A H-REV107 Peptide Inhibits Tumor Growth and Interacts Directly with Oncogenic KRAS Mutants


Abstract
1. Introduction
2. Results
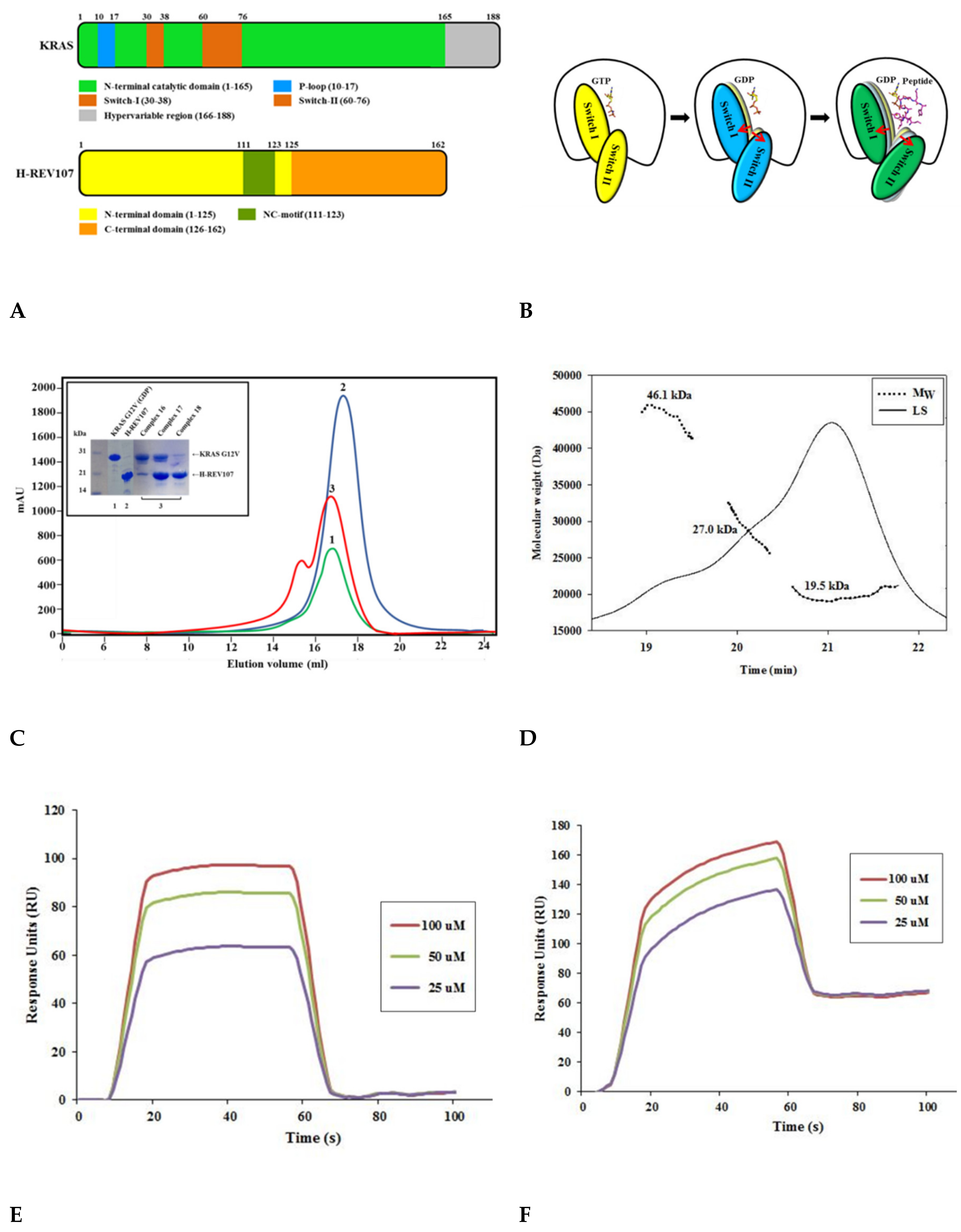
2.1. The H-REV107 Peptide Bound to KRAS G12V with High Binding Affinity
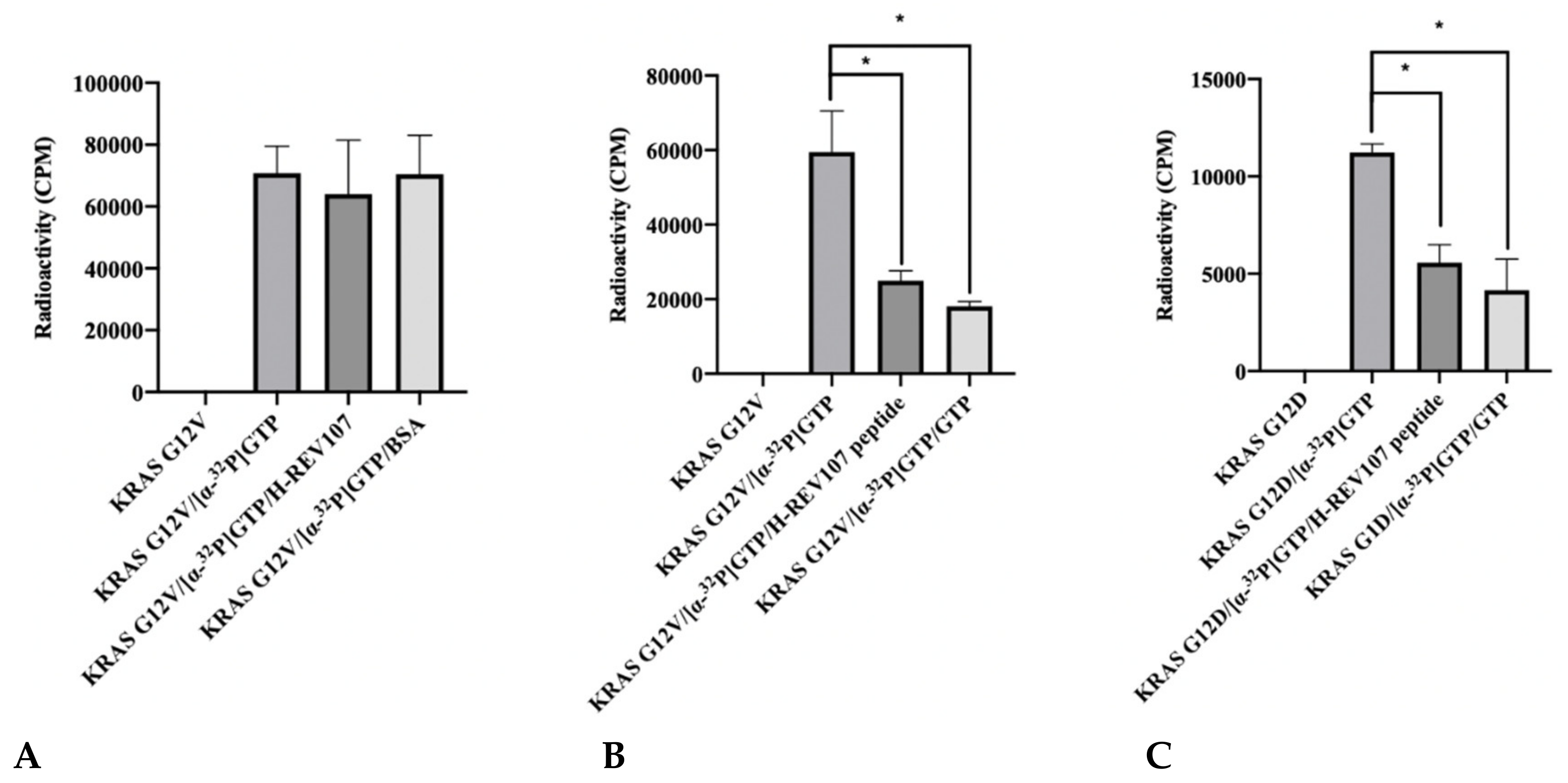
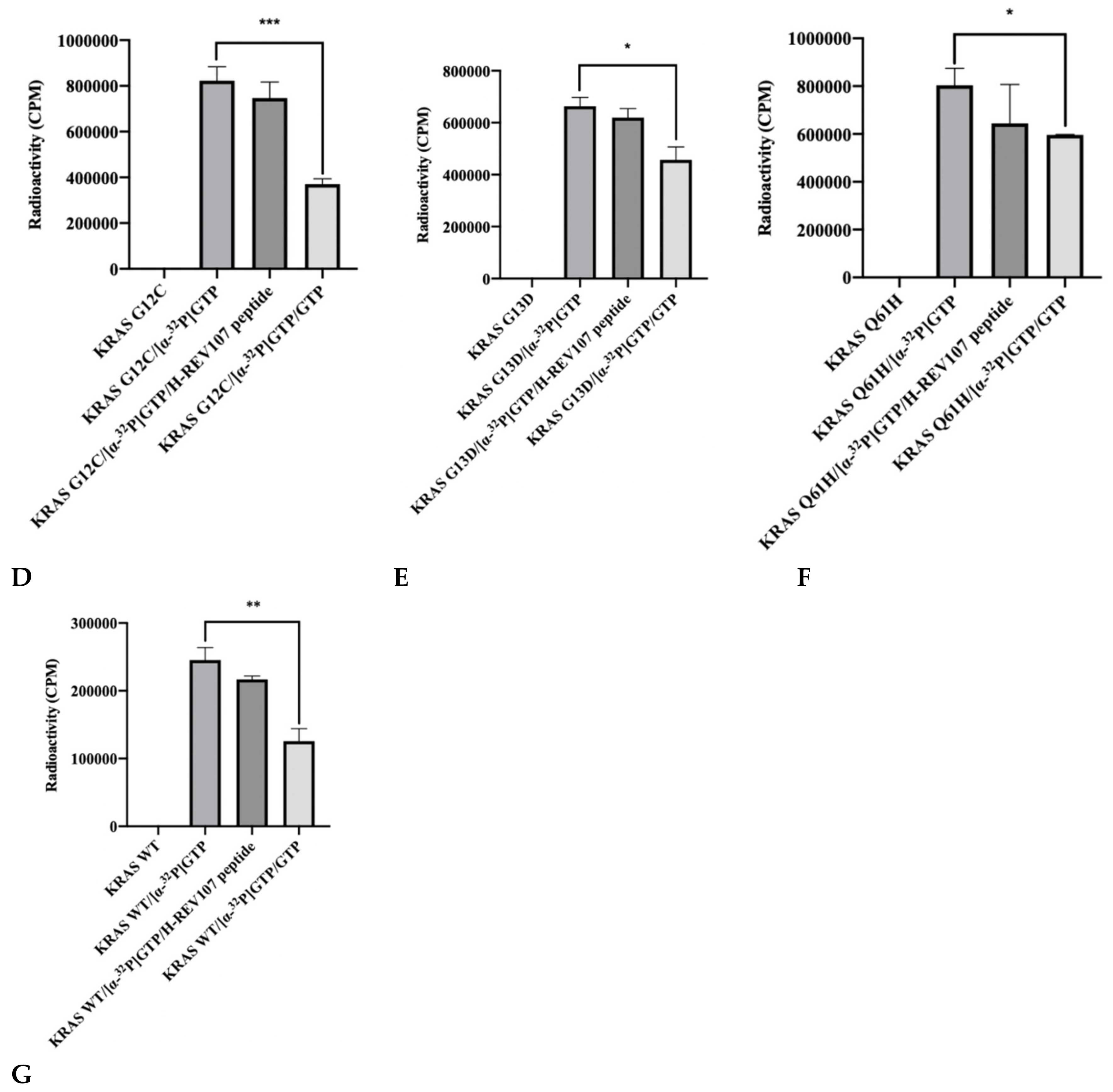
2.2. The GTP Binding to the KRAS Mutants Was Greatly Decreased in the Presence of H-REV107 Peptide
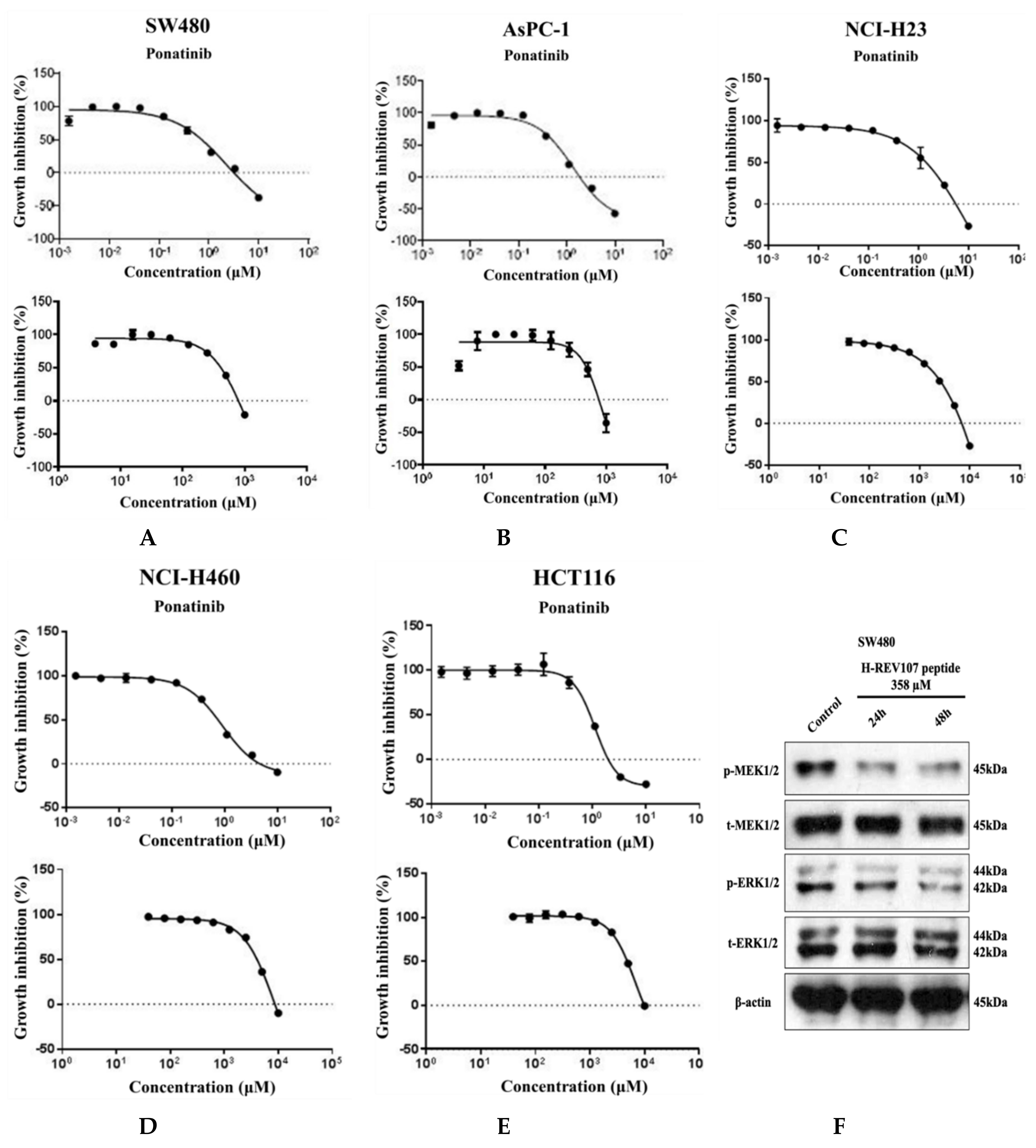
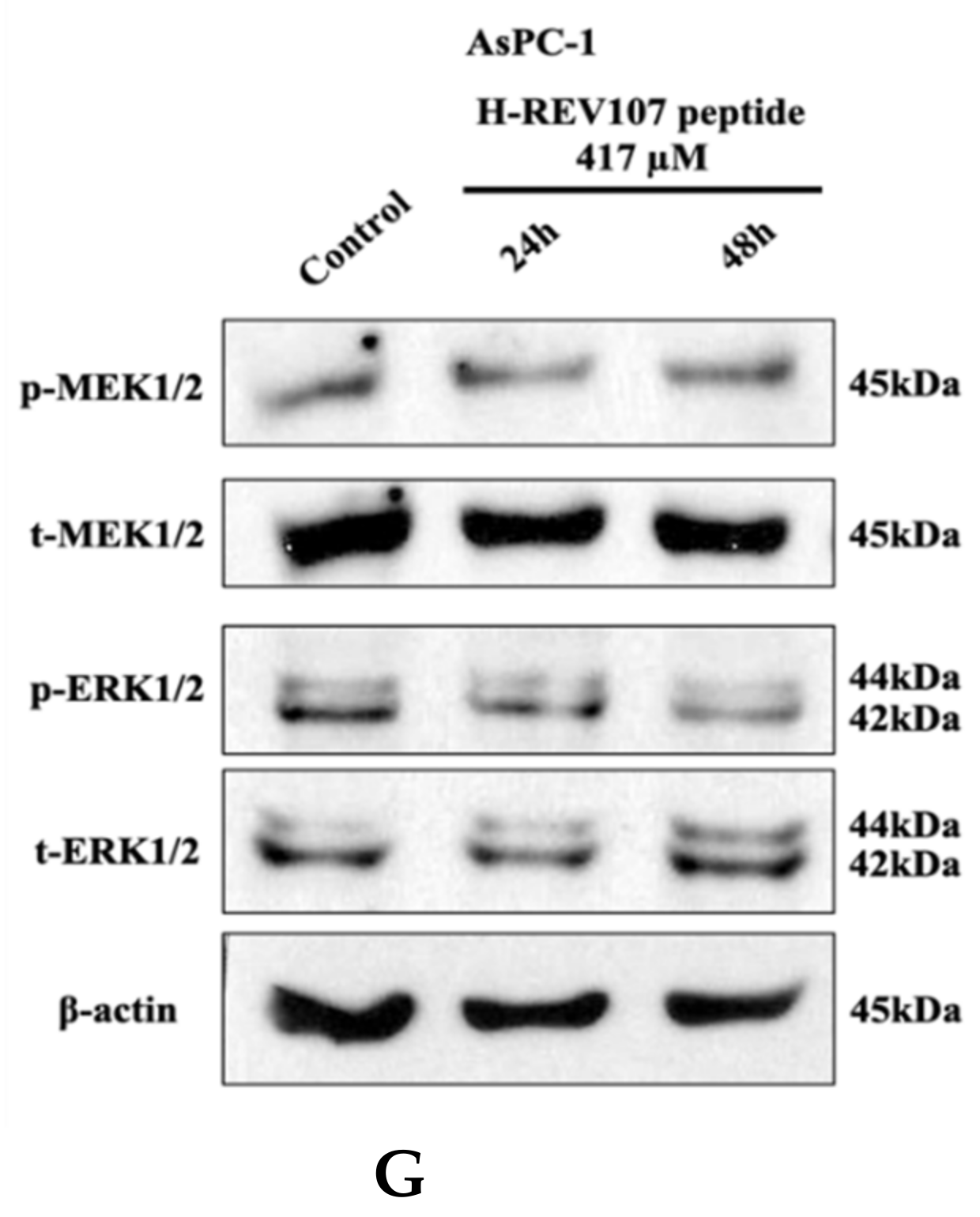
2.3. Inhibitory Effect of H-REV107 Peptide Was Observed on Tumor Cells
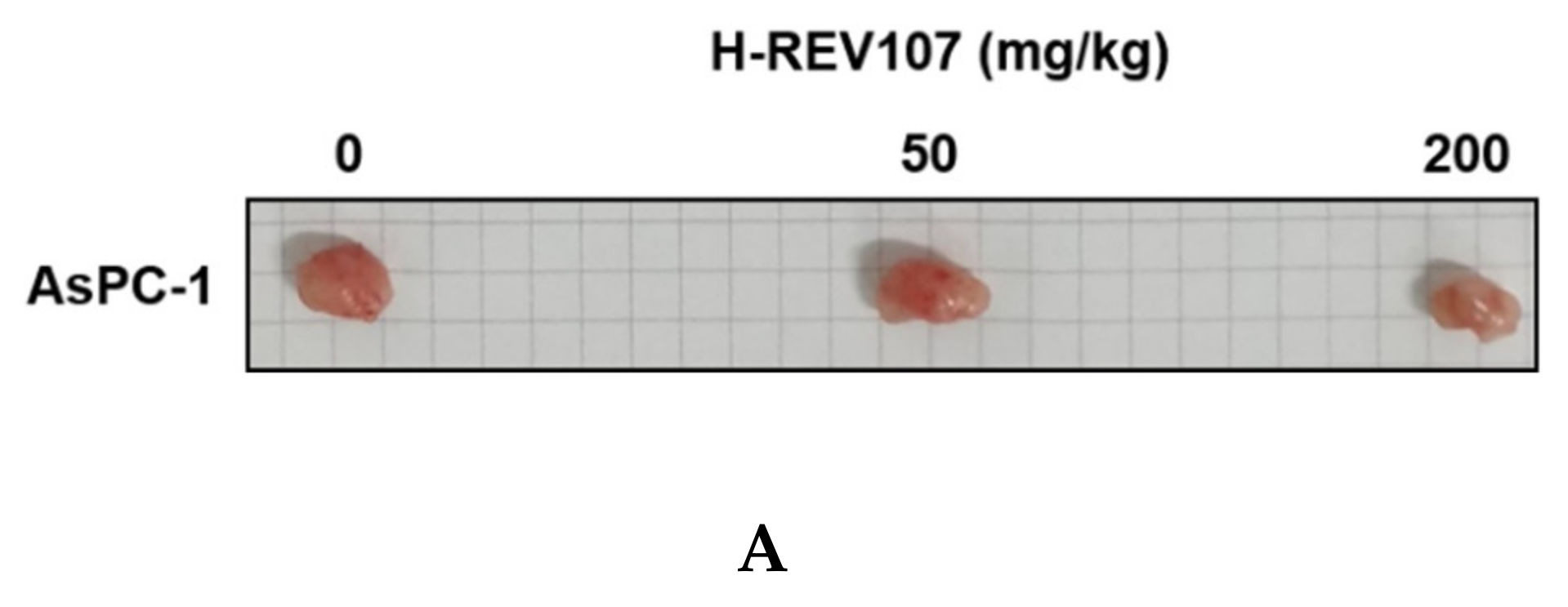
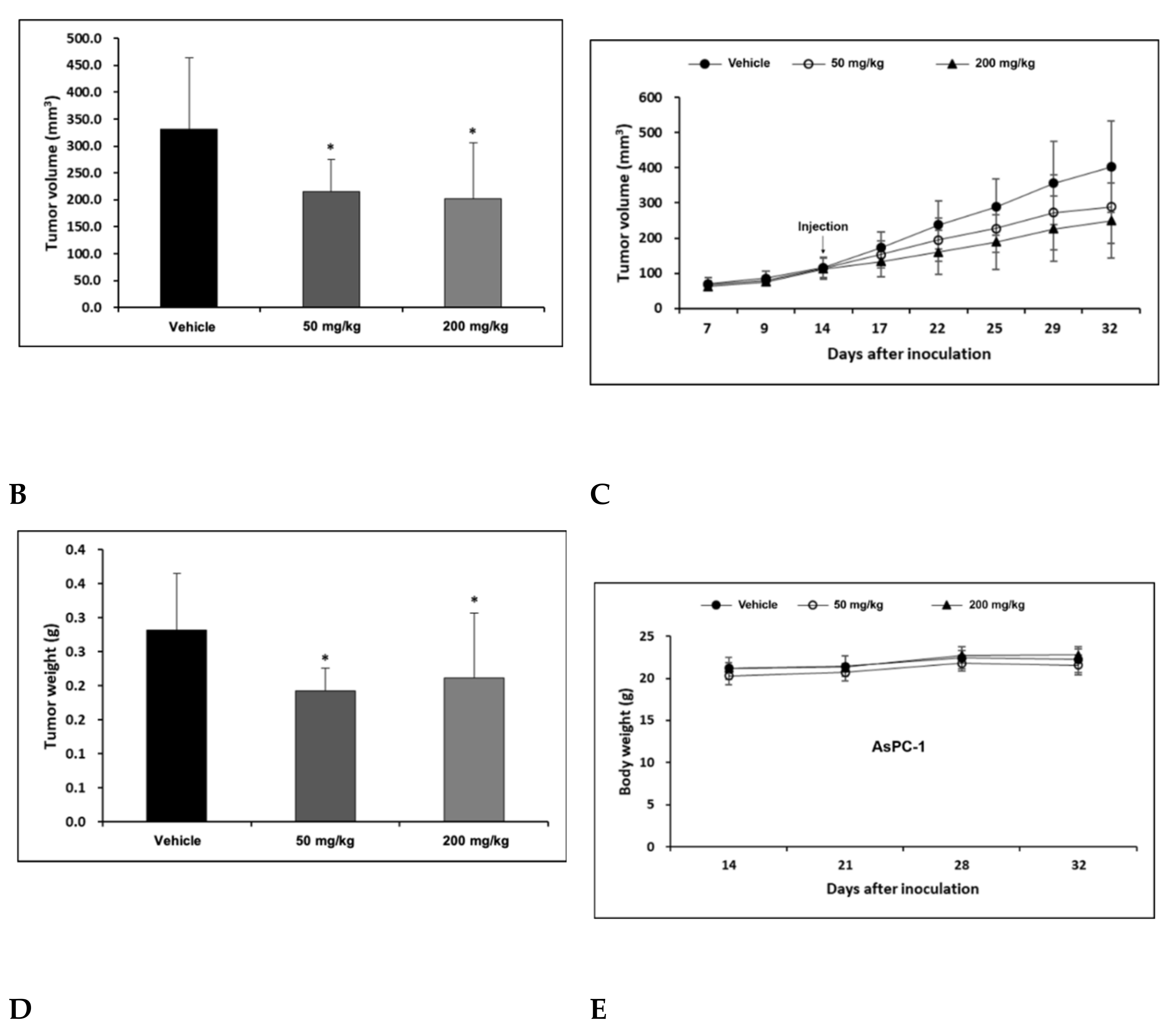
2.4. H-REV107 Peptide Can Suppress Pancreatic Tumor Growth
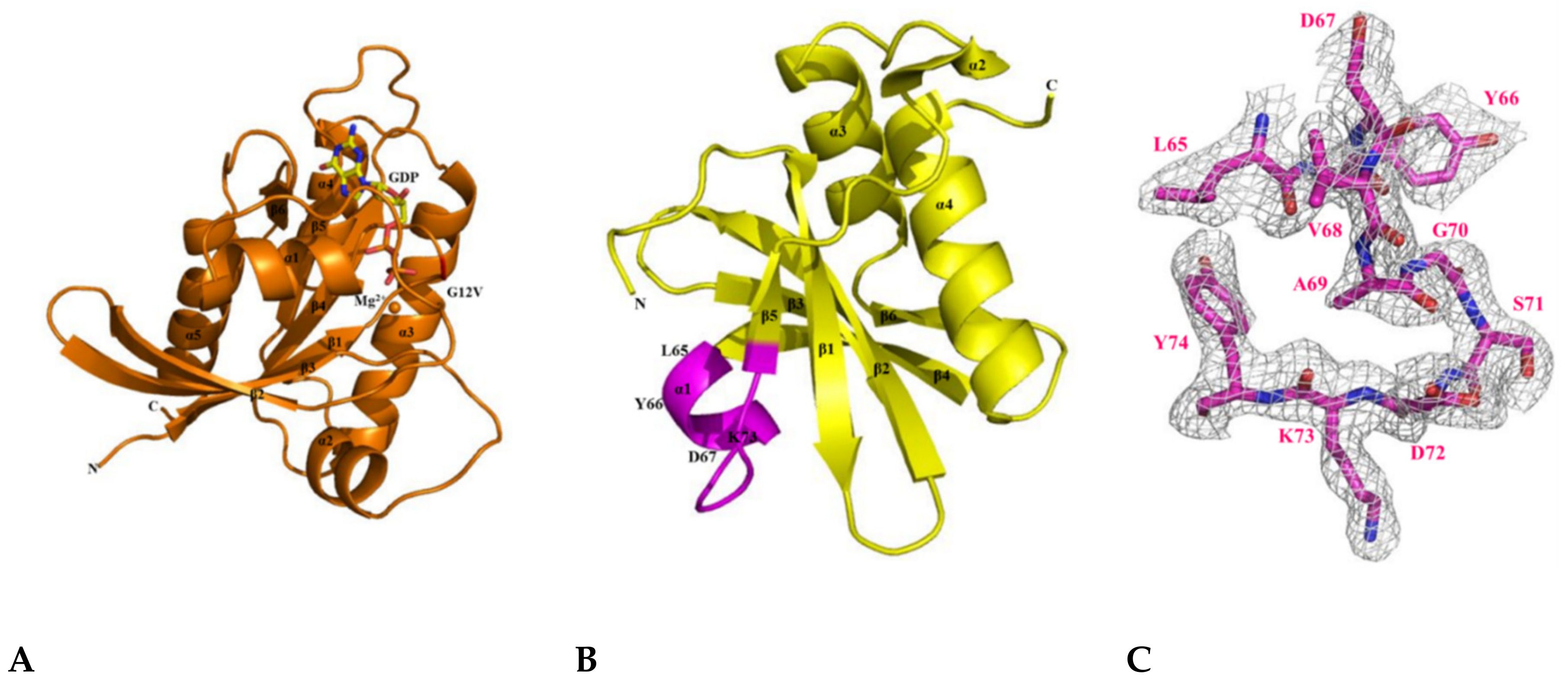
2.5. The Crystal Structure of KRAS G12V-H-REV107 Peptide Complex Was Determined
2.6. H-REV107 Peptide Was Strongly Docking in the Switch I and II Binding Pockets and the P-Loop of KRAS G12V
2.7. H-REV107 Peptide Stabilized the KRAS G12V in An Irreversible Inactive GDP Binding as An Open Conformation
3. Discussion
4. Materials and Methods
4.1. Recombinant Protein Expression and Purification
4.2. Size-Exclusion Chromatography–Multi-Angle Light Scattering (SEC-MALS)
4.3. Peptide Synthesis
4.4. Biacore Biosensor Analysis
4.5. Auto-Isothermal Titration Calorimetry (Auto-ITC)
4.6. Circular Dichroism (CD) Spectrometer Analysis
4.7. Guanine Nucleotide Binding Assay
4.8. Cell Proliferation Assay and Western Blot Analysis
