c-Src and EGFR Inhibition in Molecular Cancer Therapy: What Else Can We Improve?

 ,
,  , and
, and 
Abstract
1. Introduction
2. c-Src and EGFR Physical and Functional Interaction
3. c-Src and EGFR Activation and Cooperation in Cancer Onset and Maintenance
4. Current Status of c-Src Inhibitors and Their Effects in Drug Resistance
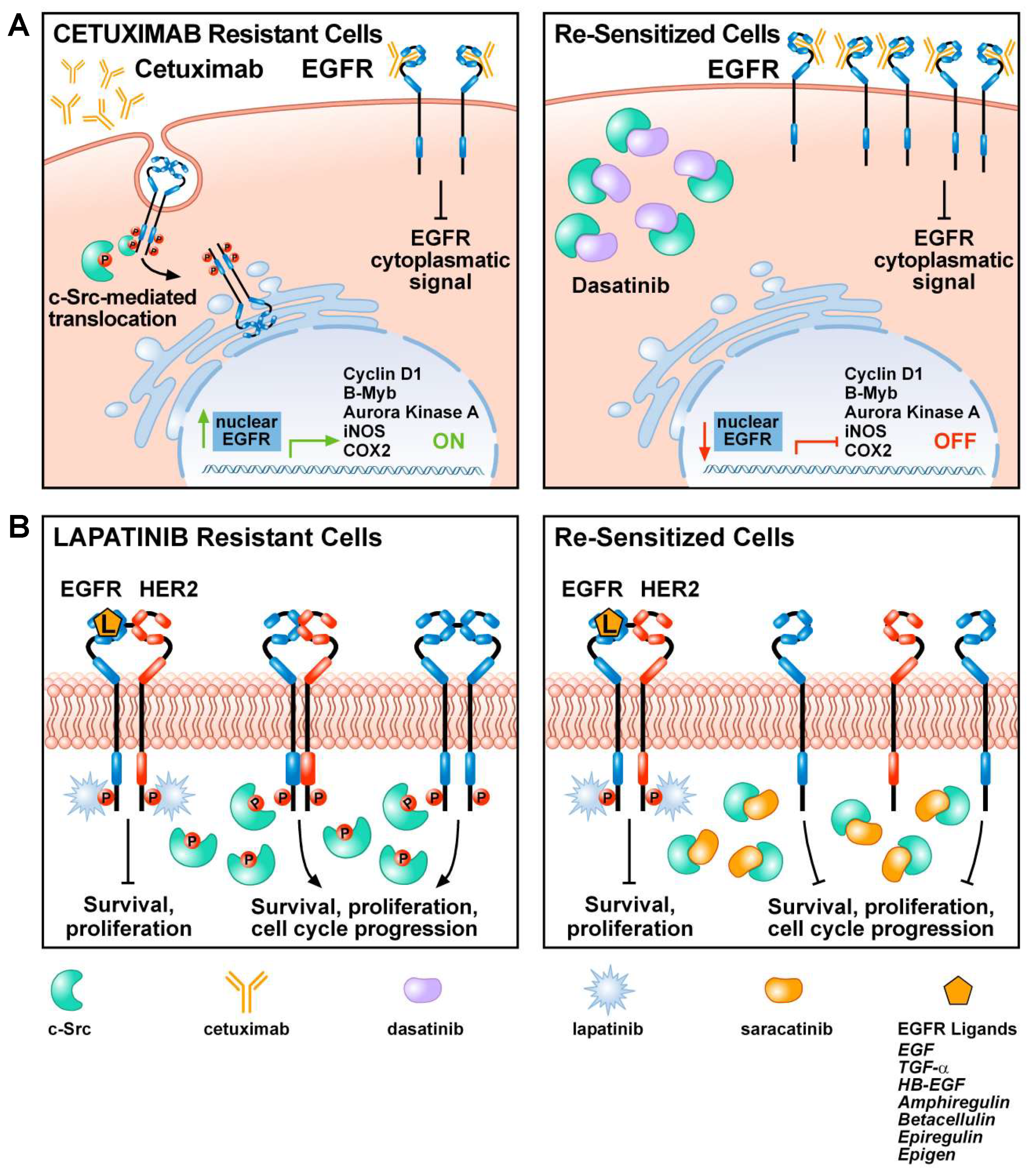
5. The Role of c-Src in Tumor Resistance to EGFR Inhibitors
6. Combined Therapy of c-Src and EGFR Inhibitors in Recent Clinical Trials
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Brown, M.T.; Cooper, J.A. Regulation, substrates and functions of src. Biochim. Biophys. Acta Rev. Cancer 1996. [Google Scholar] [CrossRef]
- Courtneidge, S.A. Isolation of novel Src substrates. Biochem. Soc. Trans. 2003. [Google Scholar] [CrossRef] [PubMed]
- Belsches, A.P.; Haskell, M.D.; Parsons, S.J. Role of c-Src tyrosine kinase in EGF-induced mitogenesis. Front. Biosci. J. Virtual Libr. 1997. [Google Scholar] [CrossRef]
- Roskoski, R. Src protein-tyrosine kinase structure and regulation. Biochem. Biophys. Res. Commun. 2004. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.T.; Weber, M.J. Genetics of src: Structure and functional organization of a protein tyrosine kinase. Curr. Top. Microbiol. Immunol. 1989. [Google Scholar] [CrossRef]
- Boggon, T.J.; Eck, M.J. Structure and regulation of Src family kinases. Oncogene 2004. [Google Scholar] [CrossRef]
- Cooper, J.A.; Gould, K.L.; Cartwright, C.A.; Hunter, T. Tyr527 is phosphorylated in pp60c-src: Implications for regulation. Science 1986. [Google Scholar] [CrossRef]
- Bjorge, J.D.; Pang, A.; Fujita, D.J. Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J. Biol. Chem. 2000. [Google Scholar] [CrossRef]
- Schaller, M.D.; Hildebrand, J.D.; Shannon, J.D.; Fox, J.W.; Vines, R.R.; Parsons, J.T. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol. Cell. Biol. 1994. [Google Scholar] [CrossRef]
- Thomas, J.W.; Ellis, B.; Boerner, R.J.; Knight, W.B.; White, G.C.; Schaller, M.D. SH2- and SH3-mediated interactions between focal adhesion kinase and Src. J. Biol. Chem. 1998. [Google Scholar] [CrossRef]
- Yeatman, T.J. A renaissance for SRC. Nat. Rev. Cancer 2004. [Google Scholar] [CrossRef] [PubMed]
- Irby, R.B.; Mao, W.; Coppola, D.; Kang, J.; Loubeau, J.M.; Trudeau, W.; Karl, R.; Fujita, D.J.; Jove, R.; Yeatman, T.J. Activating SRC mutation in a subset of advanced human colon cancers. Nat. Genet. 1999. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, M.; Kobayashi, K.; Sagae, S.; Nishioka, Y.; Ishioka, S.I.; Terasawa, K.; Tokino, T.; Kudo, R. Mutation of the SRC gene in endometrial carcinoma. Jpn. J. Cancer Res. 2000. [Google Scholar] [CrossRef] [PubMed]
- Biscardi, J.S.; Ishizawar, R.C.; Silva, C.M.; Parsons, S.J. Tyrosine kinase signalling in breast cancer: Epidermal growth factor receptor and c-Src interactions in breast cancer. Breast Cancer Res. 2000. [Google Scholar] [CrossRef] [PubMed]
- Irby, R.B.; Yeatman, T.J. Role of Src expression and activation in human cancer. Oncogene 2000. [Google Scholar] [CrossRef] [PubMed]
- Frame, M.C. Src in cancer: Deregulation and consequences for cell behaviour. Biochim. Biophys. Acta Rev. Cancer 2002. [Google Scholar] [CrossRef]
- Levin, V.A. Basis and importance of Src as a target in cancer. Cancer Treat. Res. 2004. [Google Scholar] [CrossRef]
- Tice, D.A.; Biscardi, J.S.; Nickles, A.L.; Parsons, S.J. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc. Natl. Acad. Sci. USA 1999. [Google Scholar] [CrossRef]
