Renin-Angiotensin System in Lung Tumor and Microenvironment Interactions

 ,
,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Renin-Angiotensin System: An Intricate Regulatory Mechanism in Lung Disease Physiopathology?
3. Renin-Angiotensin System and the Hallmarks of Cancer: Application to Lung Tumors
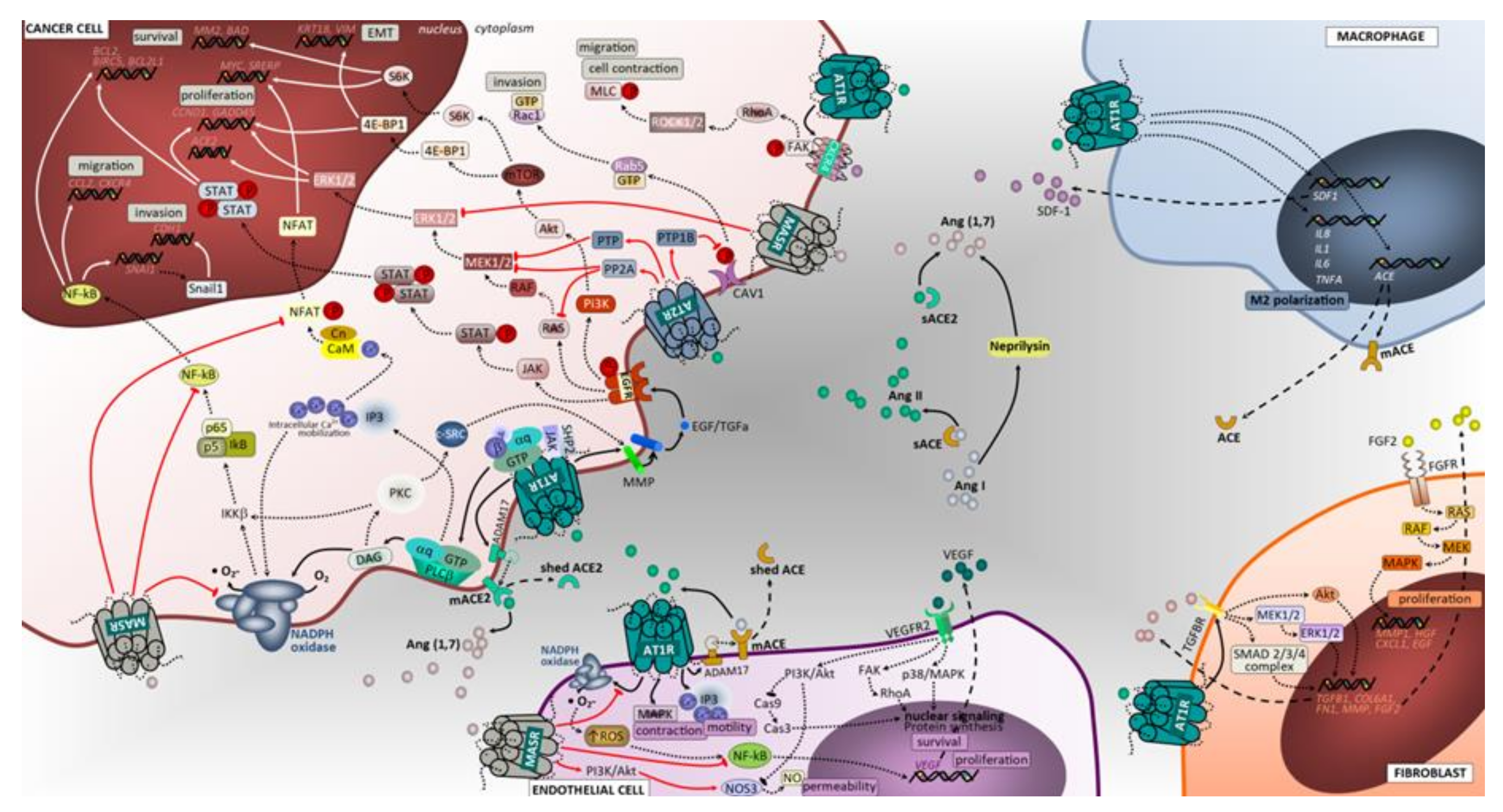
3.1. Cell Proliferation, Invasion and Migration
3.1.1. ACE/Ang II/AT1 Receptor Axis
3.1.2. AT2 Receptor
3.1.3. ACE2/Ang (1–7)/Mas Receptor Axis
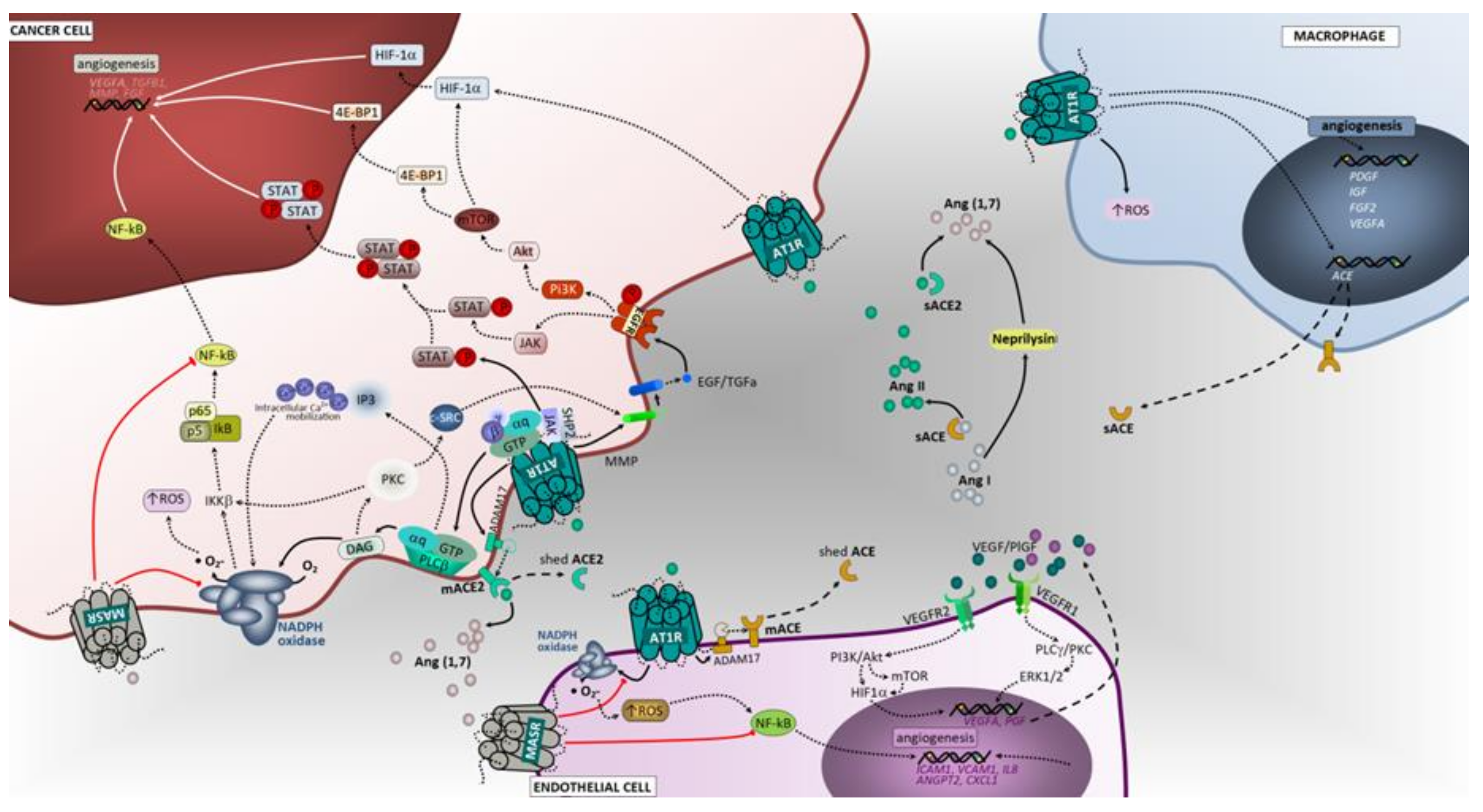
3.2. Hypoxia and Promotion of Tumor Angiogenesis
3.2.1. ACE/Ang II/AT1 Receptor Axis and AT2 Receptor
3.2.2. ACE2/Ang (1–7)/Mas Receptor Axis
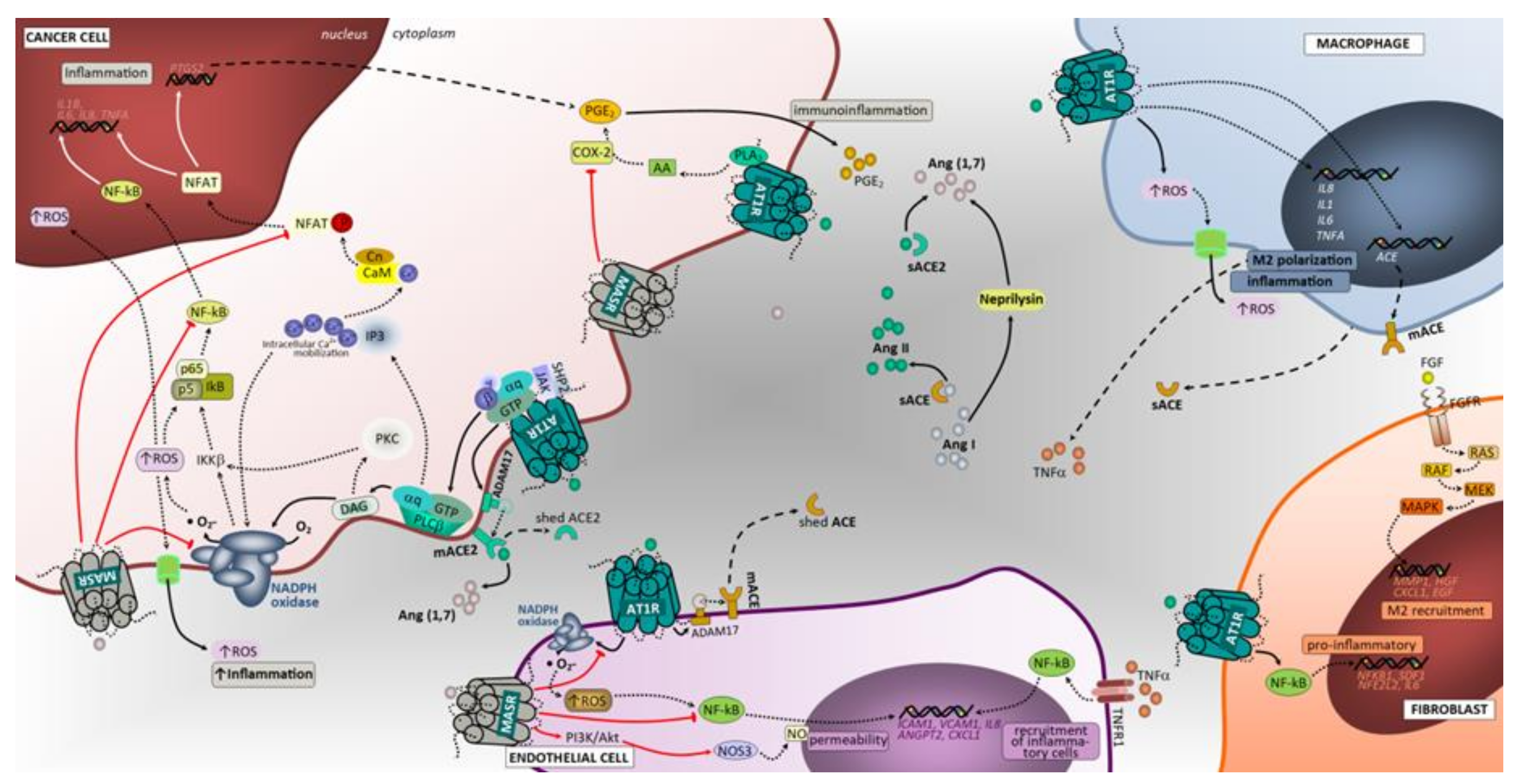
3.3. Inflammation
3.3.1. ACE/Ang II/AT1 Receptor and AT2 Receptor
3.3.2. ACE2/Ang (1–7)/Mas Receptor Axis
3.4. Tumor Immunological Response
Targeting RAS to Improve Anti-Tumor Immunity
4. Clinical Studies
4.1. Clinical Trials
4.2. Observational Studies
5. Future Perspectives
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, N.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2014, 136, E359–E386. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.C.S.; Lao, X.Q.; Ho, K.-F.; Goggins, W.B.; Tse, S.L.A. Incidence and mortality of lung cancer: global trends and association with socioeconomic status. Sci. Rep. 2017, 7, 14300. [Google Scholar] [CrossRef] [PubMed]
- Thethi, T.; Kamiyama, M.; Kobori, H. The Link Between the Renin-Angiotensin-Aldosterone System and Renal Injury in Obesity and the Metabolic Syndrome. Curr. Hypertens. Rep. 2012, 14, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Wegman-Ostrosky, T.; Soto-Reyes, E.; Vidal-Millán, S.; Corona, J.S. The renin-angiotensin system meets the hallmarks of cancer. J. Renin Angiotensin Aldosterone Syst. 2013, 16, 227–233. [Google Scholar] [CrossRef]
- Putnam, K.; Shoemaker, R.; Yiannikouris, F.; Cassis, L.A. The renin-angiotensin system: A target of and contributor to dyslipidemias, altered glucose homeostasis, and hypertension of the metabolic syndrome. Am. J. Physiol. Circ. Physiol. 2012, 302, H1219–H1230. [Google Scholar] [CrossRef]
- Lavoie, J.; Sigmund, C.D. Minireview: Overview of the Renin-Angiotensin System—An Endocrine and Paracrine System. Endocrinology 2003, 144, 2179–2183. [Google Scholar] [CrossRef]
- Kaschina, E.; Unger, T. Angiotensin AT1/AT2 receptors: Regulation, signalling and function. Blood Press 2003, 12, 70–88. [Google Scholar] [CrossRef]
- Swanson, G.N.; Hanesworth, J.M.; Sardinia, M.F.; Coleman, J.K.; Wright, J.W.; Hall, K.L.; Miller-Wing, A.V.; Stobb, J.W.; Cook, V.I.; Harding, E.C. Discovery of a distinct binding site for angiotensin II (3-8), a putative angiotensin IV receptor. Regul. Pept. 1992, 40, 409–419. [Google Scholar] [CrossRef]
- Marshall, R.P. The pulmonary renin-angiotensin system. Curr. Pharm. Des. 2003, 9, 715–722. [Google Scholar] [CrossRef]
- George, A.; Thomas, W.G.; Hannan, R.D. The renin–angiotensin system and cancer: Old dog, new tricks. Nat. Rev. Cancer 2010, 10, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Matsusaka, T.; Niimura, F.; Shimizu, A.; Pastan, I.; Saito, A.; Kobori, H.; Nishiyama, A.; Ichikawa, I. Liver Angiotensinogen Is the Primary Source of Renal Angiotensin II. J. Am. Soc. Nephrol. 2012, 23, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Vickers, C. Hydrolysis of Biological Peptides by Human Angiotensin-converting Enzyme-related Carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [PubMed]
- Orte, C.; Polak, J.M.; Haworth, S.G.; Yacoub, M.H.; Morrell, N.W. Expression of pulmonary vascular angiotensin-converting enzyme in primary and secondary plexiform pulmonary hypertension. J. Pathol. 2000, 192, 379–384. [Google Scholar] [CrossRef]
- Montes, E.; Ruiz, V.; Checa, M.; Maldonado, V.; Melendez-Zajgla, J.; Montano, M.; Ordoñez-Razo, R.M.; Cisneros, J.; De Alba, C.G.; Pardo, A.; et al. Renin is an angiotensin-independent profibrotic mediator: Role in pulmonary fibrosis. Eur. Respir. J. 2011, 39, 141–148. [Google Scholar] [CrossRef]
- Song, G.G.; Kim, J.-H.; Lee, Y.H. Associations between the angiotensin-converting enzyme insertion/deletion polymorphism and susceptibility to sarcoidosis: A meta-analysis. J. Renin-Angiotensin-Aldosterone Syst. 2013, 16, 219–226. [Google Scholar] [CrossRef]
- De Man, F.S.; Tu, L.; Handoko, M.L.; Rain, S.; Ruiter, G.; Francois, C.; Schalij, I.; Dorfmüller, P.; Simonneau, G.; Fadel, E.; et al. Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 780–789. [Google Scholar] [CrossRef]
- Wang, D.; Chai, X.; Magnussen, C.G.; Zosky, G.R.; Shu, S.-H.; Wei, X.; Hu, S.-S.; Magnnusen, C.G. Renin-angiotensin-system, a potential pharmacological candidate, in acute respiratory distress syndrome during mechanical ventilation. Pulm. Pharmacol. Ther. 2019, 58, 101833. [Google Scholar] [CrossRef]
- Rosenthal, T.; Gavras, I. Renin-Angiotensin Inhibition in Combating Malignancy: A Review. Anticancer. Res. 2019, 39, 4597–4602. [Google Scholar] [CrossRef]
- Uhal, B.D.; Li, X.; Piasecki, C.C.; Molina-Molina, M. Angiotensin signalling in pulmonary fibrosis. Int. J. Biochem. Cell Boil. 2011, 44, 465–468. [Google Scholar] [CrossRef]
