Pre-Treatment Mutational and Transcriptomic Landscape of Responding Metastatic Melanoma Patients to Anti-PD1 Immunotherapy

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Review
2.2. Sample Collection and Nucleic Acid Isolation
2.3. Next-Generation Whole-Exome Sequencing and Analysis
2.4. Next-Generation RNA Sequencing and Analysis
2.5. Data Availability
2.6. NFKBIE Wildtype and G34E Expression Vectors
2.7. NF-kB Luciferase Reporter Assay
2.8. Cell Surface Protein Analysis
3. Results
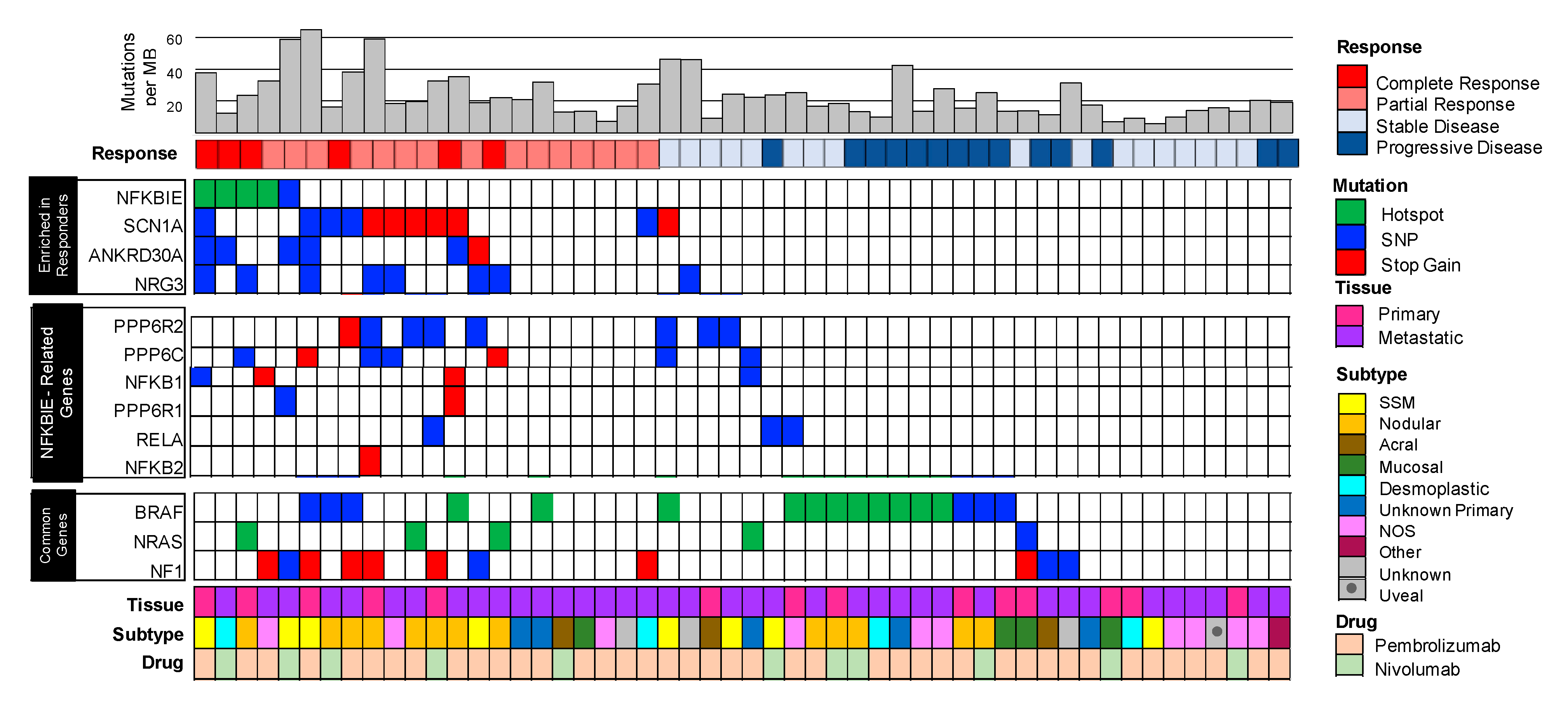
3.1. Clinical Analysis
3.2. Mutated Genes Enriched in Patients that Responded to Immunotherapy Included the Hotspot Variant G34E in NFKBIE, (NFkB Inhibitor Epsilon) the Gene that Encodes for I-kappa-B-Epsilon (IkBe), a Negative Regulator of NFkB
3.3. Enrichment of Mutations in NFKBIE-Related Genes Found in Responders Suggests Aberrations in the NFkB Signaling Pathway
3.4. Mutations in NFKBIE-Related Genes Correlate with Clinical Metrics Used to Measure Patient Responses to Immunotherapy