- Luttrell, D.K.; Lee, A.; Lansing, T.J.; Crosby, R.M.; Jung, K.D.; Willard, D.; Luther, M.; Rodriguez, M.; Berman, J.; Gilmer, T.M. Involvement of pp60(c-src) with two major signaling pathways in human breast cancer. Proc. Natl. Acad. Sci. USA 1994. [Google Scholar] [CrossRef]
- Mao, W.; Irby, R.; Coppola, D.; Fu, L.; Wloch, M.; Turner, J.; Yu, H.; Garcia, R.; Jove, R.; Yeatman, T.J. Activation of c-Src by receptor tyrosine kinases in human colon cancer cells with high metastatic potential. Oncogene 1997. [Google Scholar] [CrossRef]
- Courtneidge, S.A.; Fumagalli, S.; Koegl, M.; Superti-Furga, G.; Twamley-Stein, G.M. The Src family of protein tyrosine kinases: Regulation and functions. Dev. Suppl. 1993, 57–64. [Google Scholar]
- La Vallee, T.M.; Prudovsky, I.A.; McMahon, G.A.; Hu, X.; Maciag, T. Activation of the MAP kinase pathway by FGF-1 correlates with cell proliferation induction while activation of the Src pathway correlates with migration. J. Cell Biol. 1998. [Google Scholar] [CrossRef] [PubMed]
- Courtneidge, S.A.; Dhand, R.; Pilat, D.; Twamley, G.M.; Waterfield, M.D.; Roussel, M.F. Activation of Src family kinases by colony stimulating factor-1, and their association with its receptor. EMBO J. 1993. [Google Scholar] [CrossRef]
- Levitzki, A. SRC as a target for anti-cancer drugs. Anti Cancer Drug Des. 1996, 1, 175–182. [Google Scholar]
- Rahimi, N.; Hunglti, W.; Tremblay, E.; Saulnierl, R.; Elliott, B. c-Src kinase activity is required for hepatocyte growth factor-induced motility and anchorage-independent growth of mammary carcinoma cells. J. Biol. Chem. 1998. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, D.K.; Luttrell, L.M.; Parsons, S.J. Augmented mitogenic responsiveness to epidermal growth factor in murine fibroblasts that overexpress pp60c-src. Mol. Cell. Biol. 1988. [Google Scholar] [CrossRef]
- Maa, M.C.; Leu, T.H.; Mccarley, D.J.; Schatzman, R.C.; Parsons, S.J. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: Implications for the etiology of multiple human cancers. Proc. Natl. Acad. Sci. USA 1995. [Google Scholar] [CrossRef]
- Wasilenko, W.J.; Payne, D.M.; Fitzgerald, D.L.; Weber, M.J. Phosphorylation and activation of epidermal growth factor receptors in cells transformed by the src oncogene. Mol. Cell. Biol. 1991. [Google Scholar] [CrossRef]
- Sato, K.I.; Sato, A.; Aoto, M.; Fukami, Y. c-SRC phosphorylates epidermal growth factor receptor on tyrosine 845. Biochem. Biophys. Res. Commun. 1995. [Google Scholar] [CrossRef]
- Stover, D.R.; Becker, M.; Liebetanz, J.; Lydon, N.B. Src phosphorylation of the epidermal growth factor receptor at novel sites mediates receptor interaction with Src and P85α. J. Biol. Chem. 1995. [Google Scholar] [CrossRef]
- Biscardi, J.S.; Maa, M.C.; Tice, D.A.; Cox, M.E.; Leu, T.H.; Parsons, S.J. C-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J. Biol. Chem. 1999. [Google Scholar] [CrossRef] [PubMed]
- Sato, K. Cellular functions regulated by phosphorylation of EGFR on TYR845. Int. J. Mol. Sci. 2013, 14, 10761–10790. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Mounier, C.; Dumas, V.; Posner, B.I. Epidermal growth factor-induced DNA synthesis: Key role for Src phosphorylation of the docking protein Gab2. J. Biol. Chem. 2003. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Yu, Q.; Liu, J.H.; Zhang, J.; Wang, H.; Koul, D.; McMurray, J.S.; Fang, X.; Yung, W.K.; Siminovitch, K.A.; et al. Src family protein tyrosine kinases alter the function of PTEN to regulate PI3K/AKT cascades. J. Biol. Chem. 2003. [Google Scholar] [CrossRef]
