Hallmarks of Splicing Defects in Cancer: Clinical Applications in the Era of Personalized Medicine


Abstract
1. Introduction
2. Splicing Machinery, Splicing Code, and Splicing Catalysis
3. Splicing Alterations in Cancer
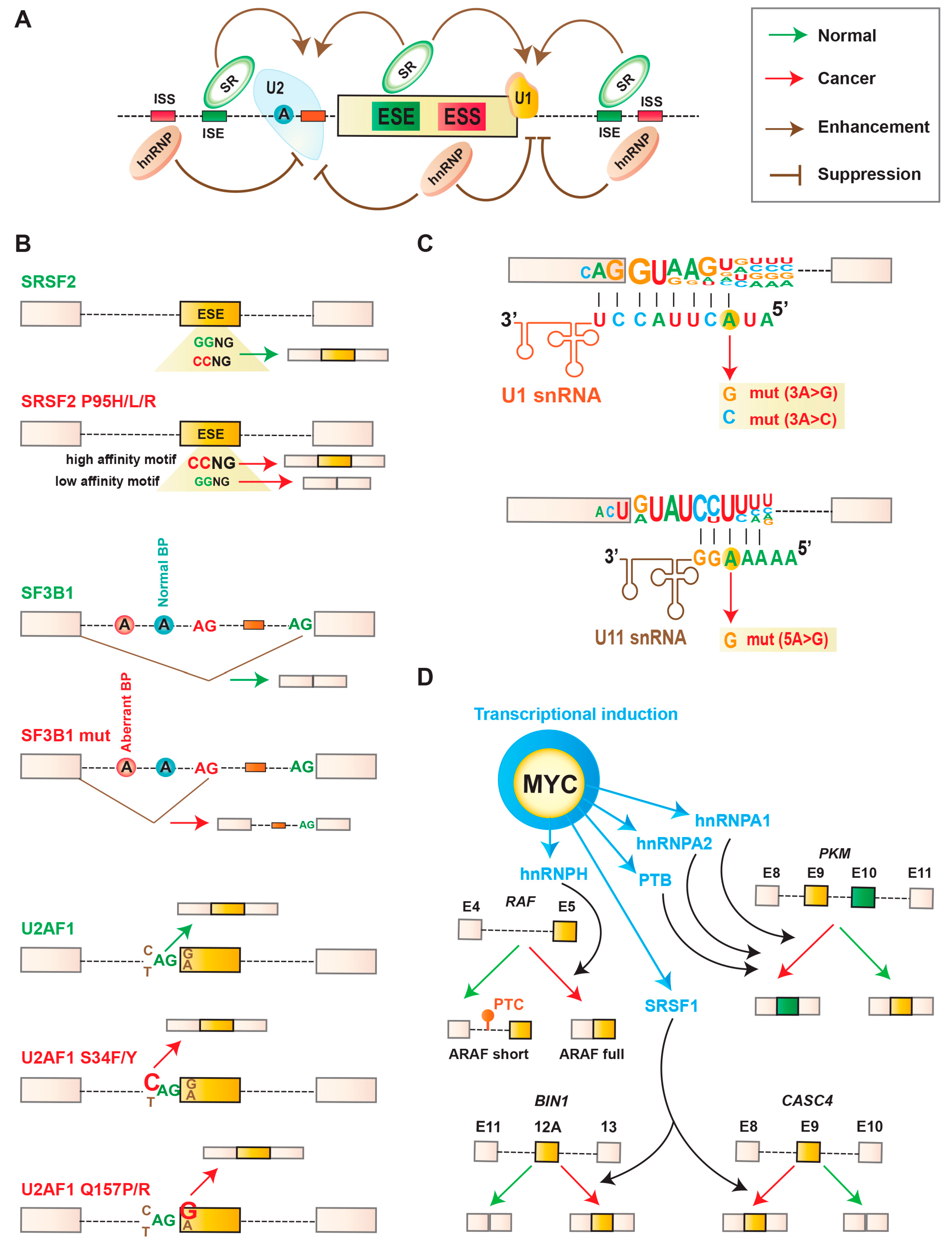
3.1. Mutations in Splicing Regulatory Cis-Elements in RNA
3.2. Mutations in Components of the Splicing Machinery
3.2.1. Mutations in Splicing Factors
Change-of-Function Mutations
Loss-of-Function Mutations
3.2.2. Mutations in snRNA
3.2.3. Abnormal Expression of Splicing Factors
3.3. Post-Translational Modification of Splicing Factors
4. Targeting Splicing Alterations for Cancer Therapies
4.1. Targeting Core Spliceosome
4.2. Targeting Splicing Regulatory Proteins
4.2.1. Sulfonamides
4.2.2. Decoy Oligonucleotides
4.3. Targeting Post-Translational Modification
4.4. Targeting MYC-Oncogene
4.5. Targeting mRNA Decay
4.6. Targeted Splicing Modulation
5. Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| ASO | Antisense oligonucleotides |
| ATR | Ataxia telangiectasia and Rad3-related protein |
| B-NHL | B-cell non-Hodgkin lymphoma |
| BP | Branch point |
| CLK | CDC2-like kinase |
| CLL | Chronic lymphocytic leukemia |
| CP | Cleavage and polyadenylation |
| EJCs | Exon junction complexes |
| ESEs | Exonic splicing enhancers |
| ESSs | Exonic splicing silencers |
| HCC | Hepatocellular carcinoma |
| hnRNPs | Heterogeneous nuclear ribonucleoproteins |
| ISEs | Intronic splicing enhancers |
| ISSs | Intronic splicing silencers |
| MB | Medulloblastoma |
| MDS | Myelodysplastic syndrome |
| METTL3 | N6-adenosine methyltransferase |
| NMD | Nonsense-mediated mRNA decay |
| PC | Pancreatic adenocarcinoma |
| PPT | Polypyrimidine tract |
| PTCs | Premature termination codons |
| PRMT5 | Protein arginine N-methyltransferase 5 |
| PKM | Pyruvate kinase |
| RNPs | Ribonucleoproteins |
| RRM | RNA-recognition motif |
| SRPK | SR protein kinase family |
| snRNPs | Small nuclear ribonucleoproteins |
| SS | Splice site |
References
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Chen, M.; Assanah, M.; Canoll, P.; Manley, J.L. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 2010, 463, 364–368. [Google Scholar] [CrossRef]
- Dvinge, H.; Kim, E.; Abdel-Wahab, O.; Bradley, R.K. RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 2016, 16, 413–430. [Google Scholar] [CrossRef]
- Rahman, M.A.; Krainer, A.R.; Abdel-Wahab, O. SnapShot: Splicing alterations in cancer. Cell 2020, 180, 208–208.e1. [Google Scholar] [CrossRef]
- Wahl, M.C.; Will, C.L.; Lührmann, R. The spliceosome: Design principles of a dynamic RNP machine. Cell 2009, 136, 701–718. [Google Scholar] [CrossRef]
- Graveley, B.R. Sorting out the complexity of SR protein functions. RNA 2000, 6, 1197–1211. [Google Scholar] [CrossRef]
- Lin, S.; Fu, X.D. SR proteins and related factors in alternative splicing. Adv. Exp. Med. Biol. 2007, 623, 107–122. [Google Scholar]
- Martinez-Contreras, R.; Cloutier, P.; Shkreta, L.; Fisette, J.F.; Revil, T.; Chabot, B. hnRNP proteins and splicing control. Adv. Exp. Med. Biol. 2007, 623, 123–147. [Google Scholar] [PubMed]
- Lee, S.C.; Abdel-Wahab, O. Therapeutic targeting of splicing in cancer. Nat. Med. 2016, 22, 976–986. [Google Scholar] [CrossRef] [PubMed]
- Anczuków, O.; Krainer, A.R. Splicing-factor alterations in cancers. RNA 2016, 22, 1285–1301. [Google Scholar] [CrossRef]