4.9. Tumor Xenograft Model
4.10. Crystallization, Data Collection, and Structure Determination
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cox, A.D.; Der, C.J. Ras history: The saga continues. Small Gtpases 2010, 1, 2–27. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C.A. Comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- van Dam, T.J.; Bos, J.L.; Snel, B. Evolution of the Ras-like small GTPases and their regulators. Small Gtpases 2011, 2, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Victor, T.; Du Toit, R.; Jordaan, A.M.; Bester, A.J.; van Helden, P.D. No evidence for point mutations in codons 12, 13, and 61 of the ras gene in a high-incidence area for esophageal and gastric cancers. Cancer Res. 1990, 50, 4911–4914. [Google Scholar] [PubMed]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell. Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A. Shokat KM. K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef]
- Morris, J.P.; Wang, S.C.; Hebrok, M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat. Rev. Cancer 2010, 10, 683–695. [Google Scholar] [CrossRef]
- Kulemann, B.; Rösch, S.; Seifert, S.; Timme, S.; Bronsert, P.; Seifert, G.; Martini, V.; Kuvendjiska, J.; Glatz, T.; Hussung, S.; et al. Pancreatic cancer: Circulating Tumor Cells and Primary Tumors show Heterogeneous KRAS Mutations. Sci. Rep. 2017, 7, 4510–4520. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589 e517. [Google Scholar] [CrossRef] [PubMed]