- Franke, T.F.; Hornik, C.P.; Segev, L.; Shostak, G.A.; Sugimoto, C. PI3K/Akt and apoptosis: Size matters. Oncogene 2003. [Google Scholar] [CrossRef] [PubMed]
- Shien, T.; Doihara, H.; Hara, H.; Takahashi, H.; Yoshitomi, S.; Taira, N.; Ishibe, Y.; Teramoto, J.; Aoe, M.; Shimizu, N. PLC and PI3K pathways are important in the inhibition of EGF-induced cell migration by gefitinib (“Iressa”, ZD1839). Breast Cancer 2004. [Google Scholar] [CrossRef]
- Jiang, T.; Qiu, Y. Interaction between Src and a C-terminal proline-rich motif of Akt is required for Akt activation. J. Biol. Chem. 2003. [Google Scholar] [CrossRef]
- Mason, C.S. Serine and tyrosine phosphorylations cooperate in Raf-1, but not B-Raf activation. EMBO J. 1999. [Google Scholar] [CrossRef]
- Bivona, T.G.; Pérez de Castro, I.; Ahearn, I.M.; Grana, T.M.; Chiu, V.K.; Lockyer, P.J.; Cullen, P.J.; Pellicer, A.; Cox, A.D.; Philips, M.R. Phospholipase Cγ activates Ras on the Golgi apparatus by means of RasGRP1. Nature 2003. [Google Scholar] [CrossRef]
- Matsuoka, H.; Nada, S.; Okada, M. Mechanism of Csk-mediated down-regulation of Src family tyrosine kinases in epidermal growth factor signaling. J. Biol. Chem. 2004. [Google Scholar] [CrossRef]
- Kloth, M.T.; Laughlin, K.K.; Biscardi, J.S.; Boerner, J.L.; Parsons, S.J.; Silva, C.M. STAT5b, a mediator of synergism between c-Src and the epidermal growth factor receptor. J. Biol. Chem. 2003. [Google Scholar] [CrossRef] [PubMed]
- Boerner, J.L.; Demory, M.L.; Silva, C.; Parsons, S.J. Phosphorylation of Y845 on the epidermal growth factor receptor mediates binding to the mitochondrial protein cytochrome c oxidase subunit II. Mol. Cell. Biol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.I.; Nagao, T.; Iwasaki, T.; Nishihira, Y.; Fukami, Y. Src-dependent phosphorylation of the EGF receptor Tyr-845 mediates Stat-p21waf1 pathway in A431 cells. Genes Cells 2003. [Google Scholar] [CrossRef] [PubMed]
- Knebel, A.; Rahmsdorf, H.J.; Ullrich, A.; Herrlich, P. Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents. EMBO J. 1996. [Google Scholar] [CrossRef]
- Prenzel, N.; Zwick, E.; Leserer, M.; Ullrich, A. Tyrosine kinase signalling in breast cancer: Epidermal growth factor receptor-Convergence point for signal integration and diversification. Breast Cancer Res. 2000. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Graves, L.M.; Gill, G.N.; Parsons, S.J.; Samet, J.M. Src-dependent phosphorylation of the epidermal growth factor receptor on tyrosine 845 is required for zinc-induced Ras activation. J. Biol. Chem. 2002. [Google Scholar] [CrossRef]
- Moro, L.; Dolce, L.; Cabodi, S.; Bergatto, E.; Erba, E.B.; Smeriglio, M.; Turco, E.; Retta, S.F.; Giuffrida, M.G.; Venturino, M.; et al. Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J. Biol. Chem. 2002. [Google Scholar] [CrossRef]
- Fischgräbe, J.; Götte, M.; Michels, K.; Kiesel, L.; Wülfing, P. Targeting endothelin A receptor enhances anti-proliferative and anti-invasive effects of the HER2 antibody trastuzumab in HER2-overexpressing breast cancer cells. Int. J. Cancer 2010. [Google Scholar] [CrossRef]
- Park, Y.J.; Lee, H.; Lee, J.H. Macrophage inhibitory cytokine-1 transactivates ErbB family receptors via the activation of Src in SK-BR-3 human breast cancer cells. BMB Rep. 2010. [Google Scholar] [CrossRef]
- Amos, S.; Martin, P.M.; Polar, G.A.; Parsons, S.J.; Hussaini, I.M. Phorbol 12-myristate 13-acetate induces epidermal growth factor receptor transactivation via protein kinase Cδ/c-Src pathways in glioblastoma cells. J. Biol. Chem. 2005. [Google Scholar] [CrossRef]