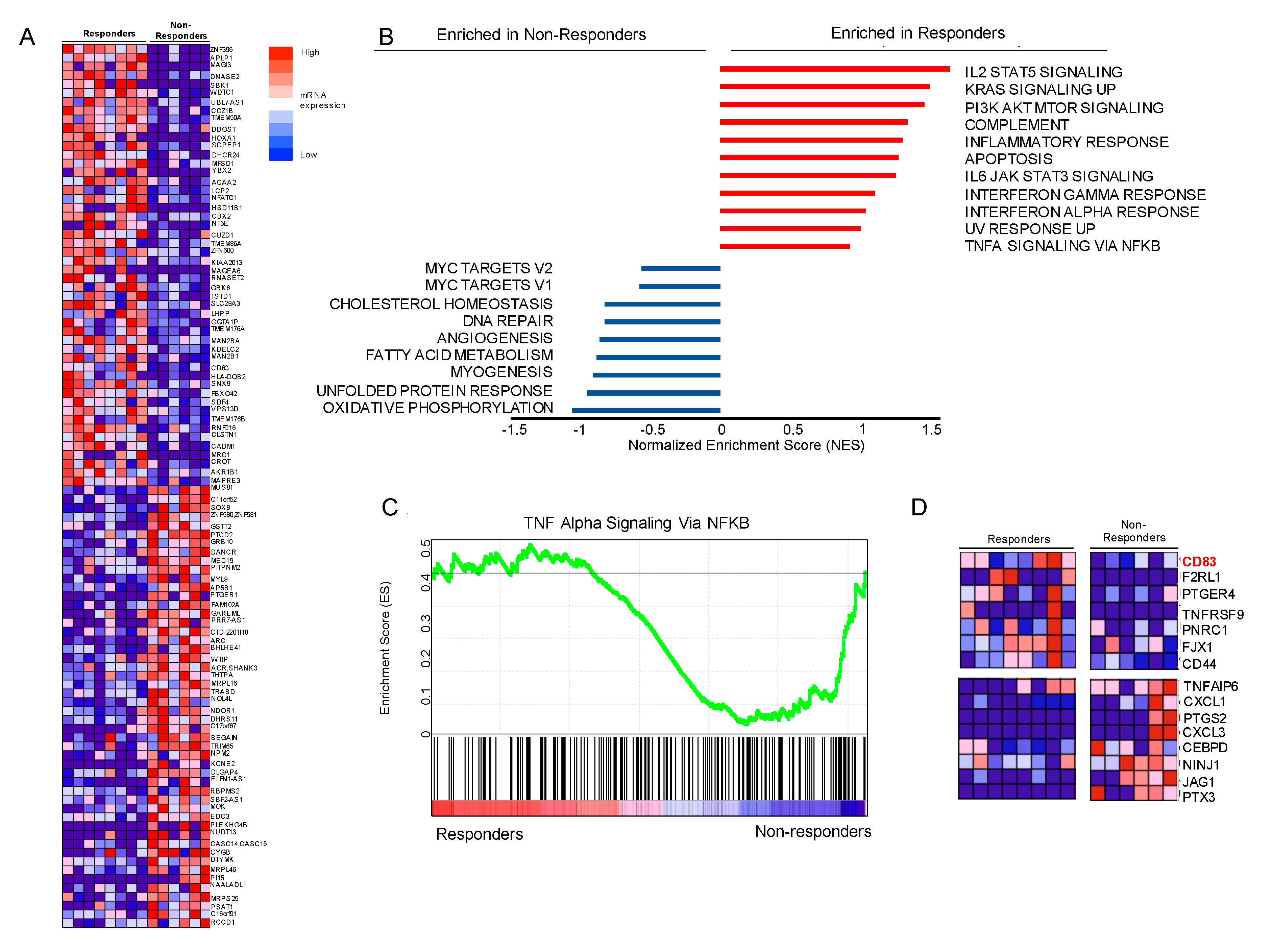
3.5. Gene Set Enrichment Analysis (GSEA) Suggests that Genes Involved in NFkB Signaling Are Differentially Expressed, Including Upregulation of CD83 in Our Responder Cohort
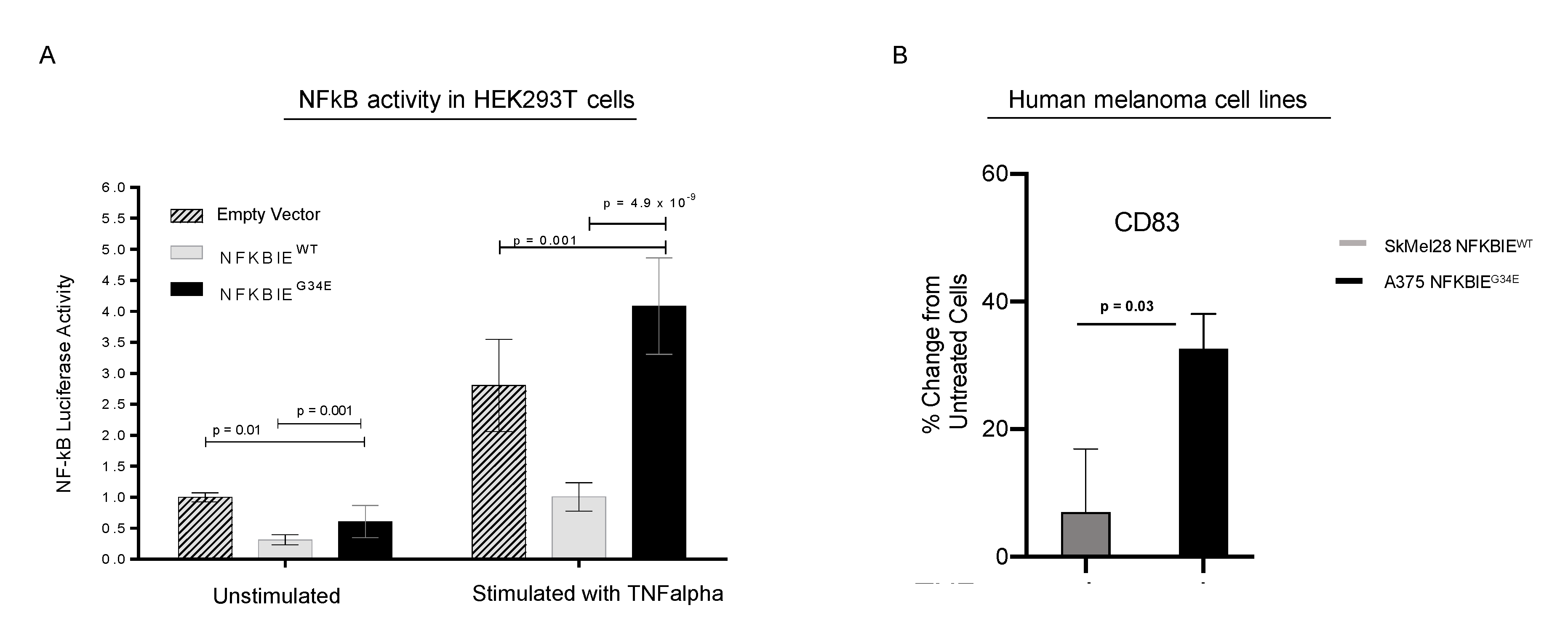
3.6. G34E of NFKBIE Is Likely a Loss-of-Function Variant, Resulting in Activation of NFkB Pathway
4. Discussion
5. Conculusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Achkar, T.; Tarhini, A.A. The use of immunotherapy in the treatment of melanoma. J. Hematol. Oncol. 2017, 10, 88. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant Pembrolizumab versus Placebo in Resected Stage III Melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Mandala, M.; Del Vecchio, M.; Gogas, H.J.; Arance, A.M.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Marquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef]