- Supek, F.; Miñana, B.; Valcárcel, J.; Gabaldón, T.; Lehner, B. Synonymous mutations frequently act as driver mutations in human cancers. Cell 2014, 156, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Lee, D.; Lee, J.; Park, D.; Kim, Y.J.; Park, W.Y.; Hong, D.; Park, P.J.; Lee, E. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat. Genet. 2015, 47, 1242–1248. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.C.; Kijima, T.; Maulik, G.; Fox, E.A.; Sattler, M.; Griffin, J.D.; Johnson, B.E.; Salgia, R. c-MET mutational analysis in small-cell lung cancer: Novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003, 63, 6272–6281. [Google Scholar] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Puente, X.S.; Beà, S.; Valdés-Mas, R.; Villamor, N.; Gutiérrez-Abril, J.; Martín-Subero, J.I.; Munar, M.; Rubio-Pérez, C.; Jares, P.; Aymerich, M.; et al. Noncoding recurrent mutations in chronic lymphocytic leukemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3’ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Nasrin, F. Human Disease-causing mutations affecting RNA splicing and NMD. J. Investig. Genomics 2016, 3, 00046. [Google Scholar] [CrossRef]
- Rahman, M.A. Oncogenic Landscapes of Splicing-Factor Mutant MDS. Genom. Gene. Ther. Int. J. 2018, 2, 000105. [Google Scholar] [CrossRef]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Watermann, D.O.; Tang, Y.; Zur Hausen, A.; Jäger, M.; Stamm, S.; Stickeler, E. Splicing factor Tra2-β1 is specifically induced in breast cancer and regulates alternative splicing of the CD44 gene. Cancer Res. 2006, 66, 4774–4780. [Google Scholar] [CrossRef]
- Jia, R.; Li, C.; McCoy, J.P.; Deng, C.X.; Zheng, Z.M. SRp20 is a proto-oncogene critical for cell proliferation and tumor induction and maintenance. Int. J. Biol. Sci. 2010, 6, 806–826. [Google Scholar] [CrossRef]
- Sen, S.; Langiewicz, M.; Jumaa, H.; Webster, N.J. Deletion of serine/arginine-rich splicing factor 3 in hepatocytes predisposes to hepatocellular carcinoma in mice. Hepatology 2015, 61, 171–183. [Google Scholar] [CrossRef]
- He, X.; Ee, P.L.; Coon, J.S.; Beck, W.T. Alternative splicing of the multidrug resistance protein 1/ATP binding cassette transporter subfamily gene in ovarian cancer creates functional splice variants and is associated with increased expression of the splicing factors PTB and SRp20. Clin. Cancer Res. 2004, 10, 4652–4660. [Google Scholar] [CrossRef]
- Huang, C.S.; Shen, C.Y.; Wang, H.W.; Wu, P.E.; Cheng, C.W. Increased expression of SRp40 affecting CD44 splicing is associated with the clinical outcome of lymph node metastasis in human breast cancer. Clin. Chim. Acta 2007, 384, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Eliav, M.; Golan-Gerstl, R.; Siegfried, Z.; Andersen, C.L.; Thorsen, K.; Orntoft, T.F.; Mu, D.; Karni, R. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J. Pathol. 2013, 229, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.A.; Wilkinson, J.E.; Krainer, A.R. Splicing factor SRSF6 promotes hyperplasia of sensitized skin. Nat. Struct. Mol. Biol. 2014, 21, 189–197. [Google Scholar] [CrossRef]
- Zhou, X.; Li, X.; Cheng, Y.; Wu, W.; Xie, Z.; Xi, Q.; Han, J.; Wu, G.; Fang, J.; Feng, Y. BCLAF1 and its splicing regulator SRSF10 regulate the tumorigenic potential of colon cancer cells. Nat. Commun. 2014, 5, 4581. [Google Scholar] [CrossRef]
- Fischer, D.C.; Noack, K.; Runnebaum, I.B.; Watermann, D.O.; Kieback, D.G.; Stamm, S.; Stickeler, E. Expression of splicing factors in human ovarian cancer. Oncol. Rep. 2004, 11, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Iborra, S.; Hirschfeld, M.; Jaeger, M.; Zur Hausen, A.; Braicu, I.; Sehouli, J.; Gitsch, G.; Stickeler, E. Alterations in expression pattern of splicing factors in epithelial ovarian cancer and its clinical impact. Int. J. Gynecol. Cancer 2013, 23, 990–996. [Google Scholar] [CrossRef]