- Reinehr, R.; Sommerfeld, A.; Häussinger, D. Insulin induces swelling-dependent activation of the epidermal growth factor receptor in rat liver. J. Biol. Chem. 2010. [Google Scholar] [CrossRef] [PubMed]
- Drube, S.; Stirnweiss, J.; Valkova, C.; Liebmann, C. Ligand-independent and EGF receptor-supported transactivation: Lessons from β2-adrenergic receptor signalling. Cell. Signal. 2006. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.; Beattie, E.C.; Lem, L.; Riethof, D.A.; Liu, S.H.; Mobley, W.C.; Soriano, P.; Brodsky, F.M. EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell 1999. [Google Scholar] [CrossRef]
- Ahn, S.; Kim, J.; Lucaveche, C.L.; Reedy, M.C.; Luttrell, L.M.; Lefkowitz, R.J.; Daaka, Y. Src-dependent tyrosine phosphorylation regulates dynamin self-assembly and ligand-induced endocytosis of the epidermal growth factor receptor. J. Biol. Chem. 2002. [Google Scholar] [CrossRef] [PubMed]
- Thien, C.B.F.; Walker, F.; Langdon, W.Y. RING finger mutations that abolish c-Cbl-directed polyubiquitination and downregulation of the EGF receptor are insufficient for cell transformation. Mol. Cell 2001. [Google Scholar] [CrossRef]
- Biscardi, J.S.; Belsches, A.P.; Parsons, S.J. Characterization of human epidermal growth factor receptor and c-Src interactions in human breast tumor cells. Mol. Carcinog. 1998. [Google Scholar] [CrossRef]
- Ishizawar, R.; Parsons, S.J. C-Src and cooperating partners in human cancer. Cancer Cell 2004. [Google Scholar] [CrossRef]
- Khazaie, K.; Schirrmacher, V.; Lichtner, R.B. EGF receptor in neoplasia and metastasis. Cancer Metastasis Rev. 1993. [Google Scholar] [CrossRef]
- Banker, N.; Evers, B.M.; Hellmich, M.R.; Townsend, C.M. The role of Src family kinases in the normal and neoplastic gastrointestinal tract. Surg. Oncol. 1996. [Google Scholar] [CrossRef]
- Mazurenko, N.N.; Zborovskaya, I.B.; Kisseljov, F.L.; Kogan, E.A. Expression of pp60c-src in human small cell and non-small cell lung carcinomas. Eur. J. Cancer 1992. [Google Scholar] [CrossRef]
- Masaki, T.; Igarashi, K.; Tokuda, M.; Yukimasa, S.; Han, F.; Jin, Y.J.; Li, J.Q.; Yoneyama, H.; Uchida, N.; Fujita, J.; et al. pp60c-src activation in lung adenocarcinoma. Eur. J. Cancer 2003. [Google Scholar] [CrossRef]
- Sonnweber, B.; Dlaska, M.; Skvortsov, S.; Dirnhofer, S.; Schmid, T.; Hilbe, W. High predictive value of epidermal growth factor receptor phosphorylation but not of EGFRvIII mutation in resected stage I non-small cell lung cancer (NSCLC). J. Clin. Pathol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Chang, H.H.; Lai, Y.H.; Lin, C.H.; Chen, M.H.; Chang, G.C.; Tsai, M.F.; Chen, J.J.W. Digoxin suppresses tumor malignancy through inhibiting multiple Src-related signaling pathways in non-small cell lung cancer. PLoS ONE 2015. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.H.; Chen, M.H.; Lin, S.Y.; Lin, S.Y.; Wong, Y.H.; Yu, S.L.; Chen, H.W.; Yang, C.H.; Chang, G.C.; Chen, J.J.W. Rhodomycin A, a novel Src-targeted compound, can suppress lung cancer cell progression via modulating Src-related pathways. Oncotarget 2015. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.I.; Wang, Y.C.; Hung, C.Y.; Yu, C.H.; Su, W.C.; Chang, W.C.; Hung, J.J. Positive feedback regulation between IL10 and EGFR promotes lung cancer formation. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Dimri, M.; Naramura, M.; Duan, L.; Chen, J.; Ortega-Cava, C.; Chen, G.; Goswami, R.; Fernandes, N.; Gao, Q.; Dimri, G.P.; et al. Modeling breast cancer-associated c-Src and EGFR overexpression in human MECs: C-Src and EGFR cooperatively promote aberrant three-dimensional acinar structure and invasive behavior. Cancer Res. 2007. [Google Scholar] [CrossRef]