- Gibney, G.T.; Weiner, L.M.; Atkins, M.B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet Oncol. 2016, 17, e542–e551. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Hintzsche, J.D.; Gorden, N.T.; Amato, C.M.; Kim, J.; Wuensch, K.E.; Robinson, S.E.; Applegate, A.J.; Couts, K.L.; Medina, T.M.; Wells, K.R.; et al. Whole-exome sequencing identifies recurrent SF3B1 R625 mutation and comutation of NF1 and KIT in mucosal melanoma. Melanoma Res. 2017, 27, 189–199. [Google Scholar] [CrossRef]
- Royer-Bertrand, B.; Torsello, M.; Rimoldi, D.; El Zaoui, I.; Cisarova, K.; Pescini-Gobert, R.; Raynaud, F.; Zografos, L.; Schalenbourg, A.; Speiser, D.; et al. Comprehensive Genetic Landscape of Uveal Melanoma by Whole-Genome Sequencing. Am. J. Hum. Genet. 2016, 99, 1190–1198. [Google Scholar] [CrossRef]
- Prickett, T.D.; Zerlanko, B.; Gartner, J.J.; Parker, S.C.J.; Dutton-Regester, K.; Lin, J.C.; Teer, J.K.; Wei, X.; Jiang, J.; Nisc Comparative Sequencing, P.; et al. Somatic mutations in MAP3K5 attenuate its proapoptotic function in melanoma through increased binding to thioredoxin. J. Investig. Dermatol. 2014, 134, 452–460. [Google Scholar] [CrossRef]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer (Oxf. Engl. 1990) 2009, 45, 228–247. [Google Scholar] [CrossRef] [PubMed]
- Hintzsche, J.; Kim, J.; Yadav, V.; Amato, C.; Robinson, S.E.; Seelenfreund, E.; Shellman, Y.; Wisell, J.; Applegate, A.; McCarter, M.; et al. IMPACT: A whole-exome sequencing analysis pipeline for integrating molecular profiles with actionable therapeutics in clinical samples. J. Am. Med. Inform. Assoc. 2016, 23, 721–730. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef]
- University of California Santa Cruz Genomics Institute. Available online: https://www.genome.ucsc.edu. (accessed on 11 July 2020).
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2012, 31, 46–53. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545. [Google Scholar] [CrossRef]
- Gene Set Enrichment Analysis User Guide. Available online: https://www.gsea-msigdb.org/gsea/doc/GSEAUserGuideFrame.html?Interpreting_GSEA (accessed on 11 July 2020).
