Inflammatory Cytokines and ctDNA Are Biomarkers for Progression in Advanced-Stage Melanoma Patients Receiving Checkpoint Inhibitors

 , ,
, ,
Abstract
1. Introduction
Translational Relevance
2. Materials and Methods
2.1. Patients and Treatment
2.2. Disease Characteristics and Response Assessment
2.3. Sample Collection and Preparation
2.4. Cell-Free DNA (cfDNA) Extraction
2.5. Droplet Digital PCR (ddPCR)
2.6. O-Link Analysis
2.7. Cytokine Analysis by Meso Scale Discovery
2.8. Statistical Analysis
3. Results
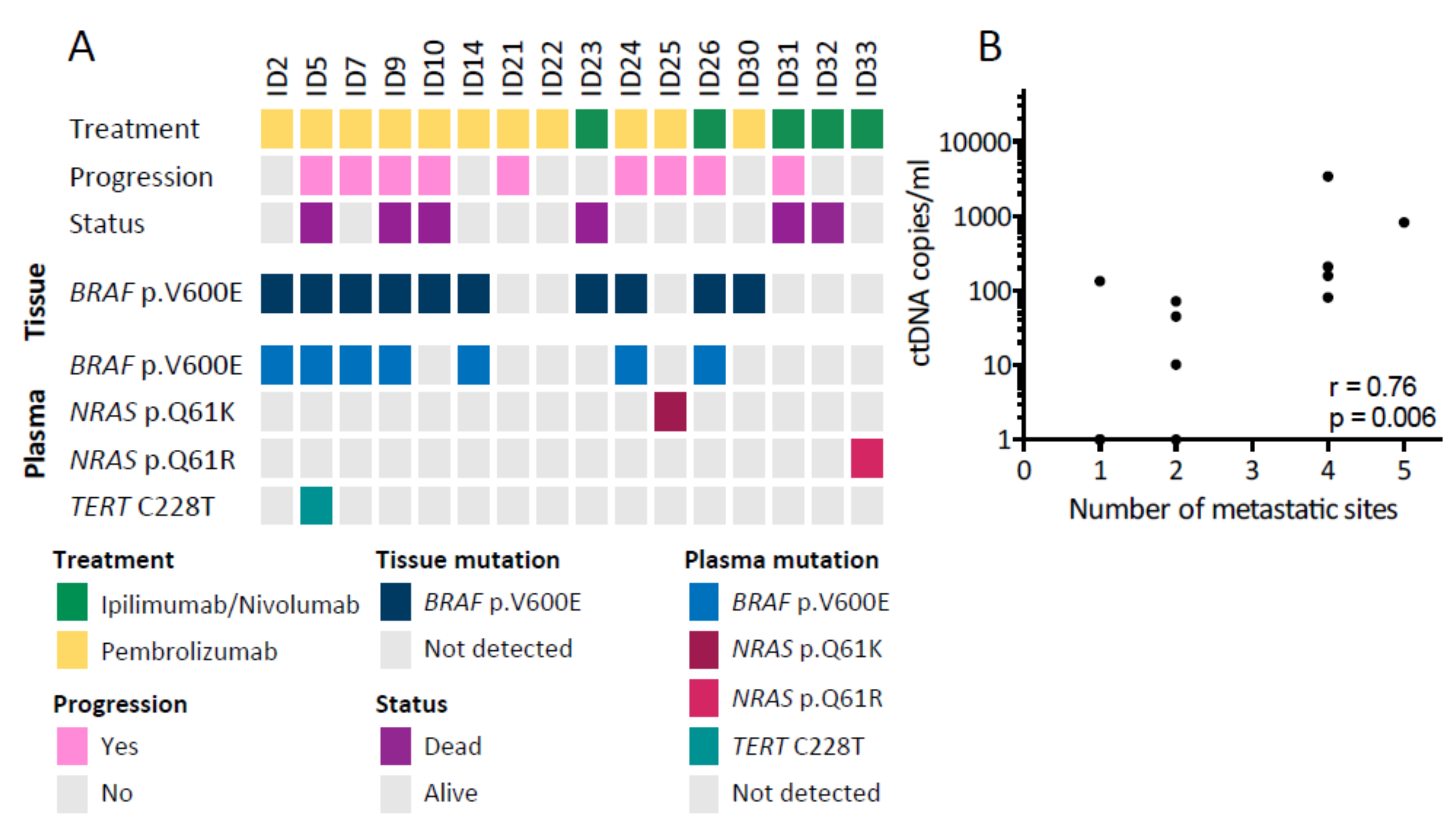
3.1. Patient Characteristics
3.2. Detection of ctDNA at Baseline
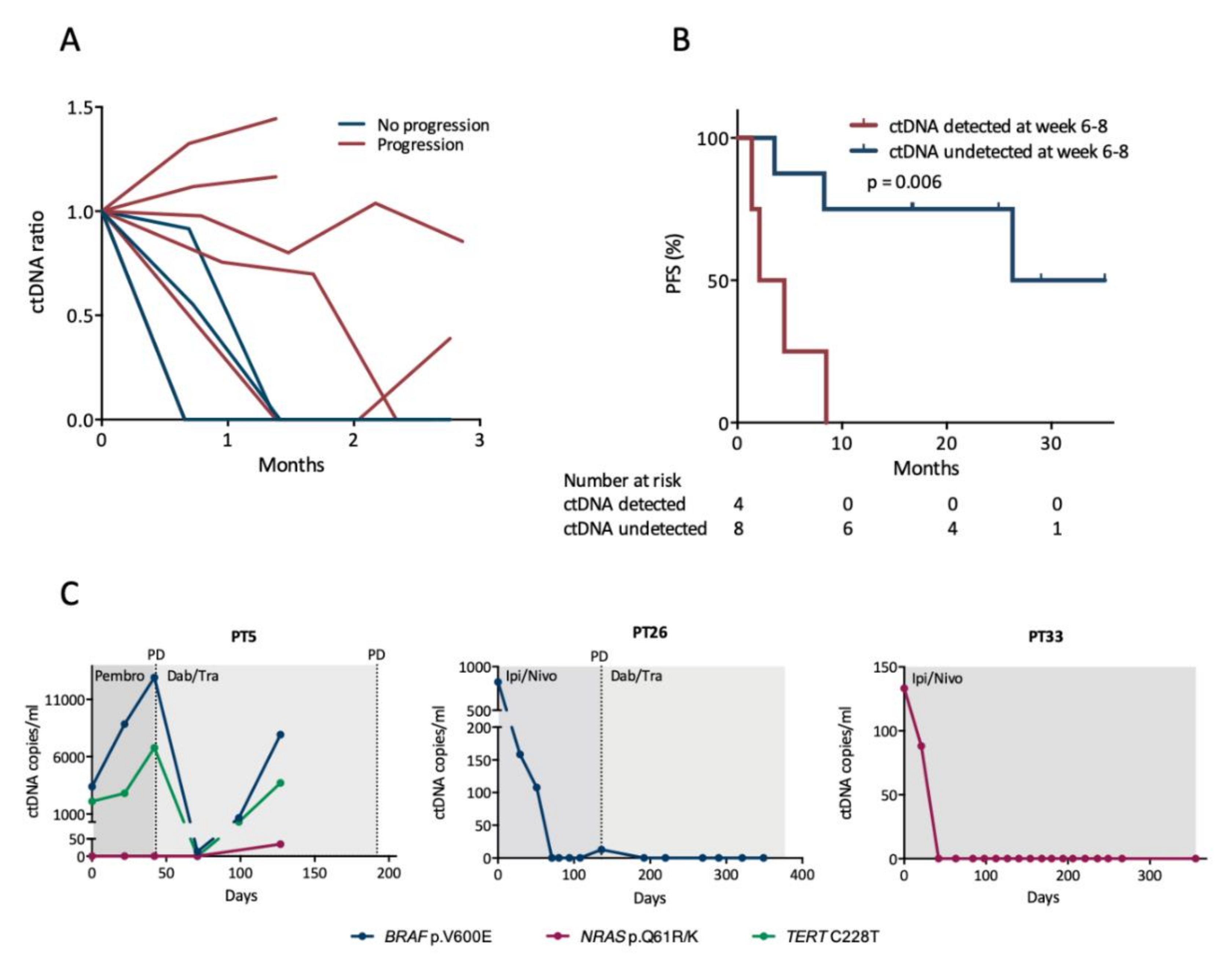