- Irwin, M.E.; Bohin, N.; Boerner, J.L. Src family kinases mediate epidermal growth factor receptor signaling from lipid rafts in breast cancer cells. Cancer Biol. Ther. 2011. [Google Scholar] [CrossRef]
- Karni, R.; Jove, R.; Levitzki, A. Inhibition of pp60(c-Src) reduces Bcl-X(L) expression and reverses the transformed phenotype of cells overexpressing EGF and HER-2 receptors. Oncogene 1999. [Google Scholar] [CrossRef]
- Belsches-Jablonski, A.P.; Biscardi, J.S.; Peavy, D.R.; Tice, D.A.; Romney, D.A.; Parsons, S.J. Src family kinases and HER2 interactions in human breast cancer cell growth and survival. Oncogene 2001. [Google Scholar] [CrossRef]
- Sheffield, L.G. C-src activation by ErbB2 leads to attachment-independent growth of human breast epithelial cells. Biochem. Biophys. Res. Commun. 1998. [Google Scholar] [CrossRef]
- Muthuswamy, S.K.; Siegel, P.M.; Dankort, D.L.; Webster, M.A.; Muller, W.J. Mammary tumors expressing the neu proto-oncogene possess elevated c-Src tyrosine kinase activity. Mol. Cell. Biol. 1994. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Li, P.; Klos, K.S.; Lu, J.; Lan, K.H.; Nagata, Y.; Fang, D.; Jing, T.; Yu, D. ErbB2 promotes Src synthesis and stability: Novel mechanisms of Src activation that confer breast cancer metastasis. Cancer Res. 2005. [Google Scholar] [CrossRef] [PubMed]
- Ishizawar, R.C.; Miyake, T.; Parsons, S.J. c-Src modulates ErbB2 and ErbB3 heterocomplex formation and function. Oncogene 2007. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015. [Google Scholar] [CrossRef]
- Guo, Y.; Higazi, A.A.; Arakelian, A.; Sachais, B.S.; Cines, D.; Goldfarb, R.H.; Jones, T.R.; Kwaan, H.; Mazar, A.P.; Rabbani, S.A. A peptide drived from the nonreceptor binding region of urokinase plasminogen activator (uPA) inhibits tumor progression and angiogenesis and induces tumor cell death in vivo. FASEB J. 2000, 14, 1400–1410. [Google Scholar] [CrossRef]
- Fan, P.; McDaniel, R.E.; Kim, H.R.; Clagett, D.; Haddad, B.; Craig Jordan, V. Modulating therapeutic effects of the c-Src inhibitor via oestrogen receptor and human epidermal growth factor receptor 2 in breast cancer cell lines. Eur. J. Cancer 2012. [Google Scholar] [CrossRef]
- Xiao, J.; Xu, M.; Hou, T.; Huang, Y.; Yang, C.; Li, J. Dasatinib enhances antitumor activity of paclitaxel in ovarian cancer through Src signaling. Mol. Med. Rep. 2015. [Google Scholar] [CrossRef]
- Jin, L.; Chun, J.; Pan, C.; Alesi, G.N.; Li, D.; Magliocca, K.R.; Kang, Y.; Chen, Z.G.; Shin, D.M.; Khuri, F.R.; et al. Phosphorylation-mediated activation of LDHA promotes cancer cell invasion and tumour metastasis. Oncogene 2017. [Google Scholar] [CrossRef]
- Lou, L.; Yu, Z.; Wang, Y.; Wang, S.; Zhao, Y. c-Src inhibitor selectively inhibits triple-negative breast cancer overexpressed Vimentin in vitro and in vivo. Cancer Sci. 2018. [Google Scholar] [CrossRef]
- Nakanishi, T.; Menju, T.; Nishikawa, S.; Takahashi, K.; Miyata, R.; Shikuma, K.; Sowa, T.; Imamura, N.; Hamaji, M.; Motoyama, H.; et al. The synergistic role of ATP-dependent drug efflux pump and focal adhesion signaling pathways in vinorelbine resistance in lung cancer. Cancer Med. 2018. [Google Scholar] [CrossRef]