- Wang, R.; Ibarra-Sunga, O.; Verlinski, L.; Pick, R.; Uhal, B.D. Abrogation of bleomycin-induced epithelial apoptosis and lung fibrosis by captopril or by a caspase inhibitor. Am. J. Physiol. Cell. Mol. Physiol. 2000, 279, L143–L151. [Google Scholar] [CrossRef] [PubMed]
- Molteni, A.; Wolfe, L.F.; Ward, W.F.; Ts’Ao, C.H.; Molteni, L.B.; Veno, P.; Fish, B.L.; Taylor, J.M.; Quintanilla, N.; Herndon, B.; et al. Effect of an angiotensin II receptor blocker and two angiotensin converting enzyme inhibitors on transforming growth factor-beta (TGF-beta) and alpha-actomyosin (alpha SMA), important mediators of radiation-induced pneumopathy and lung fibrosis. Curr. Pharm. Des. 2007, 13, 1307–1316. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi-Karakani, A.; Ghazi-Khansari, M.; Sotoudeh, M. Lisinopril ameliorates paraquat-induced lung fibrosis. Clin. Chim. Acta 2006, 367, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, M.; Lederer, D.J.; Molina-Molina, M.; Noth, I.; Valenzuela, C.; Frankenstein, L.; Weycker, D.; Atwood, M.; Kirchgaessler, K.-U.; Cottin, V. Association of Angiotensin Modulators With the Course of Idiopathic Pulmonary Fibrosis. Chest 2019, 156, 706–714. [Google Scholar] [CrossRef]
- Takai, S.; Shiota, N.; Yamamoto, D.; Okunishi, H.; Miyazaki, M. Purification and characterization of angiotensin II-generating chymase from hamster cheek pouch. Life Sci. 1996, 58, 591–597. [Google Scholar] [CrossRef]
- Dell’Italia, L.J.; Collawn, J.F.; Ferrario, C.M. Multifunctional Role of Chymase in Acute and Chronic Tissue Injury and Remodeling. Circ. Res. 2018, 122, 319–336. [Google Scholar] [CrossRef]
- Kosanovic, D.; Luitel, H.; Dahal, B.K.; Cornitescu, T.; Janssen, W.; Danser, A.J.; Garrelds, I.M.; De Mey, J.G.; Fazzi, G.; Schiffers, P.; et al. Chymase: A multifunctional player in pulmonary hypertension associated with lung fibrosis. Eur. Respir. J. 2015, 46, 1084–1094. [Google Scholar] [CrossRef]
- Ibaraki, T.; Orino, T.; Katsumata, T.; Muramatsu, M.; Takai, S.; Jin, D.; Maruyama, H.; Miyazaki, M. The relationship of tryptase and chymase-positive mast cells to angiogenesis in stage I non-small cell lung cancer. Eur. J. Cardio-Thoracic Surg. 2005, 28, 617–621. [Google Scholar] [CrossRef]
- Takai, S.; Jin, D.; Miyazaki, M. New Approaches to Blockade of the Renin–Angiotensin–Aldosterone System: Chymase as an Important Target to Prevent Organ Damage. J. Pharmacol. Sci. 2010, 113, 301–309. [Google Scholar] [CrossRef]
- Jiang, Y.; Wu, Y.; Hardie, W.J.; Zhou, X. Mast cell chymase affects the proliferation and metastasis of lung carcinoma cells in vitro. Oncol. Lett. 2017, 14, 3193–3198. [Google Scholar] [CrossRef]
- Dahmani, A.; Delisle, J.-S. TGF-β in T Cell Biology: Implications for Cancer Immunotherapy. Cancers 2018, 10, 194. [Google Scholar] [CrossRef] [PubMed]
- De Gasparo, M.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 2000, 52, 415–472. [Google Scholar] [PubMed]
- Bullock, G.R.; Steyaert, I.; Bilbe, G.; Carey, R.M.; Kips, J.; De Paepe, B.; Pauwels, R.; Praet, M.; Siragy, H.M.; De Gasparo, M. Distribution of type-1 and type-2 angiotensin receptors in the normal human lung and in lungs from patients with chronic obstructive pulmonary disease. Histochem. Cell Boil. 2001, 115, 117–124. [Google Scholar] [CrossRef]
- Ager, E.; Neo, J.; Christophi, C. The renin-angiotensin system and malignancy. Carcinog. 2008, 29, 1675–1684. [Google Scholar] [CrossRef]
- Goldstein, B.; Trivedi, M.S.; Speth, R.C. Alterations in Gene Expression of Components of the Renin-Angiotensin System and Its Related Enzymes in Lung Cancer. Lung Cancer Int. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chung, O.; Stoll, M.; Unger, T. Physiologic and pharmacologic implications of AT1 versus AT2 receptors. Blood Press. Suppl. 1996, 2, 47–52. [Google Scholar]
- Kaparianos, A.; Argyropoulou, E. Local renin-angiotensin II systems, angiotensin-converting enzyme and its homologue ACE2: Their potential role in the pathogenesis of chronic obstructive pulmonary diseases, pulmonary hypertension and acute respiratory distress syndrome. Curr. Med. Chem. 2011, 18, 3506–3515. [Google Scholar] [CrossRef]
- Uhal, B.D.; Kim, J.K.; Li, X.; Molina-Molina, M. Angiotensin-TGF-beta 1 crosstalk in human idiopathic pulmonary fibrosis: Autocrine mechanisms in myofibroblasts and macrophages. Curr. Pharm Des. 2007, 3, 1247–1256. [Google Scholar] [CrossRef]
- Renzoni, E.; Abraham, D.J.; Howat, S.; Shi-Wen, X.; Sestini, P.; Bou-Gharios, G.; Wells, A.U.; Veeraraghavan, S.; Nicholson, A.G.; Denton, C.P.; et al. Gene expression profiling reveals novel TGFβ targets in adult lung fibroblasts. Respir. Res. 2004, 5, 24. [Google Scholar] [CrossRef]
- Abdul-Hafez, A.; Shu, R.; Uhal, B.D. JunD and HIF-1α mediate transcriptional activation of angiotensinogen by TGF-β1 in human lung fibroblasts. FASEB J. 2009, 23, 1655–1662. [Google Scholar] [CrossRef]
- Ferrario, C.M.; Trask, A.J.; Jessup, J.A. Advances in biochemical and functional roles of angiotensin-converting enzyme 2 and angiotensin-(1–7) in regulation of cardiovascular function. Am. J. Physiol. Circ. Physiol. 2005, 289, H2281–H2290. [Google Scholar] [CrossRef]