3.3. Early Changes in ctDNA Level Are Associated with Progression-Free Survival
3.4. ctDNA Levels Mirror Clinical Status during Longitudinal Monitoring
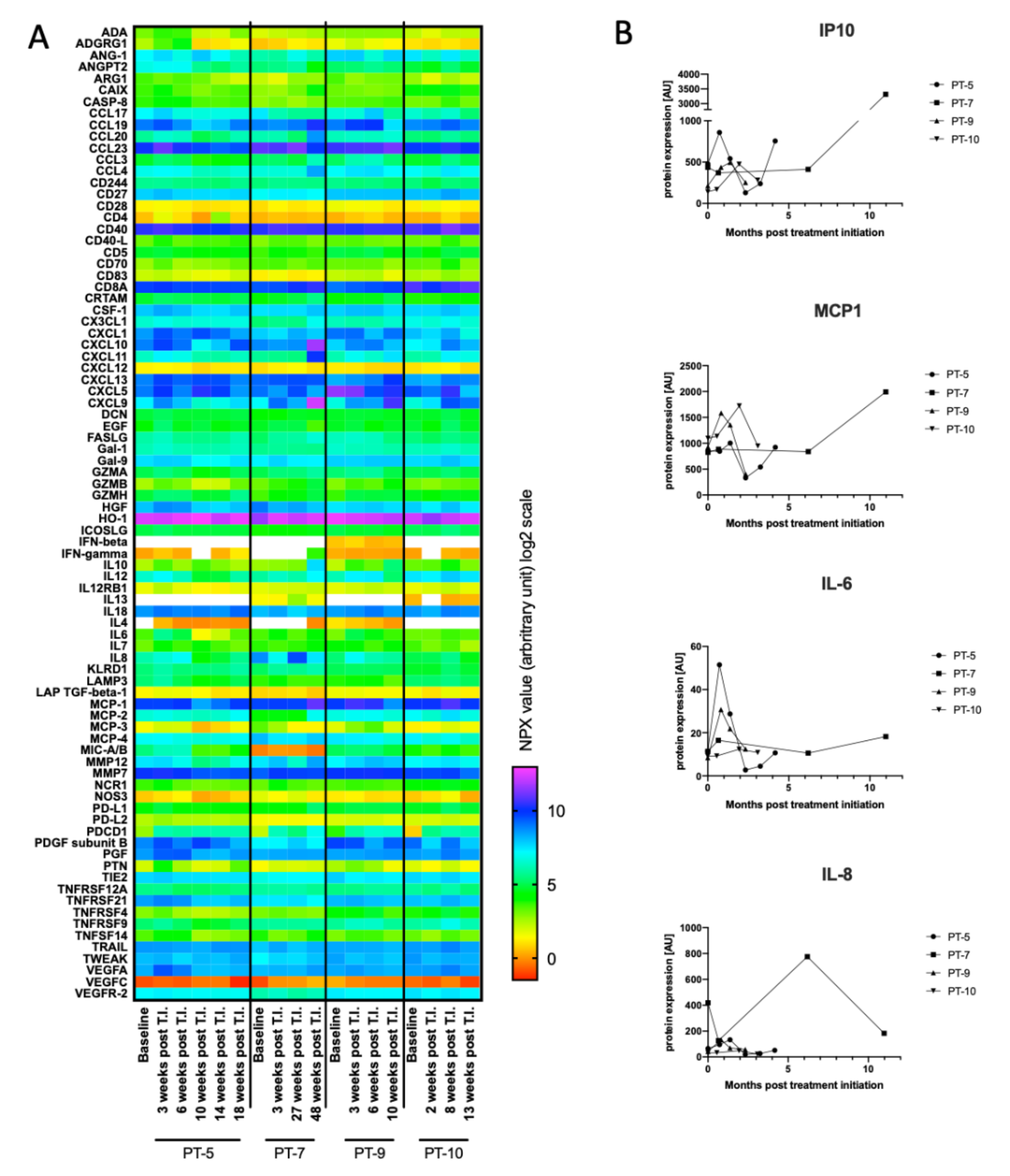
3.5. Initial Characterization of Cytokines as Biomarkers for Treatment Response
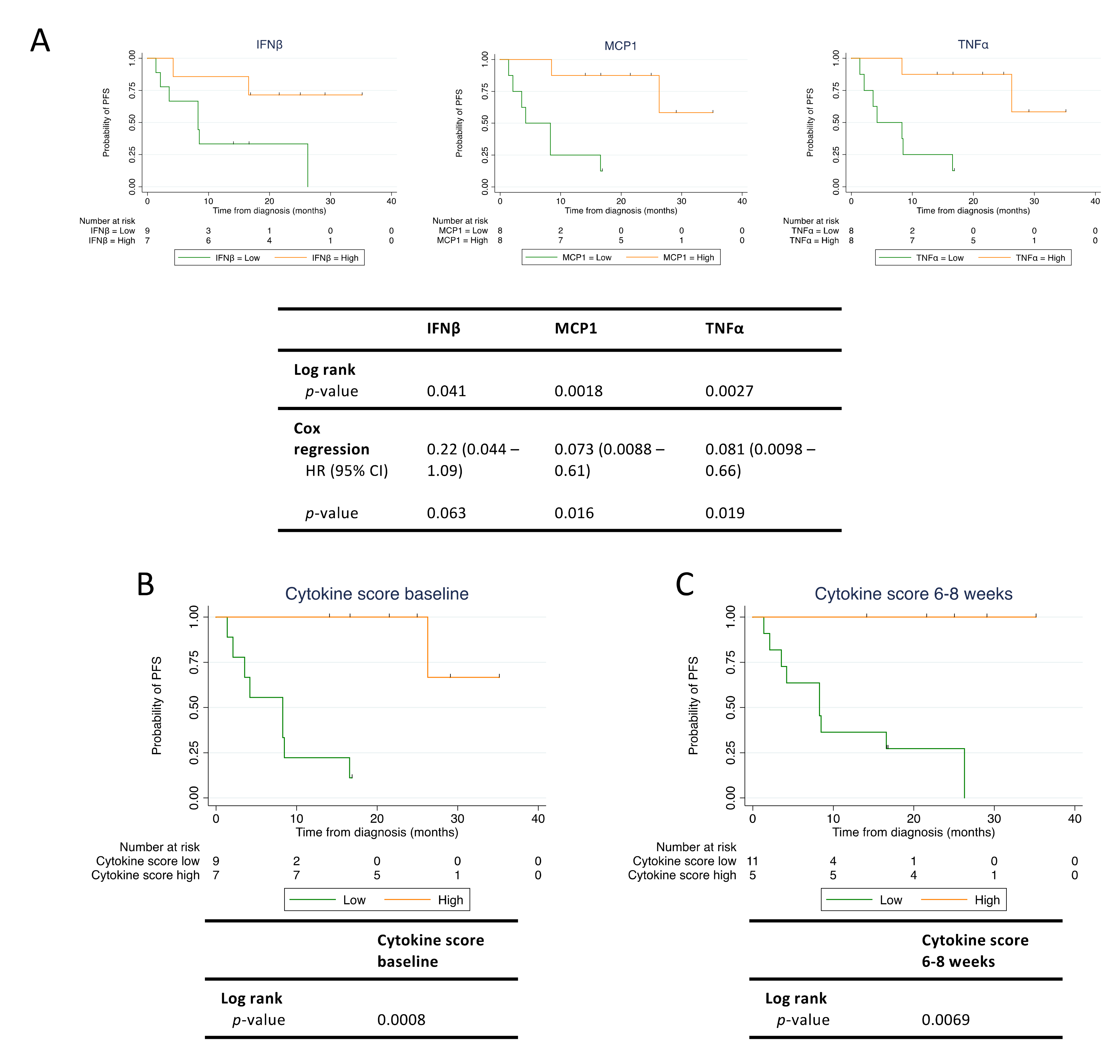
3.6. Baseline Levels of MCP1 and TNFα Correlate with Progression-Free Survival
3.7. The Immune Cytokine Score Strengthens the Prediction of PFS
3.8. ctDNA and Cytokines Are Independent Predictors of PFS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cancer Facts & Figures. 2019. Available online: http://files/761/cancer-facts-and-figures-2019.pdf (accessed on 28 January 2020).