- Perez, M.; Lucena-Cacace, A.; Marín-Gómez, L.M.; Padillo-Ruiz, J.; Robles-Frias, M.J.; Saez, C.; Garcia-Carbonero, R.; Carnero, A. Dasatinib, a Src inhibitor, sensitizes liver metastatic colorectal carcinoma to oxaliplatin in tumors with high levels of phospho-Src. Oncotarget 2016. [Google Scholar] [CrossRef]
- Duan, Z.; Zhang, J.; Ye, S.; Shen, J.; Choy, E.; Cote, G.; Harmon, D.; Mankin, H.; Hua, Y.; Zhang, Y.; et al. A-770041 reverses paclitaxel and doxorubicin resistance in osteosarcoma cells. BMC Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Min, H.Y.; Yun, J.H.; Lee, J.S.; Lee, H.J.; Cho, J.; Jang, H.J.; Park, S.H.; Liu, D.; Oh, S.H.; Lee, J.S.H.; et al. Targeting the insulin-like growth factor receptor and Src signaling network for the treatment of non-small cell lung cancer. Mol. Cancer 2015. [Google Scholar] [CrossRef]
- Formisano, L.; Nappi, L.; Rosa, R.; Marciano, R.; D’Amato, C.; D’Amato, V.; Damiano, V.; Raimondo, L.; Iommelli, F.; Scorziello, A.; et al. Epidermal growth factor-receptor activation modulates Src-dependent resistance to lapatinib in breast cancer models. Breast Cancer Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, N.S.; Washington, M.K.; Merchant, N.B. Combined blockade of Src kinase and epidermal growth factor receptor with gemcitabine overcomes STAT3-mediated resistance of inhibition of pancreatic tumor growth. Clin. Cancer Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Iida, M.; Kruser, T.J.; Nechrebecki, M.M.; Dunn, E.F.; Armstrong, E.A.; Huang, S.; Harari, P.M. Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer Biol. Ther. 2009. [Google Scholar] [CrossRef] [PubMed]
- Dunn, E.F.; Iida, M.; Myers, R.A.; Campbell, D.A.; Hintz, K.A.; Armstrong, E.A.; Li, C.; Wheeler, D.L. Dasatinib sensitizes KRAS mutant colorectal tumors to cetuximab. Oncogene 2011. [Google Scholar] [CrossRef]
- Li, C.; Iida, M.; Dunn, E.F.; Ghia, A.J.; Wheeler, D.L. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene 2009. [Google Scholar] [CrossRef]
- Murakami, Y.; Sonoda, K.; Abe, H.; Watari, K.; Kusakabe, D.; Azuma, K.; Kawahara, A.; Akiba, J.; Oneyama, C.; Pachter, J.A.; et al. The activation of SRC family kinases and focal adhesion kinase with the loss of the amplified, mutated EGFR gene contributes to the resistance to afatinib, erlotinib and osimertinib in human lung cancer cells. Oncotarget 2017. [Google Scholar] [CrossRef]
- Formisano, L.; D’Amato, V.; Servetto, A.; Brillante, S.; Raimondo, L.; Di Mauro, C.; Marciano, R.; Orsini, R.C.; Cosconati, S.; Randazzo, A.; et al. Src inhibitors act through different mechanisms in Non-Small Cell Lung Cancer models depending on EGFR and RAS mutational status. Oncotarget 2015. [Google Scholar] [CrossRef]
- Yoshida, T.; Zhang, G.; Smith, M.A.; Lopez, A.S.; Bai, Y.; Li, J.; Fang, B.; Koomen, J.; Rawal, B.; Fisher, K.J.; et al. Tyrosine phosphoproteomics identifies both codrivers and cotargeting strategies for T790M-related EGFR-TKI resistance in non-small cell lung cancer. Clin. Cancer Res. 2014. [Google Scholar] [CrossRef]
- Nozaki, M.; Yasui, H.; Ohnishi, Y. Ligand-independent EGFR activation by anchorage-stimulated Src promotes cancer cell proliferation and cetuximab resistance via ErbB3 phosphorylation. Cancers 2019, 11, 1552. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.H.; Nam, A.R.; Park, J.E.; Bang, J.H.; Bang, Y.J.; Oh, D.Y. Resistance mechanism against trastuzumab in HER2-positive cancer cells and its negation by Src inhibition. Mol. Cancer Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Campiglio, M.; De Luca, A.; Somenzi, G.; Maiello, M.; Ciardiello, F.; Gianni, L.; Salomon, D.S.; Menard, S. Cooperative inhibitory effect of ZD1839 (Iressa) in combination with trastuzumab (Herceptin) on human breast cancer cell growth. Ann. Oncol. 2002. [Google Scholar] [CrossRef] [PubMed]