- Wan, H.; Ma, Q.; Che, J.; Cao, H.; Fei, X.; Qiu, W.; Feng, Y.; Wan, H.; Liu, J.; Zhang, R.; et al. The angiotensin-converting enzyme 2 in tumor growth and tumor-associated angiogenesis in non-small cell lung cancer. Oncol. Rep. 2010, 23, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.-C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef]
- Santos, R.A.S.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M. The ACE2/angiotensin-(1–7)/MAS axis of the renin-angiotensin system: Focus on angiotensin-(1–7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [PubMed]
- Bayes-Genis, A.; Barallat, J.; Richards, A.M. A Test in context: Neprilysin: Function, inhibition, and biomarker. J. Am. Coll. Cardiol. 2016, 68, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Li, Y.; Zhang, Y.; Gerbes, A.L.; Liu, H.; Swain, M.G.; Lee, S.S. Effects of the neutral endopeptidase inhibitor thiorphan on cardiovascular and renal function in cirrhotic rats. Br. J. Pharmacol. 2003, 139, 81–88. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Papandreou, C.N.; Usmani, B.; Geng, Y.; Bogenrieder, T.; Freeman, R.; Wilk, S.; Finstad, C.L.; Reuter, V.E.; Powell, C.T.; Scheinberg, D.; et al. Neutral endopeptidase 24.11 loss in metastatic human prostate cancer contributes to androgen-independent progression. Nat. Med. 1998, 4, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.S.; Silva, A.C.S.E.; Maric, C.; Silva, D.M.R.; Machado, R.P.; De Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.B.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef]
- Chen, L.-N.; Yang, X.-H.; Nissen, D.H.; Chen, Y.-Y.; Wang, L.-J.; Wang, J.; Gao, J.-L.; Zhang, L.-Y. Dysregulated Renin-AngioteNsin System Contributes to acute Lung Injury Caused by Hind-limb Ischemia-Reperfusion in Mice. Shock 2013, 40, 420–429. [Google Scholar] [CrossRef]
- Asperen, R.M.W.-V.; Lutter, R.; Specht, P.A.; Moll, G.N.; Van Woensel, J.B.; Van Der Loos, C.M.; Van Goor, H.; Kamilic, J.; Florquin, S.; Bos, A.P. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J. Pathol. 2011, 225, 618–627. [Google Scholar] [CrossRef]
- Meng, Y.; Li, T.; Zhou, G.S.; Chen, Y.; Yu, C.H.; Pang, M.X.; Li, W.; Li, Y.; Zhang, W.Y.; Li, X. The angiotensin-converting enzyme 2/angiotensin (1–7)/Mas axis protects against lung fibroblast migration and lung fibrosis by inhibiting the NOX4-derived ROS-mediated RhoA/Rho kinase pathway. Antioxid Redox Signal 2015, 2, 241–258. [Google Scholar] [CrossRef]
- Gallagher, P.E.; Cook, K.; Soto-Pantoja, D.; Menon, J.; Tallant, E.A. Angiotensin peptides and lung cancer. Curr. Cancer Drug Targets 2011, 11, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Tajima, H.; Ohta, T.; Nakanuma, S.; Hayashi, H.; Nakagawara, H.; Onishi, I.; Takamura, H.; Ninomiya, I.; Kitagawa, H.; et al. Angiotensin II induces tumor progression and fibrosis in intrahepatic cholangiocarcinoma through an interaction with hepatic stellate cells. Int. J. Oncol. 2010, 37, 1251–1259. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rodrigues-Ferreira, S.; Abdelkarim, M.; Dillenburg-Pilla, P.; Luissint, A.-C.; Di-Tommaso, A.; Deshayes, F.; Pontes, C.L.S.; Molina, A.; Cagnard, N.; Letourneur, F.; et al. Angiotensin II Facilitates Breast Cancer Cell Migration and Metastasis. PLoS ONE 2012, 7, e35667. [Google Scholar] [CrossRef] [PubMed]
- Molina-Molina, M.; Xaubet, A.; Li, X.; Abdul-Hafez, A.; Friderici, K.; Jernigan, K.; Fu, W.; Ding, Q.; Pereda, J.; Serrano-Mollar, A.; et al. Angiotensinogen gene G-6A polymorphism influences idiopathic pulmonary fibrosis disease progression. Eur. Respir. J. 2008, 32, 1004–1008. [Google Scholar] [CrossRef]
- Wang, N.; Yang, D.; Ji, B.; Li, J. Angiotensin-converting enzyme insertion/deletion gene polymorphism and lung cancer risk: A meta-analysis. J. Renin-Angiotensin-Aldosterone Syst. 2014, 16, 189–194. [Google Scholar] [CrossRef]
- Freitas-Silva, M.F.; Pereira, D.; Coelho, C.; Bicho, M.; Lopes, C.; Medeiros, R. Angiotensin I–converting enzyme gene insertion/deletion polymorphism and endometrial human cancer in normotensive and hypertensive women. Cancer Genet. Cytogenet. 2004, 155, 42–46. [Google Scholar] [CrossRef]
- Medeiros, R.; Vasconcelos, A.; Costa, S.; Pinto, D.; Lobo, F.; Morais, A.; Medeiros, R.; Lopes, C. Linkage of angiotensin I-converting enzyme gene insertion/deletion polymorphism to the progression of human prostate cancer. J. Pathol. 2004, 202, 330–335. [Google Scholar] [CrossRef]
- Yu, C.; Tang, W.; Wang, Y.; Shen, Q.; Wang, B.; Cai, C.; Meng, X.J.; Zou, F. Downregulation of ACE2/Ang-(1–7)/Mas axis promotes breast cancer metastasis by enhancing store-operated calcium entry. Cancer Lett. 2016, 376, 268–277. [Google Scholar] [CrossRef]
- Lewandowska, U.; Lachowicz-Ochędalska, A.; Domińska, K.; Kaszewska, D.; Rebas, E. Angiotensin II as a factor modulating protein tyrosine kinase activity in two breast cancer lines-MCF-7 and MDA-MB-231. Endokrynol. Polska 2011, 62, 151–158. [Google Scholar]