- Mok, S.C.; Bell, D.A.; Knapp, R.C.; Fishbaugh, P.M.; Welch, W.R.; Muto, M.G.; Berkowitz, R.S.; Tsao, S.W. Mutation of K-ras protooncogene in human ovarian epithelial tumors of borderline malignancy. Cancer Res. 1993, 53, 1489–1492. [Google Scholar] [PubMed]
- Mayr, D.; Hirschmann, A.; Lohrs, U.; Diebold, J. KRAS and BRAF mutations in ovarian tumors: A comprehensive study of invasive carcinomas, borderline tumors and extraovarian implants. Gynecol. Oncol. 2006, 103, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Tsang, Y.T.; Deavers, M.T.; Sun, C.C.; Kwan, S.Y.; Kuo, E.; Malpica, A.; Mok, S.C.; Gershenson, D.M.; Wong, K.K. KRAS (but not BRAF) mutations in ovarian serous borderline tumour are associated with recurrent low-grade serous carcinoma. J. Pathol. 2013, 231, 449–456. [Google Scholar] [CrossRef]
- Andreyev, H.J.; Norman, A.R.; Cunningham, D.; Oates, J.R.; Clarke, P.A. Kirsten ras mutations in patients with colorectal cancer: The multicenter “RASCAL” study. J Natl. Cancer Inst. 1998, 90, 675–684. [Google Scholar] [CrossRef]
- Marshall, M.S. The effector interactions of p21ras. Trends Biochem. Sci. 1993, 18, 250–254. [Google Scholar] [CrossRef]
- Bollag, G.; McCormick, F. Differential regulation of rasGAP and neurofibromatosis gene product activities. Nature 1991, 351, 576–579. [Google Scholar] [CrossRef]