- Rintala-Maki, N.D.; Goard, C.A.; Langdon, C.E.; Wall, V.E.; Traulsen, K.E.; Morin, C.D.; Bonin, M.; Sutherland, L.C. Expression of RBM5-related factors in primary breast tissue. J. Cell Biochem. 2007, 100, 1440–1458. [Google Scholar] [CrossRef]
- Oh, J.J.; West, A.R.; Fishbein, M.C.; Slamon, D.J. A candidate tumor suppressor gene, H37, from the human lung cancer tumor suppressor locus 3p21.3. Cancer Res. 2002, 62, 3207–3213. [Google Scholar] [PubMed]
- Zhao, L.; Li, R.; Shao, C.; Li, P.; Liu, J.; Wang, K. 3p21.3 tumor suppressor gene RBM5 inhibits growth of human prostate cancer PC-3 cells through apoptosis. World J. Surg. Oncol. 2012, 10, 247. [Google Scholar] [CrossRef]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Yae, T.; Tsuchihashi, K.; Ishimoto, T.; Motohara, T.; Yoshikawa, M.; Yoshida, G.J.; Wada, T.; Masuko, T.; Mogushi, K.; Tanaka, H.; et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat. Commun. 2012, 3, 883. [Google Scholar] [CrossRef]
- Ishii, H.; Saitoh, M.; Sakamoto, K.; Kondo, T.; Katoh, R.; Tanaka, S.; Motizuki, M.; Masuyama, K.; Miyazawa, K. Epithelial splicing regulatory proteins 1 (ESRP1) and 2 (ESRP2) suppress cancer cell motility via different mechanisms. J. Biol. Chem. 2014, 289, 27386–27399. [Google Scholar] [CrossRef] [PubMed]
- Golan-Gerstl, R.; Cohen, M.; Shilo, A.; Suh, S.S.; Bakacs, A.; Coppola, L.; Karni, R. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011, 71, 4464–4472. [Google Scholar] [CrossRef]
- Zhou, J.; Allred, D.C.; Avis, I.; Martinez, A.; Vos, M.D.; Smith, L.; Treston, A.M.; Mulshine, J.L. Differential expression of the early lung cancer detection marker, heterogeneous nuclear ribonucleoprotein-A2/B1 (hnRNP-A2/B1) in normal breast and neoplastic breast cancer. Breast Cancer Res. Treat. 2001, 66, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Fielding, P.; Turnbull, L.; Prime, W.; Walshaw, M.; Field, J.K. Heterogeneous nuclear ribonucleoprotein A2/B1 up-regulation in bronchial lavage specimens: A clinical marker of early lung cancer detection. Clin. Cancer Res. 1999, 5, 4048–4052. [Google Scholar] [PubMed]
- Xu, Y.; Gao, X.D.; Lee, J.H.; Huang, H.; Tan, H.; Ahn, J.; Reinke, L.M.; Peter, M.E.; Feng, Y.; Gius, D.; et al. Cell type-restricted activity of hnRNPM promotes breast cancer metastasis via regulating alternative splicing. Genes Dev. 2014, 28, 1191–1203. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, B.; McKay, M.; Dundas, S.R.; Lawrie, L.C.; Telfer, C.; Murray, G.I. Heterogeneous nuclear ribonucleoprotein K is over expressed, aberrantly localised and is associated with poor prognosis in colorectal cancer. Br. J. Cancer 2006, 95, 921–927. [Google Scholar] [CrossRef]
- Zhou, R.; Shanas, R.; Nelson, M.A.; Bhattacharyya, A.; Shi, J. Increased expression of the heterogeneous nuclear ribonucleoprotein K in pancreatic cancer and its association with the mutant p53. Int. J. Cancer 2010, 126, 395–404. [Google Scholar] [CrossRef]
- Lefave, C.V.; Squatrito, M.; Vorlova, S.; Rocco, G.L.; Brennan, C.W.; Holland, E.C.; Pan, Y.X.; Cartegni, L. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J. 2011, 30, 4084–4097. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; McCutcheon, I.E.; Fuller, G.N.; Huang, E.S.; Cote, G.J. Fibroblast growth factor receptor-1 α-exon exclusion and polypyrimidine tract-binding protein in glioblastoma multiforme tumors. Cancer Res. 2000, 60, 1221–1224. [Google Scholar]
- Takahashi, H.; Nishimura, J.; Kagawa, Y.; Kano, Y.; Takahashi, Y.; Wu, X.; Hiraki, M.; Hamabe, A.; Konno, M.; Haraguchi, N.; et al. Significance of polypyrimidine tract-binding protein 1 expression in colorectal cancer. Mol. Cancer Ther. 2015, 14, 1705–1716. [Google Scholar] [CrossRef]