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ju, X.; Lee, K.; Clarke, C.; Hsu, J.L.; Abadir, E.; Bryant, C.E.; Pears, S.; Sunderland, N.; Heffernan, S.; et al. CD83 is a new potential biomarker and therapeutic target for Hodgkin lymphoma. Haematologica 2018, 103, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Ananiev, J.; Gulubova, M.V.; Manolova, I.M. Prognostic significance of CD83 positive tumor-infiltrating dendritic cells and expression of TGF-beta 1 in human gastric cancer. Hepatogastroenterology 2011, 58, 1834–1840. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Gaynor, R.B. IkappaB kinases: Key regulators of the NF-kappaB pathway. Trends Biochem. Sci. 2004, 29, 72–79. [Google Scholar] [CrossRef]
- Whiteside, S.T.; Epinat, J.C.; Rice, N.R.; Israël, A. I kappa B epsilon, a novel member of the IκB family, controls RelA and cRel NF-κB activity. EMBO J. 1997, 16, 1413–1426. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Gide, T.N.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Madore, J.; Lim, S.Y.; Velickovic, R.; Wongchenko, M.; et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255.e6. [Google Scholar] [CrossRef]
- Liu, D.; Schilling, B.; Sucker, A.; Livingstone, E.; Jerby-Amon, L.; Zimmer, L.; Gutzmer, R.; Satzger, I.; Loquai, C.; Grabbe, S.; et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med. 2019, 25, 1916–1927. [Google Scholar] [CrossRef]
- Maio, M.; Gulwani, B.; Morgano, A.; Ferrone, S. Differential modulation by tumor necrosis factor and immune interferon of HLA class-II antigens expressed by melanoma cells. Int. J. Cancer 1989, 44, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Jongsma, M.L.M.; Guarda, G.; Spaapen, R.M. The regulatory network behind MHC class I expression. Mol. Immunol. 2019, 113, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Ledoux, A.C.; Perkins, N.D. NF-κB and the cell cycle. Biochem. Soc. Trans. 2014, 42, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, R.; Ozono, E.; Fujisawa, J.; Ikeda, M.A.; Okamura, N.; Huang, Y.; Ohtani, K. Activation of the cyclin D2 and cdk6 genes through NF-kappaB is critical for cell-cycle progression induced by HTLV-I Tax. Oncogene 2008, 27, 5635–5642. [Google Scholar] [CrossRef]
- Mei, P.; Freitag, C.E.; Wei, L.; Zhang, Y.; Parwani, A.V.; Li, Z. High tumor mutation burden is associated with DNA damage repair gene mutation in breast carcinomas. Diagn. Pathol. 2020, 15, 50. [Google Scholar] [CrossRef]
- Shain, A.H.; Garrido, M.; Botton, T.; Talevich, E.; Yeh, I.; Sanborn, J.Z.; Chung, J.; Wang, N.J.; Kakavand, H.; Mann, G.J.; et al. Exome sequencing of desmoplastic melanoma identifies recurrent NFKBIE promoter mutations and diverse activating mutations in the MAPK pathway. Nat. Genet. 2015, 47, 1194–1199. [Google Scholar] [CrossRef]
- Eroglu, Z.; Zaretsky, J.M.; Hu-Lieskovan, S.; Kim, D.W.; Algazi, A.; Johnson, D.B.; Liniker, E.; Ben, K.; Munhoz, R.; Rapisuwon, S.; et al. High response rate to PD-1 blockade in desmoplastic melanomas. Nature 2018, 553, 347–350. [Google Scholar] [CrossRef]
- Mansouri, L.; Noerenberg, D.; Young, E.; Mylonas, E.; Abdulla, M.; Frick, M.; Asmar, F.; Ljungstrom, V.; Schneider, M.; Yoshida, K.; et al. Frequent NFKBIE deletions are associated with poor outcome in primary mediastinal B-cell lymphoma. Blood 2016, 128, 2666–2670. [Google Scholar] [CrossRef]
