oncotarget 2014, 5, 2355–2371. [Google Scholar] [CrossRef]
- Pastori, C.; Daniel, M.; Penas, C.; Volmar, C.H.; Johnstone, A.L.; Brothers, S.P.; Graham, R.M.; Allen, B.; Sarkaria, J.N.; Komotar, R.J.; et al. Bet bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics 2014, 9, 611–620. [Google Scholar] [CrossRef]
- Wyce, A.; Ganji, G.; Smitheman, K.N.; Chung, C.W.; Korenchuk, S.; Bai, Y.; Barbash, O.; Le, B.; Craggs, P.D.; McCabe, M.T.; et al. Bet inhibition silences expression of mycn and bcl2 and induces cytotoxicity in neuroblastoma tumor models. PLoS ONE 2013, 8, e72967. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Frumm, S.M.; Alexe, G.; Bassil, C.F.; Qi, J.; Chanthery, Y.H.; Nekritz, E.A.; Zeid, R.; Gustafson, W.C.; Greninger, P.; et al. Targeting mycn in neuroblastoma by bet bromodomain inhibition. Cancer Discov. 2013, 3, 308–323. [Google Scholar] [CrossRef]
- Cheng, Z.; Gong, Y.; Ma, Y.; Lu, K.; Lu, X.; Pierce, L.A.; Thompson, R.C.; Muller, S.; Knapp, S.; Wang, J. Inhibition of bet bromodomain targets genetically diverse glioblastoma. Clin. Cancer Res. 2013, 19, 1748–1759. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of hedgehog pathway transcriptional output through bet bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; McKeown, M.R.; Lin, C.Y.; Monti, S.; Roemer, M.G.; Qi, J.; Rahl, P.B.; Sun, H.H.; Yeda, K.T.; Doench, J.G.; et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large b cell lymphoma. Cancer Cell 2013, 24, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Goundiam, O.; Gestraud, P.; Popova, T.; De la Motte Rouge, T.; Fourchotte, V.; Gentien, D.; Hupe, P.; Becette, V.; Houdayer, C.; Roman-Roman, S.; et al. Histo-genomic stratification reveals the frequent amplification/overexpression of ccne1 and brd4 genes in non-brcaness high grade ovarian carcinoma. Int. J. Cancer 2015, 137, 1890–1900. [Google Scholar] [CrossRef]
- French, C.A. Pathogenesis of nut midline carcinoma. Annu. Rev. Pathol. 2012, 7, 247–265. [Google Scholar] [CrossRef]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of bet recruitment to chromatin as an effective treatment for mll-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef]
- Grayson, A.R.; Walsh, E.M.; Cameron, M.J.; Godec, J.; Ashworth, T.; Ambrose, J.M.; Aserlind, A.B.; Wang, H.; Evan, G.I.; Kluk, M.J.; et al. Myc, a downstream target of brd-nut, is necessary and sufficient for the blockade of differentiation in nut midline carcinoma. Oncogene 2014, 33, 1736–1742. [Google Scholar] [CrossRef]
- Shi, J.; Vakoc, C.R. The mechanisms behind the therapeutic activity of bet bromodomain inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of bet bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Mirguet, O.; Gosmini, R.; Toum, J.; Clement, C.A.; Barnathan, M.; Brusq, J.M.; Mordaunt, J.E.; Grimes, R.M.; Crowe, M.; Pineau, O.; et al. Discovery of epigenetic regulator i-bet762: Lead optimization to afford a clinical candidate inhibitor of the bet bromodomains. J. Med. Chem. 2013, 56, 7501–7515. [Google Scholar] [CrossRef] [PubMed]
- Berthon, C.; Raffoux, E.; Thomas, X.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Roumier, C.; Herait, P.; et al. Bromodomain inhibitor otx015 in patients with acute leukaemia: A dose-escalation, phase 1 study. Lancet Haematol. 2016, 3, e186–e195. [Google Scholar] [CrossRef]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. Bet inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Lin, C.Y.; He, H.H.; Witwicki, R.M.; Tabassum, D.P.; Roberts, J.M.; Janiszewska, M.; Huh, S.J.; Liang, Y.; Ryan, J.; et al. Response and resistance to bet bromodomain inhibitors in triple-negative breast cancer. Nature 2016, 529, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Rathert, P.; Roth, M.; Neumann, T.; Muerdter, F.; Roe, J.S.; Muhar, M.; Deswal, S.; Cerny-Reiterer, S.; Peter, B.; Jude, J.; et al. Transcriptional plasticity promotes primary and acquired resistance to bet inhibition. Nature 2015, 525, 543–547. [Google Scholar] [CrossRef]