- Creelan, B.C.; Gray, J.E.; Tanvetyanon, T.; Chiappori, A.A.; Yoshida, T.; Schell, M.J.; Antonia, S.J.; Haura, E.B. Phase 1 trial of dasatinib combined with afatinib for epidermal growth factor receptor-(EGFR-) mutated lung cancer with acquired tyrosine kinase inhibitor (TKI) resistance. Br. J. Cancer 2019. [Google Scholar] [CrossRef] [PubMed]
- Cardin, D.B.; Goff, L.W.; Chan, E.; Whisenant, J.G.; Dan Ayers, G.; Takebe, N.; Arlinghaus, L.R.; Yankeelov, T.E.; Berlin, J.; Merchant, N. Dual Src and EGFR inhibition in combination with gemcitabine in advanced pancreatic cancer: Phase I results: A phase I clinical trial. Investig. New Drugs 2018. [Google Scholar] [CrossRef] [PubMed]
- Parseghian, C.M.; Parikh, N.U.; Wu, J.Y.; Jiang, Z.Q.; Henderson, L.; Tian, F.; Pastor, B.; Ychou, M.; Raghav, K.; Dasari, A.; et al. Dual inhibition of EGFR and c-Src by cetuximab and dasatinib combined with FOLFOX chemotherapy in patients with metastatic colorectal cancer. Clin. Cancer Res. 2017. [Google Scholar] [CrossRef]
- Stabile, L.P.; Egloff, A.M.; Gibson, M.K.; Gooding, W.E.; Ohr, J.; Zhou, P.; Rothenberger, N.J.; Wang, L.; Geiger, J.L.; Flaherty, J.T.; et al. IL6 is associated with response to dasatinib and cetuximab: Phase II clinical trial with mechanistic correlatives in cetuximab-resistant head and neck cancer. Oral Oncol. 2017. [Google Scholar] [CrossRef]
- Ocana, A.; Gil-Martin, M.; Antolín, S.; Atienza, M.; Montaño, Á.; Ribelles, N.; Urruticoechea, A.; Falcón, A.; Pernas, S.; Orlando, J.; et al. Efficacy and safety of dasatinib with trastuzumab and paclitaxel in first line HER2-positive metastatic breast cancer: Results from the phase II GEICAM/2010-04 study. Breast Cancer Res. Treat. 2019. [Google Scholar] [CrossRef]
- Stabile, L.P.; He, G.; Lui, V.W.Y.; Henry, C.; Gubish, C.T.; Joyce, S.; Quesnelle, K.M.; Siegfried, J.M.; Grandis, J.R. C-Src activation mediates erlotinib resistance in head and neck cancer by stimulating c-Met. Clin. Cancer Res. 2013. [Google Scholar] [CrossRef]
- Wheeler, S.E.; Shi, H.; Lin, F.; Dasari, S.; Bednash, J.; Thorne, S.; Watkins, S.; Joshi, R.; Thomas, S.M. Enhancement of head and neck squamous cell carcinoma proliferation, invasion, and metastasis by tumor-associated fibroblasts in preclinical models. Head Neck 2014. [Google Scholar] [CrossRef] [PubMed]
- Gross, N.D.; Bauman, J.E.; Gooding, W.E.; Denq, W.; Thomas, S.M.; Wang, L.; Chiosea, S.; Hood, B.L.; Flint, M.S.; Sun, M.; et al. Erlotinib, erlotinib-sulindac versus placebo: A randomized, double-blind, placebo-controlled window trial in operable head and neck cancer. Clin. Cancer Res. 2014. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.E.; Duvvuri, U.; Gooding, W.E.; Rath, T.J.; Gross, N.D.; Song, J.; Jimeno, A.; Yarbrough, W.G.; Johnson, F.M.; Wang, L.; et al. Randomized, placebo-controlled window trial of EGFR, Src, or combined blockade in head and neck cancer. JCI Insight 2017. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Lester, J.F. Tyrosine kinase inhibitors for the treatment of EGFR mutation-positive non–small-cell lung cancer: A clash of the generations. Clin. Lung Cancer 2020. [Google Scholar] [CrossRef]


{kind=link}
| Drugs | Molecular Targets | Clinical Applications |
|---|---|---|
| Bosutinib | BCR-Abl, c-Src, Lyn, Hck, Kit, PDGFR | CML, ALL + clinical trials for breast cancer, glioblastoma |