- Wang, Z. Transactivation of Epidermal Growth Factor Receptor by G Protein-Coupled Receptors: Recent Progress, Challenges and Future Research. Int. J. Mol. Sci. 2016, 17, 95. [Google Scholar] [CrossRef] [PubMed]
- Yahata, Y.; Shirakata, Y.; Tokumaru, S.; Yang, L.; Dai, X.; Tohyama, M.; Tsuda, T.; Sayama, K.; Iwai, M.; Horiuchi, M.; et al. A novel function of angiotensin II in skin wound healing. Induction of fibroblast and keratinocyte migration by angiotensin II via heparin-binding epidermal growth factor (EGF)-like growth factor-mediated EGF receptor transactivation. J. Biol. Chem. 2006, 281, 13209–13216. [Google Scholar] [CrossRef] [PubMed]
- Greco, S.; Muscella, A.; Elia, M.; Salvatore, P.; Storelli, C.; Mazzotta, A.; Manca, C.; Marsigliante, S. Angiotensin II activates extracellular signal regulated kinases via protein kinase C and epidermal growth factor receptor in breast cancer cells. J. Cell. Physiol. 2003, 196, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Wilop, S.; Von Hobe, S.; Crysandt, M.; Esser, A.; Osieka, R.; Jost, E. Impact of angiotensin I converting enzyme inhibitors and angiotensin II type 1 receptor blockers on survival in patients with advanced non-small-cell lung cancer undergoing first-line platinum-based chemotherapy. J. Cancer Res. Clin. Oncol. 2009, 135, 1429–1435. [Google Scholar] [CrossRef]
- Miao, L.; Chen, W.; Zhou, L.; Wan, H.; Gao, B.; Feng, Y. Impact of Angiotensin I-converting Enzyme Inhibitors and Angiotensin II Type-1 Receptor Blockers on Survival of Patients with NSCLC. Sci. Rep. 2016, 6, 21359. [Google Scholar] [CrossRef]
- Glauser, D.A.; Schlegel, W. Sequential actions of ERK1/2 on the AP-1 transcription factor allow temporal integration of metabolic signals in pancreatic beta cells. FASEB J. 2007, 21, 3240–3249. [Google Scholar] [CrossRef]
- Touyz, R.M.; He, G.; Mabrouk, M.; Diep, Q.; Mardigyan, V.; Schiffrin, E.L. Differential activation of extracellular signal-regulated protein kinase 1/2 and p38 mitogen activated-protein kinase by AT1 receptors in vascular smooth muscle cells from Wistar-Kyoto rats and spontaneously hypertensive rats. J. Hypertens 2001, 19, 553–559. [Google Scholar] [CrossRef]
- Manna, P.R.; Stocco, D. The Role of Specific Mitogen-Activated Protein Kinase Signaling Cascades in the Regulation of Steroidogenesis. J. Signal Transduct. 2011, 2011, 1–13. [Google Scholar] [CrossRef]
- Meza, M.-S.; Díaz, J.; Bórquez, S.; Valderrama, V.-M.; Valdivia, D.-N.; Celis, R.-V.; Contreras, P.; Huilcaman, R.; Ocaranza, M.P.; Chiong, M.; et al. AT2 Receptor Mediated Activation of the Tyrosine Phosphatase PTP1B Blocks Caveolin-1 Enhanced Migration, Invasion and Metastasis of Cancer Cells. Cancers 2019, 11, 1299. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Y. Telmisartan inhibits NSCLC A549 cell proliferation and migration by regulating the PI3K/AKT signaling pathway. Oncol. Lett. 2018, 15, 5859–5864. [Google Scholar] [CrossRef]
- Attoub, S.; Gaben, A.M.; Al Sultan, M.; John, A.; Nicholls, M.G.; Mester, J.; Petroianu, G.; Al-Salam, S. Captopril as a Potential Inhibitor of Lung Tumor Growth and Metastasis. Ann. N. Y. Acad. Sci. 2008, 1138, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. The organ microenvironment and cancer metastasis. Differentiation 2002, 70, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Nie, B.; Pienta, K.J.; Morgan, T.M.; Taichman, R.S. Cancer stem cells and their role in metastasis. Pharmacol. Ther. 2013, 138, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, Q. Cancer stem cells and tumor metastasis (Review). Int. J. Oncol. 2014, 44, 1806–1812. [Google Scholar] [CrossRef] [PubMed]
- Fessler, E.; Dijkgraaf, F.E.; Melo, F.D.S.E.; Medema, J.P. Cancer stem cell dynamics in tumor progression and metastasis: Is the microenvironment to blame? Cancer Lett. 2013, 341, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Tajima, H.; Nakanuma, S.; Sakai, S.; Makino, I.; Kinoshita, J.; Hayashi, H.; Nakamura, K.; Oyama, K.; Nakagawara, H.; et al. Angiotensin II enhances epithelial-to-mesenchymal transition through the interaction between activated hepatic stellate cells and the stromal cell-derived factor-1/CXCR4 axis in intrahepatic cholangiocarcinoma. Int. J. Oncol. 2012, 41, 573–582. [Google Scholar] [CrossRef]
- Tawinwung, S.; Ninsontia, C.; Chanvorachote, P. Angiotensin II Increases Cancer Stem Cell-like Phenotype in Lung Cancer Cells. Anticancer. Res. 2015, 35, 4789–4797. [Google Scholar]
- Ma, Y.; Xia, Z.; Ye, C.; Lu, C.; Zhou, S.; Pan, J.; Liu, C.; Zhang, J.; Liu, T.; Hu, T.; et al. AGTR1 promotes lymph node metastasis in breast cancer by upregulating CXCR4/SDF-1alpha and inducing cell migration and invasion. Aging 2019, 11, 3969–3992. [Google Scholar] [CrossRef]
- Qi, Y.; Li, H.; Shenoy, V.; Li, Q.; Wong, F.; Zhang, L.; Raizada, M.K.; Sumners, C.; Katovich, M.J. Moderate cardiac-selective overexpression of angiotensin II type 2 receptor protects cardiac functions from ischaemic injury. Exp. Physiol. 2011, 97, 89–101. [Google Scholar] [CrossRef]
- Sun, L.; Wang, W.; Xiao, W.; Liang, H.; Yang, Y.; Yang, H. Angiotensin II induces apoptosis in intestinal epithelial cells through the AT2 receptor, GATA-6 and the Bax pathway. Biochem. Biophys. Res. Commun. 2012, 424, 663–668. [Google Scholar] [CrossRef]