- Kumar, K.; Raza, S.S.; Knab, L.M.; Chow, C.R.; Kwok, B.; Bentrem, D.J.; Popovic, R.; Ebine, K.; Licht, J.D.; Munshi, H.G. Gli2-dependent c-myc upregulation mediates resistance of pancreatic cancer cells to the bet bromodomain inhibitor jq1. Sci. Rep. 2015, 5, 9489. [Google Scholar] [CrossRef]
- Liu, W.; Stein, P.; Cheng, X.; Yang, W.; Shao, N.Y.; Morrisey, E.E.; Schultz, R.M.; You, J. Brd4 regulates nanog expression in mouse embryonic stem cells and preimplantation embryos. Cell Death Differ. 2014, 21, 1950–1960. [Google Scholar] [CrossRef]
- Wu, T.; Pinto, H.B.; Kamikawa, Y.F.; Donohoe, M.E. The bet family member brd4 interacts with oct4 and regulates pluripotency gene expression. Stem Cell Rep. 2015, 4, 390–403. [Google Scholar] [CrossRef]
- Di Micco, R.; Fontanals-Cirera, B.; Low, V.; Ntziachristos, P.; Stephanie, K.Y.; Lovell, C.D.; Dolgalev, I.; Yonekubo, Y.; Zhang, G.; Rusinova, E.; et al. Control of embryonic stem cell identity by brd4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep. 2014, 9, 234–247. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Dawn, S.N.; Lee, A.Y.; Simanski, S.; Kodadek, T.; Chiang, C.-M. Brd4 phosphorylation regulates hpv e2-mediated viral transcription, origin replication, and cellular mmp-9 expression. Cell Rep. 2016, 16, 1733–1748. [Google Scholar] [CrossRef] [PubMed]
- Andrieu, A.G.; Belkina, A.C.; Denis, G.V. Clinical trials for bet inhibitors run ahead of the science. Drug Discov. Today Technol. 2016, 19, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Cao, X.J.; Kulej, K.; Liu, W.; Ma, T.; MacDonald, M.; Chiang, C.M.; Garcia, B.A.; You, J. Uncovering brd4 hyperphosphorylation associated with cellular transformation in nut midline carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, E5352–E5361. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; You, J. Mechanistic analysis of the role of bromodomain-containing protein 4 (brd4) in brd4-nut oncoprotein-induced transcriptional activation. J. Biol. Chem. 2015, 290, 2744–2758. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Liu, W.; Helfer, C.M.; Bradner, J.E.; Hornick, J.L.; Janicki, S.M.; French, C.A.; You, J. Activation of sox2 expression by brd4-nut oncogenic fusion drives neoplastic transformation in nut midline carcinoma. Cancer Res. 2014, 74, 3332–3343. [Google Scholar] [CrossRef]
- Yan, J.; Diaz, J.; Jiao, J.; Wang, R.; You, J. Perturbation of brd4 protein function by brd4-nut protein abrogates cellular differentiation in nut midline carcinoma. J. Biol. Chem. 2011, 286, 27663–27675. [Google Scholar] [CrossRef]
- Valent, P.; Zuber, J. Brd4: A bet(ter) target for the treatment of aml? Cell Cycle 2014, 13, 689–690. [Google Scholar] [CrossRef][Green Version]
- Fiskus, W.; Sharma, S.; Qi, J.; Valenta, J.A.; Schaub, L.J.; Shah, B.; Peth, K.; Portier, B.P.; Rodriguez, M.; Devaraj, S.G.; et al. Highly active combination of brd4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Mol. Cancer Ther. 2014, 13, 1142–1154. [Google Scholar] [CrossRef]
- Dawson, M.A.; Gudgin, E.J.; Horton, S.J.; Giotopoulos, G.; Meduri, E.; Robson, S.; Cannizzaro, E.; Osaki, H.; Wiese, M.; Putwain, S.; et al. Recurrent mutations, including npm1c, activate a brd4-dependent core transcriptional program in acute myeloid leukemia. Leukemia 2014, 28, 311–320. [Google Scholar] [CrossRef]
- Herrmann, H.; Blatt, K.; Shi, J.; Gleixner, K.V.; Cerny-Reiterer, S.; Mullauer, L.; Vakoc, C.R.; Sperr, W.R.; Horny, H.P.; Bradner, J.E.; et al. Small-molecule inhibition of brd4 as a new potent approach to eliminate leukemic stem- and progenitor cells in acute myeloid leukemia aml. Oncotarget 2012, 3, 1588–1599. [Google Scholar] [CrossRef]