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.-J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Sileni, V.C.; Gonzalez, R.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef]
- Hodi, F.S.; Sileni, V.C.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Daud, A.; Wolchok, J.D.; Robert, C.; Hwu, W.-J.; Weber, J.S.; Ribas, A.; Hodi, F.S.; Joshua, A.M.; Kefford, R.; Hersey, P.; et al. Programmed Death-Ligand 1 Expression and Response to the Anti-Programmed Death 1 Antibody Pembrolizumab in Melanoma. J. Clin. Oncol. 2016, 34, 4102–4109. [Google Scholar] [CrossRef]
- Madore, J.; Vilain, R.E.; Menzies, A.M.; Kakavand, H.; Wilmott, J.S.; Hyman, J.; Yearley, J.H.; Kefford, R.F.; Thompson, J.F.; Long, G.V.; et al. PD-L1 expression in melanoma shows marked heterogeneity within and between patients: Implications for anti-PD-1/PD-L1 clinical trials. Pigment Cell Melanoma Res. 2015, 28, 245–253. [Google Scholar]
- McEvoy, A.C.; Warburton, L.; Al-Ogaili, Z.; Celliers, L.; Calapre, L.; Pereira, M.R.; Khattak, M.A.; Meniawy, T.; Millward, M.; Ziman, M.; et al. Correlation between circulating tumour DNA and metabolic tumour burden in metastatic melanoma patients. BMC Cancer 2018, 18, 726. [Google Scholar] [CrossRef]
- Winther-Larsen, A.; DeMuth, C.; Fledelius, J.; Madsen, A.T.; Hjorthaug, K.; Meldgaard, P.; Sorensen, B. Correlation between circulating mutant DNA and metabolic tumour burden in advanced non-small cell lung cancer patients. Br. J. Cancer 2017, 117, 704–709. [Google Scholar] [CrossRef]
- Cabel, L.; Riva, F.; Servois, V.; Livartowski, A.; Daniel, C.; Rampanou, A.; Lantz, O.; Romano, E.; Milder, M.; Buecher, B.; et al. Circulating tumor DNA changes for early monitoring of anti-PD1 immunotherapy: A proof-of-concept study. Ann. Oncol. 2017, 28, 1996–2001. [Google Scholar] [CrossRef]
- Gray, E.S.; Rizos, H.; Reid, A.L.; Boyd, S.C.; Pereira, M.R.; Lo, J.; Tembe, V.; Freeman, J.; Lee, J.H.; Scolyer, R.; et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015, 6, 42008–42018. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Long, G.V.; Boyd, S.; Lo, S.; Menzies, A.M.; Tembe, V.; Guminski, A.; Jakrot, V.; Scolyer, R.A.; Mann, G.J.; et al. Circulating tumour DNA predicts response to anti-PD1 antibodies in metastatic melanoma. Ann. Oncol. 2017, 28, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Lipson, E.J.; Velculescu, V.E.; Pritchard, T.S.; Sausen, M.; Pardoll, D.M.; Topalian, S.L.; Diaz, L.A. Circulating tumor DNA analysis as a real-time method for monitoring tumor burden in melanoma patients undergoing treatment with immune checkpoint blockade. J. Immunother. Cancer 2014, 2, 42. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Walker, A.; Gagnon, R.; Mazumdar, J.; Casey, M.; Long, G.V.; Schadendorf, D.; Flaherty, K.; Kefford, R.; Hauschild, A.; Hwu, P.; et al. Correlation of BRAF Mutation Status in Circulating-Free DNA and Tumor and Association with Clinical Outcome across Four BRAFi and MEKi Clinical Trials. Clin. Cancer Res. 2016, 22, 567–574. [Google Scholar] [CrossRef]