- Zlobec, I.; Kovac, M.; Erzberger, P.; Molinari, F.; Bihl, M.P.; Rufle, A.; Foerster, A.; Frattini, M.; Terracciano, L.; Heinimann, K.; et al. Combined analysis of specific KRAS mutation, BRAF and microsatellite instability identifies prognostic subgroups of sporadic and hereditary colorectal cancer. Int. J. Cancer 2010, 127, 2569–2575. [Google Scholar] [CrossRef]
- Husmann, K.; Sers, C.; Fietze, E.; Mincheva, A.; Lichter, P.; Schafer, R. Transcriptional and translational downregulation of H-REV107, a class II tumour suppressor gene located on human chromosome 11q11-12. Oncogene 1998, 17, 1305–1312. [Google Scholar] [CrossRef]
- Sers, C.; Husmann, K.; Nazarenko, I.; Reich, S.; Wiechen, K.; Zhumabayeva, B.; Adhikari, P.; SchroÈder, K.; Gontarewicz, A.; SchaÈfer, R. The class II tumour suppressor gene H-REV107-1 is a target of interferon-regulatory factor-1 and is involved in IFNgamma-induced cell death in human ovarian carcinoma cells. Oncogene 2002, 21, 2829–2839. [Google Scholar] [CrossRef][Green Version]
- Sers, C.; Emmenegger, U.; Husmann, K.; Bucher, K.; Andres, A.C.; Schafer, R. Growth-inhibitory activity and downregulation of the class II tumor-suppressor gene H-rev107 in tumor cell lines and experimental tumors. J. Cell Biol. 1997, 136, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Hiraki, Y.; Noda, M.; Shigeno, C.; Ito, H.; Nakamura, T. Molecular cloning and biological activity of a novel Ha-Ras suppressor gene predominantly expressed in skeletal muscle, heart, brain, and bone marrow by differential display using clonal mouse EC cells, ATDC5. J. Biol. Chem. 1999, 274, 32192–32197. [Google Scholar] [CrossRef] [PubMed]