- Mansouri, L.; Sutton, L.A.; Ljungstrom, V.; Bondza, S.; Arngarden, L.; Bhoi, S.; Larsson, J.; Cortese, D.; Kalushkova, A.; Plevova, K.; et al. Functional loss of IkappaBepsilon leads to NF-kappaB deregulation in aggressive chronic lymphocytic leukemia. J. Exp. Med. 2015, 212, 833–843. [Google Scholar] [CrossRef]
- Myouzen, K.; Kochi, Y.; Okada, Y.; Terao, C.; Suzuki, A.; Ikari, K.; Tsunoda, T.; Takahashi, A.; Kubo, M.; Taniguchi, A.; et al. Functional variants in NFKBIE and RTKN2 involved in activation of the NF-kappaB pathway are associated with rheumatoid arthritis in Japanese. PLoS Genet. 2012, 8, e1002949. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFkappaB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef]
- Andreakos, E.; Williams, R.O.; Wales, J.; Foxwell, B.M.; Feldmann, M. Activation of NF-kappaB by the intracellular expression of NF-kappaB-inducing kinase acts as a powerful vaccine adjuvant. Proc. Natl. Acad. Sci. USA 2006, 103, 14459–14464. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Doebbeler, M.; Koenig, C.; Krzyzak, L.; Seitz, C.; Wild, A.; Ulas, T.; Baßler, K.; Kopelyanskiy, D.; Butterhof, A.; Kuhnt, C.; et al. CD83 expression is essential for Treg cell differentiation and stability. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Hirano, N.; Butler, M.O.; Xia, Z.; Ansen, S.; von Bergwelt-Baildon, M.S.; Neuberg, D.; Freeman, G.J.; Nadler, L.M. Engagement of CD83 ligand induces prolonged expansion of CD8+ T cells and preferential enrichment for antigen specificity. Blood 2006, 107, 1528–1536. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ju, X.; Silveira, P.A.; Abadir, E.; Hsu, W.H.; Hart, D.N.J.; Clark, G.J. CD83: Activation Marker for Antigen Presenting Cells and Its Therapeutic Potential. Front. Immunol. 2019, 10, 1312. [Google Scholar] [CrossRef] [PubMed]
- Baleeiro, R.B.; Bergami-Santos, P.C.; Tomiyoshi, M.Y.; Gross, J.L.; Haddad, F.; Pinto, C.A.; Soares, F.A.; Younes, R.N.; Barbuto, J.A. Expression of a dendritic cell maturation marker CD83 on tumor cells from lung cancer patients and several human tumor cell lines: Is there a biological meaning behind it? Cancer Immunol. Immunother. 2008, 57, 265–270. [Google Scholar] [CrossRef]
- Baleeiro, R.B.; Barbuto, J.A. Local secretion/shedding of tumor-derived CD83 molecules as a novel tumor escape mechanism. Mol. Immunol. 2008, 45, 3502–3504. [Google Scholar] [CrossRef]
- Juric, D.; Lacayo, N.J.; Ramsey, M.C.; Racevskis, J.; Wiernik, P.H.; Rowe, J.M.; Goldstone, A.H.; O’Dwyer, P.J.; Paietta, E.; Sikic, B.I. Differential gene expression patterns and interaction networks in BCR-ABL-positive and -negative adult acute lymphoblastic leukemias. J. Clin. Oncol. 2007, 25, 1341–1349. [Google Scholar] [CrossRef]
- Huynh, M.Q.; Wacker, H.H.; Wündisch, T.; Sohlbach, K.; Kim, T.D.; Krause, M.; Stabla, K.; Roth, P.; Fischbach, W.; Stolte, M.; et al. Expression profiling reveals specific gene expression signatures in gastric MALT lymphomas. Leuk. Lymphoma 2008, 49, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Mareschal, S.; Pham-Ledard, A.; Viailly, P.J.; Dubois, S.; Bertrand, P.; Maingonnat, C.; Fontanilles, M.; Bohers, E.; Ruminy, P.; Tournier, I.; et al. Identification of somatic mutations in primary cutaneous diffuse large b-cell lymphoma, leg type by massive parallel sequencing. J. Investig. Dermatol. 2017, 137, 1984–1994. [Google Scholar] [CrossRef] [PubMed]