- Pickel, L.; Matsuzuka, T.; Doi, C.; Ayuzawa, R.; Maurya, D.K.; Xie, S.-X.; Berkland, C.; Tamura, M. Over-expression of angiotensin II type 2 receptor gene induces cell death in lung adenocarcinoma cells. Cancer Boil. Ther. 2009, 9, 277–285. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kawabata, A.; Baoum, A.; Ohta, N.; Jacquez, S.; Seo, G.-M.; Berkland, C.; Tamura, M.; Baoum, A. Intratracheal administration of a nanoparticle-based therapy with the angiotensin II type 2 receptor gene attenuates lung cancer growth. Cancer Res. 2012, 72, 2057–2067. [Google Scholar] [CrossRef] [PubMed]
- Pei, N.; Mao, Y.; Wan, P.; Chen, X.; Li, A.; Chen, H.; Li, J.; Wan, R.; Zhang, Y.; Du, H.; et al. Angiotensin II type 2 receptor promotes apoptosis and inhibits angiogenesis in bladder cancer. J. Exp. Clin. Cancer Res. 2017, 36, 77. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Qi, Y.; Li, C.; Braseth, L.N.; Gao, Y.; Shabashvili, A.E.; Katovich, M.J.; Sumners, C. Angiotensin type 2 receptor-mediated apoptosis of human prostate cancer cells. Mol. Cancer Ther. 2009, 8, 3255–3265. [Google Scholar] [CrossRef]
- Murphy, A.M.; Wong, A.L.; Bezuhly, M. Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenesis Tissue Repair 2015, 8, 7. [Google Scholar] [CrossRef]
- Wan, H.; Feng, Y.; Ni, L.; Fan, L.; Fei, X.; Ma, Q.; Gao, B.; Xiang, Y.; Che, J.; Li, Q. Overexpression of ACE2 produces antitumor effects via inhibition of angiogenesis and tumor cell invasion in vivo and in vitro. Oncol. Rep. 2011, 26, 1157–1164. [Google Scholar] [CrossRef]
- Menon, J.; Soto-Pantoja, D.R.; Callahan, M.F.; Cline, J.M.; Ferrario, C.M.; Tallant, E.A.; Gallagher, P.E. Angiotensin-(1-7) Inhibits Growth of Human Lung Adenocarcinoma Xenografts in Nude Mice through a Reduction in Cyclooxygenase-2. Cancer Res. 2007, 67, 2809–2815. [Google Scholar] [CrossRef]
- Wan, H.; Ni, L.; Feng, Y.; Ma, Q.; Fan, L.; Qian, Y.; Li, Q.; Xiang, Y.; Gao, B. Angiotensin-(1-7) inhibits the migration and invasion of A549 human lung adenocarcinoma cells through inactivation of the PI3K/Akt and MAPK signaling pathways. Oncol. Rep. 2011, 27, 783–790. [Google Scholar] [CrossRef]
- Gomes, E.R.; Lara, A.A.; Almeida, P.W.; Guimarães, D.; Resende, R.R.; Campagnole-Santos, M.J.; Bader, M.; Santos, R.A.S.; Guatimosim, S. Angiotensin-(1-7) Prevents Cardiomyocyte Pathological Remodeling Through a Nitric Oxide/Guanosine 3′,5′-Cyclic Monophosphate–Dependent Pathway. Hypertension 2010, 55, 153–160. [Google Scholar] [CrossRef]
- Shou, J.; Jing, J.; Xie, J.; You, L.; Jing, Z.; Yao, J.; Han, W.; Pan, H. Nuclear factor of activated T cells in cancer development and treatment. Cancer Lett. 2015, 361, 174–184. [Google Scholar] [CrossRef]
- Vaupel, P.; Kelleher, D.K.; Höckel, M. Oxygen status of malignant tumors: Pathogenesis of hypoxia and significance for tumor therapy. Semin. Oncol. 2001, 28, 29–35. [Google Scholar] [CrossRef]
- Pagé, E.L.; Robitaille, G.A.; Pouysségur, J.; Richard, D.E. Induction of Hypoxia-inducible Factor-1 by Transcriptional and Translational Mechanisms. J. Boil. Chem. 2002, 277, 48403–48409. [Google Scholar] [CrossRef] [PubMed]
- M-L, F.; Lam, S.; Dong, X.; Chen, Y.; Leung, P.S. Postnatal hypoxemia increases angiotensin II sensitivity and up-regulates AT1a angiotensin receptors in rat carotid body chemoreceptors. J. Endocrinol. 2002, 173, 305–313. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, R.; Wu, Y.; Zhao, M.; Liu, C.; Zhou, L.; Shen, S.; Liao, S.; Yang, K.; Li, Q.; Wan, H. Role of HIF-1α in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L631–L640. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Feng, Y.; Wan, H.; Ni, L.; Qian, Y.; Guo, Y.; Xiang, Y.; Li, Q. Hypoxia induces dysregulation of local renin-angiotensin system in mouse Lewis lung carcinoma cells. Genet. Mol. Res. 2014, 13, 10562–10573. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.E. Non-hypoxic pathway mediates the induction of hypoxia inducible factor 1 alpha (HIF-1 alpha ) in vascular smooth muscle cells. J. Boil. Chem. 2000, 275, 26765–26771. [Google Scholar] [CrossRef]
- Sanchez-Lopez, E.; López, A.F.; Esteban, V.; Yagüe, S.; Egido, J.; Ruiz-Ortega, M.; Alvarez-Arroyo, M.V. Angiotensin II Regulates Vascular Endothelial Growth Factor via Hypoxia-Inducible Factor-1α Induction and Redox Mechanisms in the Kidney. Antioxidants Redox Signal. 2005, 7, 1275–1284. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, J.W.; Hu, L.; Song, Y.C.; Zhou, L.; Fan, Y.; Zhu, H.-Y.; Wang, Y.; Li, Q.-P. Activation of the AT1R/HIF-1 alpha /ACE axis mediates angiotensin II-induced VEGF synthesis in mesenchymal stem cells. Biomed. Res. Int. 2014, 2014, 627380. [Google Scholar] [CrossRef]
- Imai, N.; Hashimoto, T.; Kihara, M.; Yoshida, S.-I.; Kawana, I.; Yazawa, T.; Kitamura, H.; Umemura, S. Roles for host and tumor angiotensin II type 1 receptor in tumor growth and tumor-associated angiogenesis. Lab. Investig. 2006, 87, 189–198. [Google Scholar] [CrossRef]