- Hajnal, A.; Klemenz, R.; Schafer, R. Subtraction cloning of H-rev107, a gene specifically expressed in H-ras resistant fibroblasts. Oncogene 1994, 9, 479–490. [Google Scholar] [PubMed]
- Han, C.W.; Jeong, M.S.; Jang, S.B. Molecular Interaction Between K-Ras and H-REV107 in the Ras Signaling Pathway. Biochem. Biophys. Res. Commun. 2017, 491, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.H.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.S.; et al. Multivalent Small-Molecule Pan-RAS Inhibitors. Cell 2017, 168, 878–889.e29. [Google Scholar] [CrossRef] [PubMed]
- Golczak, M.; Kiser, P.D.; Sears, A.E.; Lodowski, D.T.; Blaner, W.S.; Palczewski, K. Structural basis for the acyltransferase activity of lecithin:retinol acyltransferase-like proteins. J. Biol. Chem. 2012, 287, 23790–23807. [Google Scholar] [CrossRef]
- John, J.; Rensland, H.; Schlichting, I.; Vetter, I.; Borasio, G.D.; Goody, R.S.; Wittinghofer, A. Kinetic and structural analysis of the Mg(2+)-binding site of the guanine nucleotide-binding protein p21H-ras. J. Biol. Chem. 1993, 268, 923–929. [Google Scholar]
- Feig, L.A.; Cooper, G.M. Inhibition of NIH 3T3 cell proliferation by a mutant ras protein with preferential affinity for GDP. Mol. Cell Biol. 1988, 8, 3235–3243. [Google Scholar] [CrossRef]