- Lu, W.; Feng, F.; Xu, J.; Lu, X.; Wang, S.; Wang, L.; Lu, H.; Wei, M.; Yang, G.; Wang, L.; et al. QKI impairs self-renewal and tumorigenicity of oral cancer cells via repression of SOX2. Cancer Biol. Ther. 2014, 15, 1174–1184. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, G.; Wei, M.; Lu, X.; Fu, H.; Feng, F.; Wang, S.; Lu, W.; Wu, N.; Lu, Z.; et al. The tumor suppressing effects of QKI-5 in prostate cancer: A novel diagnostic and prognostic protein. Cancer Biol. Ther. 2014, 15, 108–118. [Google Scholar] [CrossRef]
- Thol, F.; Kade, S.; Schlarmann, C.; Löffeld, P.; Morgan, M.; Krauter, J.; Wlodarski, M.W.; Kölking, B.; Wichmann, M.; Görlich, K.; et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 2012, 119, 3578–3584. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lieu, Y.K.; Ali, A.M.; Penson, A.; Reggio, K.S.; Rabadan, R.; Raza, A.; Mukherjee, S.; Manley, J.L. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc. Natl. Acad. Sci. USA 2015, 112, E4726–E4734. [Google Scholar] [CrossRef]
- Komeno, Y.; Huang, Y.-J.; Qiu, J.; Lin, L.; Xu, Y.; Zhou, Y.; Chen, L.; Monterroza, D.D.; Li, H.; DeKelver, R.C.; et al. SRSF2 Is Essential for Hematopoiesis, and Its Myelodysplastic Syndrome-Related Mutations Dysregulate Alternative Pre-mRNA Splicing. Mol. Cell. Biol. 2015, 35, 3071–3082. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.A.; Lin, K.T.; Bradley, R.K.; Abdel-Wahab, O.; Krainer, A.R. Recurrent SRSF2 Mutations in MDS Affect Both Splicing and NMD. Genes Dev. 2020, 34, 413–427. [Google Scholar] [CrossRef] [PubMed]
- DeBoever, C.; Ghia, E.M.; Shepard, P.J.; Rassenti, L.; Barrett, C.L.; Jepsen, K.; Jamieson, C.H.M.; Carson, D.; Kipps, T.J.; Frazer, K.A. Transcriptome Sequencing Reveals Potential Mechanism of Cryptic 3’ Splice Site Selection in SF3B1-mutated Cancers. PLoS Comput. Biol. 2015, 11, e1004105. [Google Scholar] [CrossRef]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic Expression of Sf3b1K700E Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 2016, 30, 404–417. [Google Scholar] [CrossRef]
- Dolatshad, H.; Pellagatti, A.; Liberante, F.G.; Llorian, M.; Repapi, E.; Steeples, V.; Roy, S.; Scifo, L.; Armstrong, R.N.; Shaw, J.; et al. Cryptic splicing events in the iron transporter ABCB7 and other key target genes in SF3B1-mutant myelodysplastic syndromes. Leukemia 2016, 30, 2322–2331. [Google Scholar] [CrossRef]
- Conte, S.; Katayama, S.; Vesterlund, L.; Karimi, M.; Dimitriou, M.; Jansson, M.; Mortera-Blanco, T.; Unneberg, P.; Papaemmanuil, E.; Sander, B.; et al. Aberrant splicing of genes involved in haemoglobin synthesis and impaired terminal erythroid maturation in SF3B1 mutated refractory anaemia with ring sideroblasts. Br. J. Haematol. 2015, 171, 478–490. [Google Scholar] [CrossRef]
- Visconte, V.; Avishai, N.; Mahfouz, R.; Tabarroki, A.; Cowen, J.; Sharghi-Moshtaghin, R.; Hitomi, M.; Rogers, H.J.; Hasrouni, E.; Phillips, J.; et al. Distinct iron architecture in SF3B1-mutant myelodysplastic syndrome patients is linked to an SLC25A37 splice variant with a retained intron. Leukemia 2015, 29, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Furney, S.J.; Pedersen, M.; Gentien, D.; Dumont, A.G.; Rapinat, A.; Desjardins, L.; Turajlic, S.; Piperno-Neumann, S.; de la Grange, P.; Roman-Roman, S.; et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013, 3, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Inoue, D.; Chew, G.L.; Liu, B.; Michel, B.C.; Pangallo, J.; D’Avino, A.R.; Hitchman, T.; North, K.; Lee, S.C.W.; Bitner, L.; et al. Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature 2019, 574, 432–436. [Google Scholar] [CrossRef]