- Berchtold, S.; Muhl-Zurbes, P.; Maczek, E.; Golka, A.; Schuler, G.; Steinkasserer, A. Cloning and characterization of the promoter region of the human CD83 gene. Immunobiology 2002, 205, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef]
- Armand, P.; Engert, A.; Younes, A.; Fanale, M.; Santoro, A.; Zinzani, P.L.; Timmerman, J.M.; Collins, G.P.; Ramchandren, R.; Cohen, J.B.; et al. Nivolumab for relapsed/refractory classic hodgkin lymphoma after failure of autologous hematopoietic cell transplantation: Extended follow-up of the multicohort single-arm phase ii checkmate 205 trial. J. Clin. Oncol. 2018, 36, 1428–1439. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Cohort | 52 |
|---|---|
| Sex | |
| Male | 34 (65%) |
| Female | 18 (35%) |
| Age (year) | |
| Median | 56 |
| Range | 22–87 |
| Mutational Analysis | |
| BRAF (V600E/K) | 11 (21%) |
| NRAS (G13_/Q61_) | 4 (7%) |
| NF1 | 11 (21%) |
| Immune Checkpoint Therapy | |
| Nivolomab | 11 (21%) |
| Pembrolizumab | 41 (79%) |
| Response | |
| Complete Response | 6 (11%) |
| Partial Response | 15 (28%) |
| Stable Disease | 16 (30%) |
| Progressive Disease | 15 (28%) |
| Melanoma Subtype | |
| Superficial Spreading | 17 (33%) |
| Nodular | 13 (25%) |
| Desmoplastic | 4 (7%) |
| Unknown Primary | 5 (9%) |
| Acral | 3 (5%) |
| Mucosal | 4 (7%) |
| Ocular | 1 (2%) |
| Other | 5 (9%) |
| Stage at Diagnosis | |
| III Unresectable | 1 (2%) |
| IIIB | 2 (3%) |
| IIIC | 10 (19%) |
| IV | 39 (75%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amato, C.M.; Hintzsche, J.D.; Wells, K.; Applegate, A.; Gorden, N.T.; Vorwald, V.M.; Tobin, R.P.; Nassar, K.; Shellman, Y.G.; Kim, J.; et al. Pre-Treatment Mutational and Transcriptomic Landscape of Responding Metastatic Melanoma Patients to Anti-PD1 Immunotherapy. Cancers 2020, 12, 1943. https://doi.org/10.3390/cancers12071943
Amato CM, Hintzsche JD, Wells K, Applegate A, Gorden NT, Vorwald VM, Tobin RP, Nassar K, Shellman YG, Kim J, et al. Pre-Treatment Mutational and Transcriptomic Landscape of Responding Metastatic Melanoma Patients to Anti-PD1 Immunotherapy. Cancers. 2020; 12(7):1943. https://doi.org/10.3390/cancers12071943
Chicago/Turabian StyleAmato, Carol M., Jennifer D. Hintzsche, Keith Wells, Allison Applegate, Nicholas T. Gorden, Victoria M. Vorwald, Richard P. Tobin, Kelsey Nassar, Yiqun G. Shellman, Jihye Kim, and et al. 2020. "Pre-Treatment Mutational and Transcriptomic Landscape of Responding Metastatic Melanoma Patients to Anti-PD1 Immunotherapy" Cancers 12, no. 7: 1943. https://doi.org/10.3390/cancers12071943
APA StyleAmato, C. M., Hintzsche, J. D., Wells, K., Applegate, A., Gorden, N. T., Vorwald, V. M., Tobin, R. P., Nassar, K., Shellman, Y. G., Kim, J., Medina, T. M., Rioth, M., Lewis, K. D., McCarter, M. D., Gonzalez, R., Tan, A.-C., & Robinson, W. A. (2020). Pre-Treatment Mutational and Transcriptomic Landscape of Responding Metastatic Melanoma Patients to Anti-PD1 Immunotherapy. Cancers, 12(7), 1943. https://doi.org/10.3390/cancers12071943