- Knihtila, R.; Holzapfel, G.; Weiss, K.; Meilleur, F.; Mattos, C. Neutron Crystal Structure of RAS GTPase Puts in Question the Protonation State of the GTP gamma-Phosphate. J. Biol. Chem. 2015, 290, 31025–31036. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Johnson, W.C. Analyzing protein circular dichroism spectra for accurate secondary structures. Proteins 1999, 35, 307–312. [Google Scholar] [CrossRef]
- Hassell, A.M.; An, G.; Bledsoe, R.K.; Bynum, J.M.; Carter, H.L.; Deng, S.J.; Gampe, R.T.; Grisard, T.E.; Madauss, K.P.; Nolte, R.T.; et al. Crystallization of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Minor, W.; Cymborowski, M.; Otwinowski, Z.; Chruszcz, M. HKL-3000: The integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Potterton, E.; Briggs, P.; Turkenburg, M.; Dodson, E. A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 2003, 59, 1131–1137. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta. Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | KRAS G12V- H-REV107 Peptide (PDB ID: 7C41) | KRAS G12V (PDB ID: 7C40) | H-REV107 (PDB ID: 7C3Z) |
|---|---|---|---|
| Space group | P1 | P63 | P212121 |
| Unit cell | |||
| a, b, c (Å) | 41.907, 84.339, 84.335 | 82.547, 82.547, 40.804 | 42.935, 52.996, 62.796 |
| α, β, γ (o) | 59.983, 89.961, 89.965 | 90.0, 90.0, 120.0 | 90.0, 90.0, 90.0 |
| Wavelength (Å) | 0.97934 | 0.97934 | 0.97934 |
| Resolution (Å) | 50.0–2.27 | 50.0–2.50 | 50.0–1.95 |
| Completeness (%) | 97.7 (81.6) | 99.9 (100) | 99.9 (100) |
| Observed reflections | 328,760 | 823,903 | 367,550 |
| Unique reflections | 45,853 | 5543 | 10,814 |