- Knoechel, B.; Roderick, J.E.; Williamson, K.E.; Zhu, J.; Lohr, J.G.; Cotton, M.J.; Gillespie, S.M.; Fernandez, D.; Ku, M.; Wang, H.; et al. An epigenetic mechanism of resistance to targeted therapy in t cell acute lymphoblastic leukemia. Nat. Genet. 2014, 46, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Roderick, J.E.; Tesell, J.; Shultz, L.D.; Brehm, M.A.; Greiner, D.L.; Harris, M.H.; Silverman, L.B.; Sallan, S.E.; Gutierrez, A.; Look, A.T.; et al. C-myc inhibition prevents leukemia initiation in mice and impairs the growth of relapsed and induction failure pediatric t-all cells. Blood 2014, 123, 1040–1050. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.E.; Rix, U.; Carlson, S.M.; Gleixner, K.V.; Grebien, F.; Gridling, M.; Muller, A.C.; Breitwieser, F.P.; Bilban, M.; Colinge, J.; et al. Systems-pharmacology dissection of a drug synergy in imatinib-resistant cml. Nat. Chem. Biol. 2012, 8, 905–912. [Google Scholar] [CrossRef][Green Version]
- Kinoshita, E.; Kinoshita-Kikuta, E.; Koike, T. Separation and detection of large phosphoproteins using phos-tag sds-page. Nat. Protoc. 2009, 4, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Chen, J.G.; Horwitz, S.B. Differential mitotic responses to microtubule-stabilizing and -destabilizing drugs. Cancer Res. 2002, 62, 1935–1938. [Google Scholar]
- Ngan, V.K.; Bellman, K.; Hill, B.T.; Wilson, L.; Jordan, M.A. Mechanism of mitotic block and inhibition of cell proliferation by the semisynthetic vinca alkaloids vinorelbine and its newer derivative vinflunine. Mol. Pharmacol. 2001, 60, 225–232. [Google Scholar] [CrossRef]
- Azarenko, O.; Smiyun, G.; Mah, J.; Wilson, L.; Jordan, M.A. Antiproliferative mechanism of action of the novel taxane cabazitaxel as compared with the parent compound docetaxel in mcf7 breast cancer cells. Mol. Cancer Ther. 2014, 13, 2092–2103. [Google Scholar] [CrossRef]
- Zieve, G.W.; Turnbull, D.; Mullins, J.M.; McIntosh, J.R. Production of large numbers of mitotic mammalian cells by use of the reversible microtubule inhibitor nocodazole. Nocodazole accumulated mitotic cells. Exp. Cell Res. 1980, 126, 397–405. [Google Scholar] [CrossRef]
- Potapova, T.A.; Daum, J.R.; Pittman, B.D.; Hudson, J.R.; Jones, T.N.; Satinover, D.L.; Stukenberg, P.T.; Gorbsky, G.J. The reversibility of mitotic exit in vertebrate cells. Nature 2006, 440, 954–958. [Google Scholar] [CrossRef]
- Obenauer, J.C.; Cantley, L.C.; Yaffe, M.B. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003, 31, 3635–3641. [Google Scholar] [CrossRef]
- Nishimura, K.; Fukagawa, T.; Takisawa, H.; Kakimoto, T.; Kanemaki, M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 2009, 6, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Fukagawa, T. An efficient method to generate conditional knockout cell lines for essential genes by combination of auxin-inducible degron tag and crispr/cas9. Chromosome Res. 2017, 25, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Natsume, T.; Kiyomitsu, T.; Saga, Y.; Kanemaki, M.T. Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donors. Cell Rep. 2016, 15, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Lambrus, B.G.; Moyer, T.C.; Holland, A.J. Applying the auxin-inducible degradation system for rapid protein depletion in mammalian cells. Methods Cell Biol. 2018, 144, 107–135. [Google Scholar] [PubMed]
- Wu, S.Y.; Lee, A.Y.; Lai, H.T.; Zhang, H.; Chiang, C.M. Phospho switch triggers brd4 chromatin binding and activator recruitment for gene-specific targeting. Mol. Cell 2013, 49, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Bednarek, K.; Kiwerska, K.; Szaumkessel, M.; Bodnar, M.; Kostrzewska-Poczekaj, M.; Marszalek, A.; Janiszewska, J.; Bartochowska, A.; Jackowska, J.; Wierzbicka, M.; et al. Recurrent cdk1 overexpression in laryngeal squamous cell carcinoma. Tumour Biol. 2016, 37, 11115–11126. [Google Scholar] [CrossRef]