- Graubert, T.A.; Shen, D.; Ding, L.; Okeyo-Owuor, T.; Lunn, C.L.; Shao, J.; Krysiak, K.; Harris, C.C.; Koboldt, D.C.; Larson, D.E.; et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat. Genet. 2012, 44, 53–57. [Google Scholar] [CrossRef]
- Przychodzen, B.; Jerez, A.; Guinta, K.; Sekeres, M.A.; Padgett, R.; Maciejewski, J.P.; Makishima, H. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood 2013, 122, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.N.; Choi, P.S.; De Waal, L.; Sharifnia, T.; Imielinski, M.; Saksena, G.; Sekhar, P.C.; Sivachenko, A.; Rosenberg, M.; Chmielecki, J.; et al. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS ONE 2014, 9, e87361. [Google Scholar] [CrossRef] [PubMed]
- Ilagan, J.O.; Ramakrishnan, A.; Hayes, B.; Murphy, M.E.; Zebari, A.S.; Bradley, P.; Bradley, R.K. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015, 25, 14–26. [Google Scholar] [CrossRef]
- Okeyo-Owuor, T.; White, B.S.; Chatrikhi, R.; Mohan, D.R.; Kim, S.; Griffith, M.; Ding, L.; Ketkar-Kulkarni, S.; Hundal, J.; Laird, K.M.; et al. U2AF1 mutations alter sequence specificity of pre-mRNA binding & splicing. Leukemia 2015, 29, 909–917. [Google Scholar] [CrossRef]
- Yip, B.H.; Steeples, V.; Repapi, E.; Armstrong, R.N.; Llorian, M.; Roy, S.; Shaw, J.; Dolatshad, H.; Taylor, S.; Verma, A.; et al. The U2AF1S34Fmutation induces lineage-specific splicing alterations in myelodysplastic syndromes. J. Clin. Investig. 2017, 127, 2206–2221. [Google Scholar] [CrossRef]
- Bejar, R.; Stevenson, K.; Abdel-Wahab, O.; Galili, N.; Nilsson, B.B.; Garcia-Manero, G.; Kantarjian, H.; Raza, A.; Levine, R.L.; Neberg, D.; et al. Clinical effect of point mutations in myelodysplastic syndromes. N. Engl. J. Med. 2011, 364, 2496–2506. [Google Scholar] [CrossRef]
- Borel, C.; Dastugue, N.; Cances Lauwers, V.; Mozziconacci, M.J.; Prebet, T.; Vey, N.; Pigneux, A.; Lippert, E.; Visanica, S.; Legrand, F.; et al. PICALM-MLLT10 acute myeloid leukemia: A French cohort of 18 patients. Leuk. Res. 2012, 36, 1365–1369. [Google Scholar] [CrossRef] [PubMed]
- Chia, N.Y.; Chan, Y.S.; Feng, B.; Lu, X.; Orlov, Y.L.; Moreau, D.; Kumar, P.; Yang, L.; Jiang, J.; Lau, M.S.; et al. A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature 2010, 468, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Gaspar-Maia, A.; Qadeer, Z.A.; Hasson, D.; Ratnakumar, K.; Leu, N.A.; Leroy, G.; Liu, S.; Costanzi, C.; Valle-Garcia, D.; Schaniel, C.; et al. Macro H2A histone variants act as a barrier upon reprogramming towards pluripotency. Nat. Commun. 2013, 4, 1565. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.D.; Yadav, T.; Giri, S.; Saez, B.; Graubert, T.A.; Zou, L. Functions of replication protein A as a sensor of R loops and a regulator of RNaseH1. Mol. Cell 2017, 65, 832–847.e4. [Google Scholar] [CrossRef]
- Chen, L.; Chen, J.Y.; Huang, Y.J.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H.; et al. The augmented R-loop is a unifying mechanism for myelodysplastic syndromes induced by high-risk splicing factor mutations. Mol. Cell 2018, 69, 412–425.e6. [Google Scholar] [CrossRef]
- Park, S.M.; Ou, J.; Chamberlain, L.; Simone, T.M.; Yang, H.; Virbasius, C.M.; Ali, A.M.; Zhu, L.J.; Mukherjee, S.; Raza, A.; et al. U2AF35 (S34F) promotes transformation by directing aberrant ATG7 pre-mRNA 3’ end formation. Mol. Cell 2016, 62, 479–490. [Google Scholar] [CrossRef]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Abu Kar, S.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, K.; Afable, M.G.; et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood 2012, 119, 3203–3210. [Google Scholar] [CrossRef]