| I/σ (I) | 16.5 (5.7) | 27.2 (9.3) | 36.9 (8.0) |
| Rmerge (%) a | 10.2 (20.0) | 12.8 (51.1) | 8.8 (21.8) |
| Redundancy | 3.9 | 21.6 | 14.0 |
| Rcryst/Rfree (%) b | 26.03/28.34 | 18.46/24.24 | 23.43/26.29 |
| Protein atoms | 5348 | 1337 | 839 |
| Solvent molecules | 83 | 14 | 56 |
| Mg2+/GDP molecules | 4 | 1 | 0 |
| Peptide | 1 | 0 | 0 |
| Bond length (Å) | 0.009 | 0.009 | 0.008 |
| Bond angle (o) | 1.160 | 1.077 | 0.890 |
| Average <B> factor (Å2) Peptide | 31.3 | 0 | 0 |
| Average <B> factor (Å2) Mg2+/GDP | 19.3 | 24.4 | 0 |
| Most favored regions (%) | 97.2 | 94.0 | 97.0 |
| Additional allowed regions (%) | 2.2 | 6.0 | 3.0 |
| Disallowed regions (%) | 0.6 | 0 | 0 |
| KRAS | H-REV107 Peptide | Interaction Distance (Å) | KRAS | H-REV107 Peptide | Interaction Distance (Å) |
|---|---|---|---|---|---|
| O (G12V) | N (K73) | 4.04 | O (N85) | NZ (K73) | 2.66 |
| N (G12V) | O (Y74) | 3.91 | O2B (GDP) | O (G70) | 2.50 |
| N (G13) | O (D72) | 3.89 | N (KRAS G13)–O1B (GDP) | 2.70 | |
| OG (S17) | OH (Y66) | 2.81 | O (KRAS V29)–O2 (GDP) | 2.59 | |
| O (D33) | OH (Y66) | 2.51 | O (KRAS D30)–O2 (GDP) | 2.91 | |
| O (P34) | OD1 (D67) | 2.85 | O (KRAS D30)–O3 (GDP) | 2.76 | |
| O (T35) | OD1 (D67) | 3.21 | OD1 (KRAS N116)–O6 (GDP) | 3.21 | |
| O (T35) | OD2 (D67) | 3.16 | OD2 (KRAS D119)–N2 (GDP) | 3.14 | |
| O (A59) | N (L65) | 3.10 | OG (KRAS S17)–Mg2+ | 2.71 | |
| O (A59) | N (Y66) | 3.13 | O (KRAS T58)–Mg2+ | 3.03 | |
| NE2 (Q61) | N (L65) | 2.74 | O3B (GDP)–Mg2+ | 2.99 | |