- Pan, P.; Fu, H.; Zhang, L.; Huang, H.; Luo, F.; Wu, W.; Guo, Y.; Liu, X. Angiotensin II upregulates the expression of placental growth factor in human vascular endothelial cells and smooth muscle cells. BMC Cell Biol. 2010, 11, 36. [Google Scholar] [CrossRef]
- Fischer, C.; Jonckx, B.; Mazzone, M.; Zacchigna, S.; Loges, S.; Pattarini, L.; Chorianopoulos, E.; Liesenborghs, L.; Koch, M.; De Mol, M.; et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell 2007, 131, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Autiero, M.; Waltenberger, J.; Communi, D.; Kranz, A.; Moons, L.; Lambrechts, D.; Kroll, J.; Plaisance, S.; De Mol, M.; Bono, F.; et al. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat. Med. 2003, 9, 936–943. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, T.; Lou, Y.; Yan, B.; Cui, S.; Jiang, L.; Han, B. Placental Growth Factor Promotes Metastases of Non-Small Cell Lung Cancer Through MMP9. Cell. Physiol. Biochem. 2015, 37, 1210–1218. [Google Scholar] [CrossRef] [PubMed]
- Clere, N.; Corre, I.; Faure, S.; Guihot, A.-L.; Vessières, E.; Chalopin, M.; Morel, A.; Coqueret, O.; Hein, L.; Delneste, Y.; et al. Deficiency or blockade of angiotensin II type 2 receptor delays tumorigenesis by inhibiting malignant cell proliferation and angiogenesis. Int. J. Cancer 2010, 127, 2279–2291. [Google Scholar] [CrossRef] [PubMed]
- Petty, W.J.; Aklilu, M.; Varela, V.A.; Lovato, J.; Savage, P.D.; Miller, A.A. Reverse translation of phase I biomarker findings links the activity of angiotensin-(1–7) to repression of hypoxia inducible factor-1alpha in vascular sarcomas. BMC Cancer 2012, 12, 404. [Google Scholar] [CrossRef] [PubMed]
- Prontera, C.; Rossi, C.; Poggi, A.; Rotilio, M.; Rossi, F.; Mariani, B. Inhibition of gelatinase A (MMP-2) by batimastat and captopril reduces tumor growth and lung metastases in mice bearing Lewis lung carcinoma. Int. J. Cancer 1999, 81, 761–766. [Google Scholar] [CrossRef]
- Cheng, Q.; Zhou, L.; Zhou, J.; Wan, H.; Li, Q.Y.; Feng, Y. ACE2 overexpression inhibits acquired platinum resistance-induced tumor angiogenesis in NSCLC. Oncol. Rep. 2016, 36, 1403–1410. [Google Scholar] [CrossRef]
- Pei, N.; Wan, R.; Chen, X.; Li, A.; Zhang, Y.; Li, J.; Du, H.; Chen, B.; Wei, W.; Qi, Y.; et al. Angiotensin-(1–7) Decreases Cell Growth and Angiogenesis of Human Nasopharyngeal Carcinoma Xenografts. Mol. Cancer Ther. 2016, 15, 37–47. [Google Scholar] [CrossRef]
- Gomes, M.; Teixeira, A.L.; Coelho, A.; Araujo, A.C.; Medeiros, R. The Role of Inflammation in Lung Cancer. Adv. Exp. Med. Biol. 2014, 816, 1–23. [Google Scholar]
- Engels, E.A. Inflammation in the development of lung cancer: Epidemiological evidence. Expert Rev. Anticancer. Ther. 2008, 8, 605–615. [Google Scholar] [CrossRef]
- Engels, E.A.; Wu, X.; Gu, J.; Dong, Q.; Liu, J.; Spitz, M.R. Systematic Evaluation of Genetic Variants in the Inflammation Pathway and Risk of Lung Cancer. Cancer Res. 2007, 67, 6520–6527. [Google Scholar] [CrossRef][Green Version]
- Durham, A.; Adcock, I.M. The relationship between COPD and lung cancer. Lung Cancer 2015, 90, 121–127. [Google Scholar] [CrossRef]
- Archontogeorgis, K.; Steiropoulos, P.; Tzouvelekis, A.; Nena, E.; Bouros, D. Lung Cancer and Interstitial Lung Diseases: A Systematic Review. Pulm. Med. 2012, 2012, 1–11. [Google Scholar] [CrossRef]
- Capettini, L.; Montecucco, F.; Mach, F.; Stergiopulos, N.; Santos, R.A.; Da Silva, R.F. Role of renin-angiotensin system in inflammation, immunity and aging. Curr. Pharm. Des. 2012, 18, 963–970. [Google Scholar] [CrossRef]
- Husain, K.; Hernandez, W.; Ansari, R.A.; Ferder, L. Inflammation, oxidative stress and renin angiotensin system in atherosclerosis. World J. Boil. Chem. 2015, 6, 209–217. [Google Scholar] [CrossRef]
- Montecucco, F.; Pende, A.; Mach, F. The Renin-Angiotensin System Modulates Inflammatory Processes in Atherosclerosis: Evidence from Basic Research and Clinical Studies. Mediat. Inflamm. 2009, 2009, 1–13. [Google Scholar] [CrossRef]
- Sekiguchi, K.; Coker, M.; Flesch, M.; Barger, P.M.; Sivasubramanian, N.; Li, X.; Mann, D.L. Cross-regulation between the renin?angiotensin system and inflammatory mediators in cardiac hypertrophy and failure. Cardiovasc. Res. 2004, 63, 433–442. [Google Scholar] [CrossRef]
- Nakamura, K.; Yaguchi, T.; Ohmura, G.; Kobayashi, A.; Kawamura, N.; Iwata, T.; Kiniwa, Y.; Okuyama, R.; Kawakami, Y. Involvement of local renin-angiotensin system in immunosuppression of tumor microenvironment. Cancer Sci. 2017, 109, 54–64. [Google Scholar] [CrossRef]
- Allavena, P.; Mantovani, A. Immunology in the clinic review series; focus on cancer: Tumour-associated macrophages: Undisputed stars of the inflammatory tumour microenvironment. Clin. Exp. Immunol. 2012, 167, 195–205. [Google Scholar] [CrossRef]
- Reinartz, S.; Schumann, T.; Finkernagel, F.; Wortmann, A.; Jansen, J.M.; Meissner, W.; Krause, M.; Schwörer, A.-M.; Wagner, U.; Müller-Brüsselbach, S.; et al. Mixed-polarization phenotype of ascites-associated macrophages in human ovarian carcinoma: Correlation of CD163 expression, cytokine levels and early relapse. Int. J. Cancer 2013, 134, 32–42. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Almatroodi, S.A.; McDonald, C.F.; Pouniotis, D.S. Alveolar Macrophage Polarisation in Lung Cancer. Lung Cancer Int. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Cortez-Retamozo, V.; Etzrodt, M.; Newton, A.; Ryan, R.; Pucci, F.; Sio, S.W.; Kuswanto, W.; Rauch, P.J.; Chudnovskiy, A.; Iwamoto, Y.; et al. Angiotensin II drives the production of tumor-promoting macrophages. Immunity 2013, 38, 296–308. [Google Scholar] [CrossRef]