| Dasatinib | BCR-Abl, SFKs, Arg, c-KIT, EGFR, PDGFR, DDR1, DDR2, c-FMS, ephrin receptors, TEK, BTK, EphA2 | CML + clinical trials for ALL, breast, colorectal, endometrial, head and neck, ovarian, and small cell lung cancers, glioblastoma, melanoma, and NSCLC |
| Ponatinib | BCR-Abl, SFKs, VEGFR, PDGFR, FGFR, Eph, Kit, RET, Tie2, Flt3 | CML, ALL + clinical trials for endometrial, GIST, hepatic biliary, small cell lung, and thyroid cancers |
| Vandetanib | RET, SFKs, EGFR, VEGFRs, Brk, Tie2, EphR | medullary thyroid carcinoma |
| Saracatinib (AZD0530) | c-Src, BCR-Abl | Clinical trial for SCLC, NSCLC, colorectal, gastric, ovarian and metastatic osteosarcoma |
| Authors | Disease | Clinical Trial/Phase | Drugs | Total No. of Patients | Mutational State/Drug Resistance | Results |
|---|---|---|---|---|---|---|
| Creelan BC et al. [95] | Lung Cancer, Non-small cell lung cancer (NSCLC) | NCT01999985 Phase IA, IB | saracatinib, afatinib | 25 | EGFR mut or EGFR TKI resistant | mPFS = 3.7 months (95% CI, 2.3–5.0) mOS = 14.7 months (95% CI, 8.5–20.9) |
| Cardin DB et al. [96] | MPAC, RPA, Stage III and IV Pancreatic Cancer | NCT01660971 Phase I | dasatinib, erlotinib, gengitabine | 19 | NA | DCR = 69%, mPFS = 3.6 months (95% CI, 3.8 to NA), OS = 8 months (95% CI, 4.4 to 17) |
| Parseghian CM et al. [97] | Metastatic Colon-Rectal Cancer | NCT00501410 Phase IB/II | dasatinib, FOLFOX | 77 | KRAS c12/13mut KRAS c12/13wt | ORR = 30% (95% CI, 0.17 to 0.45) only KRASwt; stable disease 23%; (95% CI, 0.12 to 0.38) mOS = 6.7 months in all patients |
| Stabile LP et al. [98] | Recurrant/metastatic HNSCC | NCT01488318 Phase II | dasatinib, cetuximab | 21 | Progression after cetuximab | SD= 36%, PD = 57%; mPFS = 1.7months (90%CI, 1.4–3.9 months); mOS = 5.1 months (90% CI, 4.2–11.5 months) |
| Ocana A et al. [99] | Metastatic Breast Cancer | NCT01306942 Phase I/II | dasatinib, paclitaxel, trastuzumab | 39 | HER2 + | ORR = 79.3% (n = 23; 95% CI 60.3 to 92.0); CBR = 82.8% (n = 24; 95% CI 64.2–94.2); mPFS = 23.9 months (95% CI 10.3–NR); TTP = 23.9 months (95% CI 14.9-NR): RD = NR |
| Clinical Trial ID | Disease Condition | Study Phase | Combination Drugs | Status at Time of Search |
|---|---|---|---|---|
| NCT00444015 | Stage IIIB/IV disease Recurrent NSCLC | Phase I | Erlotinib + Dasatinib | Completed |
| NCT02954523 | EGFR mut NSCLC | Phase I/II | Dasatinib + osimertinib | Active, not recruiting |
| NCT00996723 | Diffuse Intrinsic Pontine Glioma | Phase I | Vandetanib + dasatinib | Completed |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belli, S.; Esposito, D.; Servetto, A.; Pesapane, A.; Formisano, L.; Bianco, R. c-Src and EGFR Inhibition in Molecular Cancer Therapy: What Else Can We Improve? Cancers 2020, 12, 1489. https://doi.org/10.3390/cancers12061489
Belli S, Esposito D, Servetto A, Pesapane A, Formisano L, Bianco R. c-Src and EGFR Inhibition in Molecular Cancer Therapy: What Else Can We Improve? Cancers. 2020; 12(6):1489. https://doi.org/10.3390/cancers12061489
Chicago/Turabian StyleBelli, Stefania, Daniela Esposito, Alberto Servetto, Ada Pesapane, Luigi Formisano, and Roberto Bianco. 2020. "c-Src and EGFR Inhibition in Molecular Cancer Therapy: What Else Can We Improve?" Cancers 12, no. 6: 1489. https://doi.org/10.3390/cancers12061489
APA StyleBelli, S., Esposito, D., Servetto, A., Pesapane, A., Formisano, L., & Bianco, R. (2020). c-Src and EGFR Inhibition in Molecular Cancer Therapy: What Else Can We Improve? Cancers, 12(6), 1489. https://doi.org/10.3390/cancers12061489