- Hoffmann, T.K.; Trellakis, S.; Okulicz, K.; Schuler, P.; Greve, J.; Arnolds, J.; Bergmann, C.; Bas, M.; Lang, S.; Lehnerdt, G.; et al. Cyclin b1 expression and p53 status in squamous cell carcinomas of the head and neck. Anticancer Res. 2011, 31, 3151–3157. [Google Scholar]
- Salaun, P.; Rannou, Y.; Prigent, C. Cdk1, plks, auroras, and neks: The mitotic bodyguards. Adv. Exp. Med. Biol. 2008, 617, 41–56. [Google Scholar]
- Malumbres, M.; Barbacid, M. Cell cycle, cdks and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, D.; Kusdra, L.; Huskey, N.E.; Chandriani, S.; Lenburg, M.E.; Gonzalez-Angulo, A.M.; Creasman, K.J.; Bazarov, A.V.; Smyth, J.W.; Davis, S.E.; et al. Myc pathway activation in triple-negative breast cancer is synthetic lethal with cdk inhibition. J. Exp. Med. 2012, 209, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, A.; Mochizuki, K.; Mueller, F.; Karpova, T.; McNally, J.G.; Ozato, K. Intracellular delivery of acetyl-histone peptides inhibits native bromodomain-chromatin interactions and impairs mitotic progression. FEBS Lett. 2008, 582, 1501–1507. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Misra, R.N.; Xiao, H.; Rawlins, D.B.; Shan, W.; Kellar, K.A.; Mulheron, J.G.; Sack, J.S.; Tokarski, J.S.; Kimball, S.D.; Webster, K.R. 1h-pyrazolo[3,4-b]pyridine inhibitors of cyclin-dependent kinases: Highly potent 2,6-difluorophenacyl analogues. Bioorg. Med. Chem. Lett. 2003, 13, 2405–2408. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Tovar, C.; Chen, S.; Knezevic, D.; Zhao, X.; Sun, H.; Heimbrook, D.C.; Chen, L. Selective small-molecule inhibitor reveals critical mitotic functions of human cdk1. Proc. Natl. Acad. Sci. USA 2006, 103, 10660–10665. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Shafer, D.; Grant, S. Update on rational targeted therapy in aml. Blood Rev. 2016, 30, 275–283. [Google Scholar] [CrossRef]
- Wang, R.; Li, Q.; Helfer, C.M.; Jiao, J.; You, J. Bromodomain protein brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. J. Biol. Chem. 2012, 287, 10738–10752. [Google Scholar] [CrossRef]
- Liu, W.; Yang, R.; Payne, A.S.; Schowalter, R.M.; Spurgeon, M.E.; Lambert, P.F.; Xu, X.; Buck, C.B.; You, J. Identifying the target cells and mechanisms of merkel cell polyomavirus infection. Cell Host Microbe 2016, 19, 775–787. [Google Scholar] [CrossRef]
- Devaiah, B.N.; Lewis, B.A.; Cherman, N.; Hewitt, M.C.; Albrecht, B.K.; Robey, P.G.; Ozato, K.; Sims, R.J., 3rd; Singer, D.S. Brd4 is an atypical kinase that phosphorylates serine2 of the rna polymerase ii carboxy-terminal domain. Proc. Natl. Acad. Sci. USA 2012, 109, 6927–6932. [Google Scholar] [CrossRef]
- Fachinetti, D.; Logsdon, G.A.; Abdullah, A.; Selzer, E.B.; Cleveland, D.W.; Black, B.E. Cenp-a modifications on ser68 and lys124 are dispensable for establishment, maintenance, and long-term function of human centromeres. Dev. Cell 2017, 40, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by rna-guided crispr cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Holland, A.J.; Fachinetti, D.; Han, J.S.; Cleveland, D.W. Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc. Natl. Acad. Sci. USA 2012, 109, E3350–E3357. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.; Yang, J.F.; Ho, F.; Robertson, E.S.; You, J. Bromodomain-Containing Protein BRD4 Is Hyperphosphorylated in Mitosis. Cancers 2020, 12, 1637. https://doi.org/10.3390/cancers12061637
Wang R, Yang JF, Ho F, Robertson ES, You J. Bromodomain-Containing Protein BRD4 Is Hyperphosphorylated in Mitosis. Cancers. 2020; 12(6):1637. https://doi.org/10.3390/cancers12061637
Chicago/Turabian StyleWang, Ranran, June F. Yang, Flora Ho, Erle S. Robertson, and Jianxin You. 2020. "Bromodomain-Containing Protein BRD4 Is Hyperphosphorylated in Mitosis" Cancers 12, no. 6: 1637. https://doi.org/10.3390/cancers12061637
APA StyleWang, R., Yang, J. F., Ho, F., Robertson, E. S., & You, J. (2020). Bromodomain-Containing Protein BRD4 Is Hyperphosphorylated in Mitosis. Cancers, 12(6), 1637. https://doi.org/10.3390/cancers12061637