- Shen, X.Z.; Okwan-Duodu, D.; Blackwell, W.-L.; Ong, F.S.; Janjulia, T.; Bernstein, E.A.; Fuchs, S.; Alkan, S.; Bernstein, K. Myeloid expression of angiotensin-converting enzyme facilitates myeloid maturation and inhibits the development of myeloid-derived suppressor cells. Lab. Investig. 2014, 94, 536–544. [Google Scholar] [CrossRef]
- Benicky, J.; Sánchez-Lemus, E.; Pavel, J.; Saavedra, J.M. Anti-Inflammatory Effects of Angiotensin Receptor Blockers in the Brain and the Periphery. Cell. Mol. Neurobiol. 2009, 29, 781–792. [Google Scholar] [CrossRef]
- Abraham, F.; Sacerdoti, F.; De León, R.; Gentile, T.; Canellada, A. Angiotensin II Activates the Calcineurin/NFAT Signaling Pathway and Induces Cyclooxygenase-2 Expression in Rat Endometrial Stromal Cells. PLoS ONE 2012, 7, e37750. [Google Scholar] [CrossRef]
- Müller, M.R.; Rao, A. NFAT, immunity and cancer: A transcription factor comes of age. Nat. Rev. Immunol. 2010, 10, 645–656. [Google Scholar] [CrossRef]
- Marchesi, C.; Paradis, P.; Schiffrin, E.L. Role of the renin–angiotensin system in vascular inflammation. Trends Pharmacol. Sci. 2008, 29, 367–374. [Google Scholar] [CrossRef]
- Sabuhi, R.; Ali, Q.; Asghar, M.; Al-Zamily, N.R.H.; Hussain, T. Role of the angiotensin II AT2 receptor in inflammation and oxidative stress: Opposing effects in lean and obese Zucker rats. Am. J. Physiol. Physiol. 2011, 300, F700–F706. [Google Scholar] [CrossRef]
- Dagenais, N.J.; Jamali, F. Protective Effects of Angiotensin II Interruption: Evidence for Antiinflammatory Actions. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2005, 25, 1213–1229. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Lorenzo, O.; Ruperez, M.; Blanco, J.; Egido, J. Systemic infusion of angiotensin II into normal rats activates nuclear factor-kappaB and AP-1 in the kidney: Role of AT(1) and AT(2) receptors. Am. J. Pathol. 2001, 158, 1743–1756. [Google Scholar] [CrossRef]
- Terenzi, R.; Manetti, M.; Rosa, I.; Romano, E.; Galluccio, F.; Guiducci, S.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Angiotensin II type 2 receptor (AT2R) as a novel modulator of inflammation in rheumatoid arthritis synovium. Sci. Rep. 2017, 7, 13293. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Hu, X.; Zhang, W.; Tian, F. Angiotensin (1–7) ameliorates angiotensin II-induced inflammation by inhibiting LOX-1 expression. Inflamm. Res. 2013, 62, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Hammer, A.; Yang, G.; Friedrich, J.; Kovacs, A.; Lee, D.-H.; Grave, K.; Jörg, S.; Alenina, N.; Grosch, J.; Winkler, J.; et al. Role of the receptor Mas in macrophage-mediated inflammation in vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 14109–14114. [Google Scholar] [CrossRef]
- Padda, R.S.; Shi, Y.; Lo, C.S.; Zhang, S.L.; Chan, J.S. Angiotensin-(1–7): A Novel Peptide to Treat Hypertension and Nephropathy in Diabetes? J. Diabetes Metab. 2015, 11, 6. [Google Scholar]
- Deslypere, G.; Gullentops, D.; Wauters, E.; Vansteenkiste, J. Immunotherapy in non-metastatic non-small cell lung cancer: Can the benefits of stage IV therapy be translated into earlier stages? Ther. Adv. Med Oncol. 2018, 10. [Google Scholar] [CrossRef]
- Kim, J.M.; Chen, D.S. Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure). Ann. Oncol. 2016, 27, 1492–1504. [Google Scholar] [CrossRef]
- Yaguchi, T.Y.; Kawakami, Y. Cancer-induced heterogeneous immunosuppressive tumor microenvironments and their personalized modulation. Int. Immunol. 2016, 28, 393–399. [Google Scholar] [CrossRef]
- Milette, S.; Fiset, P.O.; Walsh, L.; Spicer, J.D.; Quail, D. The innate immune architecture of lung tumors and its implication in disease progression. J. Pathol. 2019, 247, 589–605. [Google Scholar] [CrossRef]
- Pinter, M.; Jain, R.K. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci. Transl. Med. 2017, 9, 5616. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, W.; Yuan, X.; Fu, M.; Qian, H.; Xu, W. Neutrophils in cancer development and progression: Roles, mechanisms, and implications (Review). Int. J. Oncol. 2016, 49, 857–867. [Google Scholar] [CrossRef]
- Wang, J.; Huang, X.; Li, F.-R. Impaired dendritic cell functions in lung cancer: A review of recent advances and future perspectives. Cancer Commun. 2019, 39, 43. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Cheng, T.; Lin, J.; Zhang, L.; Zheng, J.; Liu, Y.; Xie, G.; Wang, B.; Yuan, Y. Local angiotensin II contributes to tumor resistance to checkpoint immunotherapy. J. Immunother. Cancer 2018, 6, 88. [Google Scholar] [CrossRef] [PubMed]
- Vallejo-Ardila, D.L.; Fifis, T.; Burrell, L.M.; Walsh, K.; Christophi, C. Renin-angiotensin inhibitors reprogram tumor immune microenvironment: A comprehensive view of the influences on anti-tumor immunity. Oncotarget 2018, 9, 35500–35511. [Google Scholar] [CrossRef]