- Quesada, V.; Conde, L.; Villamor, N.; Ordonez, G.R.; Jares, P.; Bassaganyas, L.; Ramsay, A.J.; Bea, S.; Pinyol, M.; Martinez-Trillos, A.; et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat. Genet. 2012, 44, 47–52. [Google Scholar] [CrossRef]
- Walter, M.J.; Shen, D.; Shao, J.; Ding, L.; White, B.S.; Kandoth, C.; Miller, C.A.; Niu, B.; McLellan, M.D.; Dees, N.D.; et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia 2013, 27, 1275–1282. [Google Scholar] [CrossRef]
- Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G.; The Cancer Genome Atlas Research Network; Buonamici, S.; Yu, L. Somatic mutational landscape of splicing factor genes and their func-tional consequences across 33 cancer types. Cell Rep. 2018, 23, 282–296. [Google Scholar] [CrossRef]
- Madan, V.; Kanojia, D.; Li, J.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y.; et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat. Commun. 2015, 6, 6042. [Google Scholar] [CrossRef] [PubMed]
- Ibrahimpasic, T.; Xu, B.; Landa, I.; Dogan, S.; Middha, S.; Seshan, V.; Deraje, S.; Carlson, D.L.; Migliacci, J.; Knauf, J.A.; et al. Genomic alterations in fatal forms of nonanaplastic thyroid cancer: Identification of MED12 and RBM10 as novel thyroid cancer genes associated with tumor virulence. Clin. Cancer Res. 2017, 23, 5970–5980. [Google Scholar] [CrossRef]
- Bechara, E.G.; Sebestyen, E.; Bernardis, I.; Eyras, E.; Valcarcel, J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol. Cell 2013, 52, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kumar, S.A.; Shuai, S.; Diaz-Navarro, A.; Gutierrez-Fernandez, A.; Antonellis, P.; Cavalli, F.M.G.; Juraschka, K.; Farooq, H.; Shibahara, I.; et al. Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature 2019, 574, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Shuai, S.; Suzuki, H.; Diaz-Navarro, A.; Nadeu, F.; Kumar, S.A.; Gutierrez-Fernandez, A.; Delgado, J.; Pinyol, M.; López-Otín, C.; Puente, X.S.; et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature 2019, 574, 712–716. [Google Scholar] [CrossRef]
- Anczuków, O.; Rosenberg, A.Z.; Akerman, M.; Das, S.; Zhan, L.; Karni, R.; Muthuswamy, S.K.; Krainer, A.R. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat. Struct. Mol. Biol. 2012, 19, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Zong, F.Y.; Fu, X.; Wei, W.J.; Luo, Y.G.; Heiner, M.; Cao, L.J.; Fang, Z.; Fang, R.; Lu, D.; Ji, H.; et al. The RNA-binding protein QKI suppresses cancer-associated aberrant splicing. PLoS Genet. 2014, 10, e1004289. [Google Scholar] [CrossRef]
- Goncalves, V.; Pereira, J.F.S.; Jordan, P. Signaling pathways driving aberrant splicing in cancer cells. Genes 2017, 9, 9. [Google Scholar] [CrossRef]
- Clower, C.V.; Chatterjee, D.; Wang, Z.; Cantley, L.C.; Vander Heiden, M.G.; Krainer, A.R. The alternative splicing repressors hnRNP A1/A2 and PTBinfluence pyruvate kinase isoform expression and cell metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 1894–1899. [Google Scholar] [CrossRef]
- Das, S.; Anczukow, O.; Akerman, M.; Krainer, A.R. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep. 2012, 1, 110–117. [Google Scholar] [CrossRef]
- Ghigna, C.; Giordano, S.; Shen, H.; Benvenuto, F.; Castiglioni, F.; Comoglio, P.M.; Green, M.R.; Riva, S.; Biamonti, G. Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol. Cell 2005, 20, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Anczuków, O.; Akerman, M.; Clery, A.; Wu, J.; Shen, C.; Shirole, N.H.; Raimer, A.; Sun, S.; Jensen, M.A.; Hua, Y.; et al. SRSF1-regulated alternative splicing in breast cancer. Mol. Cell 2015, 60, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Horikawa, I.; Ajiro, M.; Robles, A.I.; Fujita, K.; Mondal, A.M.; Stauffer, J.K.; Zheng, Z.M.; Harris, C.C. Downregulation of splicing factor SRSF3 induces p53β, an alternatively spliced isoform of p53 that promotes cellular senescence. Oncogene 2013, 32, 2792–2798. [Google Scholar] [CrossRef] [PubMed]