- Váraljai, R.; Wistuba-Hamprecht, K.; Seremet, T.; Diaz, J.M.S.; Nsengimana, J.; Sucker, A.; Griewank, K.; Placke, J.-M.; Horn, P.A.; Von Neuhoff, N.; et al. Application of Circulating Cell-Free Tumor DNA Profiles for Therapeutic Monitoring and Outcome Prediction in Genetically Heterogeneous Metastatic Melanoma. JCO Precis. Oncol. 2019, 3, 1–10. [Google Scholar] [CrossRef]
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef]
- Yum, S.; Li, M.; Frankel, A.E.; Chen, Z.J. Roles of the cGAS-STING Pathway in Cancer Immunosurveillance and Immunotherapy. Annu. Rev. Cancer Biol. 2019, 3, 323–344. [Google Scholar] [CrossRef]
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 152. [Google Scholar] [CrossRef]
- Woo, S.R.; Corrales, L.; Gajewski, T.F. The STING pathway and the T cell-inflamed tumor microenvironment. Trends Immunol. 2015, 36, 250–256. [Google Scholar] [CrossRef]
- Yamazaki, N.; Kiyohara, Y.; Uhara, H.; Iizuka, H.; Uehara, J.; Otsuka, F.; Fujisawa, Y.; Takenouchi, T.; Isei, T.; Iwatsuki, K.; et al. Cytokine biomarkers to predict antitumor responses to nivolumab suggested in a phase 2 study for advanced melanoma. Cancer Sci. 2017, 108, 1022–1031. [Google Scholar] [CrossRef]
- Nonomura, Y.; Otsuka, A.; Nakashima, C.; Seidel, J.; Kitoh, A.; Dainichi, T.; Nakajima, S.; Sawada, Y.; Matsushita, S.; Aoki, M.; et al. Peripheral blood Th9 cells are a possible pharmacodynamic biomarker of nivolumab treatment efficacy in metastatic melanoma patients. Oncoimmunology 2016, 5, e1248327. [Google Scholar] [CrossRef] [PubMed]
- Tarhini, A.; Zahoor, H.; Lin, Y.; Malhotra, U.; Sander, C.; Butterfield, L.H.; Kirkwood, J.M. Baseline circulating IL-17 predicts toxicity while TGF-β1 and IL-10 are prognostic of relapse in ipilimumab neoadjuvant therapy of melanoma. J. Immunother. Cancer 2015, 3, 39. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.-H.; Aiba, S.; Bröcker, E.-B.; LeBoit, P.E.; et al. Distinct Sets of Genetic Alterations in Melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Colombino, M.; Capone, M.; Lissia, A.; Cossu, A.G.M.; Rubino, C.; De Giorgi, V.; Massi, D.; Fonsatti, E.; Staibano, S.; Nappi, O.; et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J. Clin. Oncol. 2012, 30, 2522–2529. [Google Scholar] [CrossRef] [PubMed]
- Vinagre, J.; Almeida, A.; Pópulo, H.; Batista, R.; Lyra, J.; Pinto, V.; Coelho, R.; Celestino, R.; Prazeres, H.; Lima, L.; et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013, 4, 1–6. [Google Scholar] [CrossRef]
- Griewank, K.G.; Murali, R.; Puig-Butille, J.A.; Schilling, B.; Livingstone, E.; Potrony, M.; Carrera, C.; Schimming, T.; Möller, I.; Schwamborn, M.; et al. TERT promoter mutation status as an independent prognostic factor in cutaneous melanoma. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef]
- Wong, S.Q.; Raleigh, J.M.; Callahan, J.; Vergara, I.A.; Ftouni, S.; Hatzimihalis, A.; Colebatch, A.J.; Li, J.; Semple, T.; Doig, K.; et al. Circulating Tumor DNA Analysis and Functional Imaging Provide Complementary Approaches for Comprehensive Disease Monitoring in Metastatic Melanoma. JCO Precis. Oncol. 2017, 1, 1–14. [Google Scholar] [CrossRef]