- Diop-Frimpong, B.; Chauhan, V.; Krane, S.; Boucher, Y.; Jain, R.K. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 2909–2914. [Google Scholar] [CrossRef]
- Cook, K.L.; Metheny-Barlow, L.J.; Tallant, E.A.; Gallagher, P.E. Angiotensin-(1–7) reduces fibrosis in orthotopic breast tumors. Cancer Res. 2010, 70, 8319–8328. [Google Scholar] [CrossRef]
- Tu, E.; Chia, P.Z.; Chen, W. TGFbeta in T cell biology and tumor immunity: Angel or devil? Cytokine Growth Factor Rev. 2014, 25, 423–435. [Google Scholar] [CrossRef]
- Kalinski, P. Regulation of immune responses by prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef]
- Bernstein, K.E.; Khan, Z.; Giani, J.F.; Cao, D.-Y.; Bernstein, E.A.; Shen, X.Z. Angiotensin-converting enzyme in innate and adaptive immunity. Nat. Rev. Nephrol. 2018, 14, 325–336. [Google Scholar] [CrossRef]
- Small, W.; James, L., Jr.; Moore, T.D.; Fintel, D.J.; Lutz, S.T.; Movsas, B.; Suntharalingam, M.; Garces, Y.I.; Ivker, R.; Moulder, J.; et al. Utility of the ACE Inhibitor captopril in mitigating radiation-associated pulmonary toxicity in lung cancer: Results from NRG oncology RTOG 0123. Am. J. Clin. Oncol. 2018, 41, 396–401. [Google Scholar] [CrossRef]
- Sio, T.T.; Atherton, P.J.; Pederson, L.D.; Zhen, W.K.; Mutter, R.W.; Garces, Y.I.; Ma, D.J.; Leenstra, J.L.; Rwigema, J.-C.M.; Dakhil, S.; et al. Daily Lisinopril vs Placebo for Prevention of Chemoradiation-Induced Pulmonary Distress in Patients With Lung Cancer (Alliance MC1221): A Pilot Double-Blind Randomized Trial. Int. J. Radiat. Oncol. 2018, 103, 686–696. [Google Scholar] [CrossRef]
- Couluris, M.; Kinder, B.W.; Xu, P.; Gross-King, M.; Krischer, J.; Panos, R.J. Treatment of idiopathic pulmonary fibrosis with losartan: A pilot project. Lung 2012, 190, 523–527. [Google Scholar] [CrossRef]
- Petty, W.J.; Miller, A.A.; McCoy, T.P.; Gallagher, P.E.; Tallant, E.A.; Torti, F.M. Phase I and pharmacokinetic study of angiotensin-(1–7), an endogenous antiangiogenic hormone. Clin. Cancer Res. 2009, 15, 7398–7404. [Google Scholar] [CrossRef]
- Savage, P.D.; Lovato, J.; Brosnihan, K.B.; Miller, A.A.; Petty, W.J. Phase II Trial of Angiotensin-(1–7) for the treatment of patients with metastatic sarcoma. Sarcoma 2016, 2016, 4592768. [Google Scholar] [CrossRef]
- Aydiner, A.; Ciftci, R.; Sen, F. Renin-Angiotensin System Blockers May Prolong Survival of Metastatic Non-Small Cell Lung Cancer Patients Receiving Erlotinib. Medicine 2015, 94, e887. [Google Scholar] [CrossRef]
- Zhang, W.; Liang, Z.; Li, J.; Cai, S. Angiotensin receptor blockers use and the risk of lung cancer: A meta-analysis. J. Renin-Angiotensin-Aldosterone Syst. 2015, 16, 768–773. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, J.; Chen, J.; Li, X.; Wu, Y.; Chen, H.; Wu, W.; Zhang, K.; Gu, L. Angiotensin receptor blockers (ARBs) reduce the risk of lung cancer: A systematic review and meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 12656–12660. [Google Scholar]
- Hicks, B.M.; Filion, K.B.; Yin, H.; Sakr, L.; Udell, J.A.; Azoulay, L. Angiotensin converting enzyme inhibitors and risk of lung cancer: Population based cohort study. BMJ 2018, 363, k4209. [Google Scholar] [CrossRef]
- Taddei, S.; Bortolotto, L.A. Unraveling the Pivotal Role of Bradykinin in ACE Inhibitor Activity. Am. J. Cardiovasc. Drugs 2016, 16, 309–321. [Google Scholar] [CrossRef]
- Tiret, L.; Rigat, B.; Visvikis, S.; Breda, C.; Corvol, P.; Cambien, F.; Soubrier, F. Evidence, from combined segregation and linkage analysis, that a variant of the angiotensin I-converting enzyme (ACE) gene controls plasma ACE levels. Am. J. Hum. Genet. 1992, 51, 197–205. [Google Scholar]
- Chen, J.; Guo, L.; Peiffer, D.A.; Zhou, L.; Chan, O.T.M.; Bibikova, M.; Wickham-Garcia, E.; Lu, S.-H.; Zhan, Q.; Wang-Rodriguez, J.; et al. Genomic profiling of 766 cancer-related genes in archived esophageal normal and carcinoma tissues. Int. J. Cancer 2008, 122, 2249–2254. [Google Scholar] [CrossRef]
- Fouad, Y.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036. [Google Scholar]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catarata, M.J.; Ribeiro, R.; Oliveira, M.J.; Robalo Cordeiro, C.; Medeiros, R. Renin-Angiotensin System in Lung Tumor and Microenvironment Interactions. Cancers 2020, 12, 1457. https://doi.org/10.3390/cancers12061457
Catarata MJ, Ribeiro R, Oliveira MJ, Robalo Cordeiro C, Medeiros R. Renin-Angiotensin System in Lung Tumor and Microenvironment Interactions. Cancers. 2020; 12(6):1457. https://doi.org/10.3390/cancers12061457
Chicago/Turabian StyleCatarata, Maria Joana, Ricardo Ribeiro, Maria José Oliveira, Carlos Robalo Cordeiro, and Rui Medeiros. 2020. "Renin-Angiotensin System in Lung Tumor and Microenvironment Interactions" Cancers 12, no. 6: 1457. https://doi.org/10.3390/cancers12061457
APA StyleCatarata, M. J., Ribeiro, R., Oliveira, M. J., Robalo Cordeiro, C., & Medeiros, R. (2020). Renin-Angiotensin System in Lung Tumor and Microenvironment Interactions. Cancers, 12(6), 1457. https://doi.org/10.3390/cancers12061457