- Rauch, J.; Moran-Jones, K.; Albrecht, V.; Schwarzl, T.; Hunter, K.; Gires, O.; Kolch, W. c-Myc regulates RNA splicing of the A-Raf kinase and its activation of the ERK pathway. Cancer Res. 2011, 71, 4664–4674. [Google Scholar] [CrossRef]
- Xiao, S.H.; Manley, J.L. Phosphorylation of the ASF/SF2 RS domain affects both protein-protein and protein-RNA interactions and is necessary for splicing. Genes Dev. 1997, 11, 334–344. [Google Scholar] [CrossRef]
- Tazi, J.; Kornstadt, U.; Rossi, F.; Jeanteur, P.; Cathala, G.; Brunel, C.; Lührmann, R. Thiophosphorylation of U1-70K protein inhibits pre-mRNA splicing. Nature 1993, 363, 283–286. [Google Scholar] [CrossRef]
- Cao, W.; Jamison, S.F.; Garcia-Blanco, M.A. Both phosphorylation and dephosphorylation of ASF/SF2 are required for pre-mRNA splicing in vitro. RNA 1997, 3, 1456–1467. [Google Scholar]
- Rossi, F.; Labourier, E.; Forne, T.; Divita, G.; Derancourt, J.; Riou, J.F.; Antoine, E.; Cathala, G.; Brunel, C.; Tazi, J. Specific phosphorylation of SR proteins by mammalian DNA topoisomerase I. Nature 1996, 381, 80–82. [Google Scholar] [CrossRef]
- Blaustein, M.; Pelisch, F.; Coso, O.A.; Bissell, M.J.; Kornblihtt, A.R.; Srebrow, A. Mammary epithelial-mesenchymal interaction regulates fibronectin alternative splicing via phosphatidylinositol 3-kinase. J. Biol. Chem. 2004, 279, 21029–21037. [Google Scholar] [CrossRef]
- Zhou, Z.; Qiu, J.; Liu, W.; Zhou, Y.; Plocinik, R.M.; Li, H.; Hu, Q.; Ghosh, G.; Adams, J.A.; Rosenfeld, M.G.; et al. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol. Cell 2012, 47, 422–433. [Google Scholar] [CrossRef]
- Hayes, G.M.; Carrigan, P.E.; Beck, A.M.; Miller, L.J. Targeting the RNA splicing machinery as a novel treatment strategy for pancreatic carcinoma. Cancer Res. 2006, 66, 3819–3827. [Google Scholar] [CrossRef] [PubMed]
- Hayes, G.M.; Carrigan, P.E.; Miller, L.J. Serine-arginine protein kinase 1 overexpression is associated with tumorigenic imbalance in mitogen-activated protein kinase pathways in breast, colonic, and pancreatic carcinomas. Cancer Res. 2007, 67, 2072–2080. [Google Scholar] [CrossRef] [PubMed]
- Palacino, J.; Swalley, S.E.; Song, C.; Cheung, A.K.; Shu, L.; Zhang, X.; Van Hoosear, M.; Shin, Y.; Chin, D.N.; Keller, C.G.; et al. SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat. Chem. Biol. 2015, 11, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Krishnakumar, S.; Mohan, A.; Kandalam, M.; Ramkumar, H.L.; Venkatesan, N.; Das, R.R. SRPK1: A cisplatin sensitive protein expressed in retinoblastoma. Pediatr. Blood Cancer 2008, 50, 402–406. [Google Scholar] [CrossRef]
- Amin, E.M.; Oltean, S.; Hua, J.; Gammons, M.V.; Hamdollah-Zadeh, M.; Welsh, G.I.; Cheung, M.K.; Ni, L.; Kase, S.; Rennel, E.S.; et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 2011, 20, 768–780. [Google Scholar] [CrossRef]
- Goehe, R.W.; Shultz, J.C.; Murudkar, C.; Usanovic, S.; Lamour, N.F.; Massey, D.H.; Zhang, L.; Camidge, D.R.; Shay, J.W.; Minna, J.D.; et al. hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase-9 pre-mRNA processing. J. Clin. Investig. 2010, 120, 3923–3939. [Google Scholar] [CrossRef]
- Vu, N.T.; Park, M.A.; Shultz, J.C.; Goehe, R.W.; Hoeferlin, L.A.; Shultz, M.D.; Smith, S.A.; Lynch, K.W.; Chalfant, C.E. hnRNP U enhances caspase-9 splicing and is modulated by AKT-dependent phosphorylation of hnRNP L. J. Biol. Chem. 2013, 288, 8575–8584. [Google Scholar] [CrossRef] [PubMed]