- Calapre, L.; Giardina, T.; Robinson, C.; Reid, A.L.; Al-Ogaili, Z.; Pereira, M.R.; McEvoy, A.C.; Warburton, L.; Hayward, N.K.; Khattak, M.A.; et al. Locus-specific concordance of genomic alterations between tissue and plasma circulating tumor DNA in metastatic melanoma. Mol. Oncol. 2019, 13, 171–184. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef]
- Karachaliou, N.; González-Cao, M.; Crespo, G.; Drozdowskyj, A.; Aldeguer, E.; Giménez-Capitán, A.; Teixido, C.; Molina-Vila, M.Á.; Viteri, S.; Gil, M.D.L.L.; et al. Interferon gamma, an important marker of response to immune checkpoint blockade in non-small cell lung cancer and melanoma patients. Ther. Adv. Med. Oncol. 2018, 10, 1758834017749748. [Google Scholar] [CrossRef]
- Lim, S.Y.; Lee, J.H.; Gide, T.N.; Menzies, A.M.; Guminski, A.; Carlino, M.S.; Breen, E.J.; Yang, J.Y.H.; Ghazanfar, S.; Kefford, R.; et al. Circulating cytokines predict immune-related toxicity in melanoma patients receiving anti-PD-1–based immunotherapy. Clin. Cancer Res. 2018, 25, 1557–1563. [Google Scholar] [CrossRef] [PubMed]
- DeMaria, O.; Cornen, S.; Daëron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Ayers, M.; Lunceford, J.K.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, S.S.; Keilholz, U.; Gilles, E.; Bedikian, A.Y.; Wu, J.; Kay, R.; Stein, C.A.; Itri, L.M.; Suciu, S.; Eggermont, A. LDH correlation with survival in advanced melanoma from two large, randomised trials (Oblimersen GM301 and EORTC 18951). Eur. J. Cancer 2009, 45, 1807–1814. [Google Scholar] [CrossRef]
- Eigentler, T.; Figl, A.; Rass, K.; Bostroem, A.; Heese, O.; Garbe, C.; Krex, D.; Mohr, P.; Mauch, C.; Koelbl, O.; et al. Number of metastases, serum lactate dehydrogenase level, and type of treatment are prognostic factors in patients with brain metastases of malignant melanoma. Cancer 2011, 117, 1697–1703. [Google Scholar] [CrossRef]
- Kelderman, S.; Heemskerk, B.; Van Tinteren, H.; Brom, R.R.H.V.D.; Hospers, G.A.P.; Eertwegh, A.J.M.V.D.; Kapiteijn, E.W.; De Groot, J.W.B.; Soetekouw, P.; Jansen, R.L.; et al. Lactate dehydrogenase as a selection criterion for ipilimumab treatment in metastatic melanoma. Cancer Immunol. Immunother. 2014, 3, 449–458. [Google Scholar] [CrossRef]
- Wagner, N.B.; Forschner, A.; Leiter, U.; Garbe, C.; Eigentler, T.K. S100B and LDH as early prognostic markers for response and overall survival in melanoma patients treated with anti-PD-1 or combined anti-PD-1 plus anti-CTLA-4 antibodies. Br. J. Cancer 2018, 119, 339–346. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Davis, A.A.; Patel, V.G. The role of PD-L1 expression as a predictive biomarker: An analysis of all US Food and Drug Administration (FDA) approvals of immune checkpoint inhibitors. J. Immunother. Cancer 2019, 7, 278. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Fernández-Landázuri, S.; Rodriguez, C.; Zarate, R.; Lozano, M.D.; Zubiri, L.; Pérez-Gracia, J.L.; Algarra, S.M.; Gonzalez, A.; Gracia, J.L.P. Quantitative Cell-Free Circulating BRAFV600E Mutation Analysis by Use of Droplet Digital PCR in the Follow-up of Patients with Melanoma Being Treated with BRAF Inhibitors. Clin. Chem. 2015, 61, 297–304. [Google Scholar] [CrossRef]