| KRAS Residues | RMSD (Å) | KRAS Residues | RMSD (Å) |
|---|---|---|---|
| M1 | 1.83 | E62 | 1.36 |
| V12 | 0.48 | E63 | 2.15 |
| G13 | 0.48 | Y64 | 2.31 |
| D30 | 0.97 | S65 | 1.58 |
| E31 | 0.84 | A66 | 1.26 |
| Y32 | 0.78 | M67 | 1.08 |
| D33 | 0.92 | M72 | 1.09 |
| P34 | 1.37 | R73 | 1.25 |
| T35 | 1.03 | T74 | 0.89 |
| Q61 | 1.05 | D108 | 0.99 |
| Cell Lines | Tissues | KRAS Mutations |
|---|---|---|
| SW480 | Colon | G12V |
| AsPC-1 | Pancreas | G12D |
| NCI-H23 | Lung | G12C |
| NCI-H460 | Lung | Q61H |
| HCT116 | Colon | G13D |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, C.W.; Jeong, M.S.; Ha, S.C.; Jang, S.B. A H-REV107 Peptide Inhibits Tumor Growth and Interacts Directly with Oncogenic KRAS Mutants. Cancers 2020, 12, 1412. https://doi.org/10.3390/cancers12061412
Han CW, Jeong MS, Ha SC, Jang SB. A H-REV107 Peptide Inhibits Tumor Growth and Interacts Directly with Oncogenic KRAS Mutants. Cancers. 2020; 12(6):1412. https://doi.org/10.3390/cancers12061412
Chicago/Turabian StyleHan, Chang Woo, Mi Suk Jeong, Sung Chul Ha, and Se Bok Jang. 2020. "A H-REV107 Peptide Inhibits Tumor Growth and Interacts Directly with Oncogenic KRAS Mutants" Cancers 12, no. 6: 1412. https://doi.org/10.3390/cancers12061412
APA StyleHan, C. W., Jeong, M. S., Ha, S. C., & Jang, S. B. (2020). A H-REV107 Peptide Inhibits Tumor Growth and Interacts Directly with Oncogenic KRAS Mutants. Cancers, 12(6), 1412. https://doi.org/10.3390/cancers12061412