- Ji, R.-R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012, 61, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Clemente, C.G.; Mihm, M.C., Jr.; Bufalino, R.; Zurrida, S.; Collini, P.; Cascinelli, N. Prognostic value of tumor infiltrating lymphocytes in the vertical growth phase of primary cutaneous melanoma. Cancer 1996, 77, 1303–1310. [Google Scholar] [CrossRef]
- Hamid, O.; Schmidt, H.; Nissan, A.; Ridolfi, L.; Aamdal, S.; Hansson, J.; Guida, M.; Hyams, D.M.; Gómez, H.; Bastholt, L.; et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J. Transl. Med. 2011, 9, 204. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Pembrolizumab | Ipilimumab/ Nivolumab | Total |
|---|---|---|---|
| No. of patients | 11 | 5 | 16 |
| Age-yr | |||
| Median | 54 | 62 | 57 |
| Range | 44–67 | 45–75 | 44–75 |
| Sex-no. (%) | |||
| Male | 7 (63.6) | 4 (80) | 11 (69) |
| Female | 4 (36.4) | 1 (20) | 5 (31) |
| Lactate dehydrogenase-no. (%) | |||
| Normal | 7 (63.6) | 4 (80) | 11 (69) |
| Elevated | 4 (36.4) | 1 (20) | 5 (31) |
| BRAF V600 mutation, tissue-no. (%) | 8 (72.7) | 2 (40) | 10 (62,5) |
| Tumour PD-L1 expression-no (%) | |||
| <1% | 1 (9) | 5 (100) | 6 (37.5) |
| ≥1% | 5 (45.5) | 0 (0) | 5 (31.3) |
| N/A | 5 (45.5) | 0 (0) | 5 (31.3) |
| Tumour stage-no. (%) | |||
| Stage III | 1 (9) | 0 (0) | 1 (6) |
| Stage IV | 10 (91) | 5 (100) | 15 (94) |
| Progression-no. (%) | 7 (63.6) | 2 (40) | 9 (56.3) |
| MORS-no. (%) | 3 (27.2) | 3 (60) | 6 (37.5) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedersen, J.G.; Madsen, A.T.; Gammelgaard, K.R.; Aggerholm-Pedersen, N.; Sørensen, B.S.; Øllegaard, T.H.; Jakobsen, M.R. Inflammatory Cytokines and ctDNA Are Biomarkers for Progression in Advanced-Stage Melanoma Patients Receiving Checkpoint Inhibitors. Cancers 2020, 12, 1414. https://doi.org/10.3390/cancers12061414
Pedersen JG, Madsen AT, Gammelgaard KR, Aggerholm-Pedersen N, Sørensen BS, Øllegaard TH, Jakobsen MR. Inflammatory Cytokines and ctDNA Are Biomarkers for Progression in Advanced-Stage Melanoma Patients Receiving Checkpoint Inhibitors. Cancers. 2020; 12(6):1414. https://doi.org/10.3390/cancers12061414
Chicago/Turabian StylePedersen, Jesper Geert, Anne Tranberg Madsen, Kristine Raaby Gammelgaard, Ninna Aggerholm-Pedersen, Boe Sandahl Sørensen, Trine Heide Øllegaard, and Martin Roelsgaard Jakobsen. 2020. "Inflammatory Cytokines and ctDNA Are Biomarkers for Progression in Advanced-Stage Melanoma Patients Receiving Checkpoint Inhibitors" Cancers 12, no. 6: 1414. https://doi.org/10.3390/cancers12061414
APA StylePedersen, J. G., Madsen, A. T., Gammelgaard, K. R., Aggerholm-Pedersen, N., Sørensen, B. S., Øllegaard, T. H., & Jakobsen, M. R. (2020). Inflammatory Cytokines and ctDNA Are Biomarkers for Progression in Advanced-Stage Melanoma Patients Receiving Checkpoint Inhibitors. Cancers, 12(6), 1414. https://doi.org/10.3390/cancers12061414