- Jo, O.D.; Martin, J.; Bernath, A.; Masri, J.; Lichtenstein, A.; Gera, J. Heterogeneous nuclear ribonucleoprotein A1 regulates cyclin D1 and c-myc internal ribosome entry site function through Akt signaling. J. Biol. Chem. 2008, 283, 23274–23287. [Google Scholar] [CrossRef]
- Bonnal, S.; Vigevani, L.; Valcárcel, J. The spliceosome as a target of novel antitumour drugs. Nat. Rev. Drug Discov. 2012, 11, 847–859. [Google Scholar] [CrossRef]
- Webb, T.R.; Joyner, A.S.; Potter, P.M. The development and application of small molecule modulators of SF3b as therapeutic agents for cancer. Drug Discov. Today 2013, 18, 43–49. [Google Scholar] [CrossRef]
- Yokoi, A.; Kotake, Y.; Takahashi, K.; Kadowaki, T.; Matsumoto, Y.; Minoshima, Y.; Sugi, N.H.; Sagane, K.; Hamaguchi, M.; Iwata, M.; et al. Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J. 2011, 278, 4870–4880. [Google Scholar] [CrossRef] [PubMed]
- Teng, T.; Tsai, J.H.; Puyang, X.; Seiler, M.; Peng, S.; Prajapati, S.; Aird, D.; Buonamici, S.; Caleb, B.; Chan, B.; et al. Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A-SF3b complex. Nat. Commun. 2017, 8, 15522. [Google Scholar] [CrossRef] [PubMed]
- Finci, L.I.; Zhang, X.; Huang, X.; Zhou, Q.; Tsai, J.; Teng, T.; Agrawal, A.; Chan, B.; Irwin, S.; Karr, C.; et al. The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev. 2018, 32, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Cretu, C.; Agrawal, A.A.; Cook, A.; Will, C.L.; Fekkes, P.; Smith, P.G.; Lührmann, R.; Larsen, N.; Buonamici, S.; Pena, V. Structural basis of splicing modulation by antitumor macrolide compounds. Mol. Cell 2018, 70, 265–273. [Google Scholar] [CrossRef]
- Eskens, F.A.; Ramos, F.J.; Burger, H.; O’Brien, J.P.; Piera, A.; de Jonge, M.J.; Mizui, Y.; Wiemer, E.A.; Carreras, M.J.; Baselga, J.; et al. Phase I, pharmacokinetic and pharmacodynamic study of the first- in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin. Cancer Res. 2013, 19, 6296–6304. [Google Scholar] [CrossRef]
- Hong, D.S.; Kurzrock, R.; Naing, A.; Wheler, J.J.; Falchook, G.S.; Schiffman, J.S.; Faulkner, N.; Pilat, M.J.; O’Brien, J.; LoRusso, P. A phase I, open- label, single- arm, dose- escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Investig. New Drugs 2014, 32, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Mizui, Y.; Sakai, T.; Iwata, M.; Uenaka, T.; Okamoto, K.; Shimizu, H.; Yamori, T.; Yoshimatsu, K.; Asada, M. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. III. In vitro and in vivo antitumor activities. J. Antibiot. 2004, 57, 188–196. [Google Scholar] [CrossRef]
- Sakai, T.; Asai, N.; Okuda, A.; Kawamura, N.; Mizui, Y. Pladienolides, new substances from culture of Streptomyces platensis Mer-11107. II. Physico-chemical properties and structure elucidation. J. Antibiot. 2004, 57, 180–187. [Google Scholar] [CrossRef]
- Salton, M.; Misteli, T. Small molecule modulators of pre-mRNA splicing in cancer therapy. Trends Mol. Med. 2016, 22, 28–37. [Google Scholar] [CrossRef]
- Kotake, Y.; Sagane, K.; Owa, T.; Mimori-Kiyosue, Y.; Shimizu, H.; Uesugi, M.; Ishihama, Y.; Iwata, M.; Mizui, Y. Splicing factor SF3b as a target of the antitumor natural product pladienolide. Nat. Chem. Biol. 2007, 3, 570–575. [Google Scholar] [CrossRef]
- Iwata, M.; Ozawa, Y.; Uenaka, T.; Shimizu, H.; Niijima, J.; Kanada, R.M.; Fukuda, Y.; Nagai, M.; Kotake, Y.; Yoshida, M.; et al. E7107, a new 7-urethane derivative of pladienolide D, displays curative effect against several human tumor xenografts. Cancer Res. 2004, 64, 691. [Google Scholar]
- Nakajima, H.; Hori, Y.; Terano, H.; Okuhara, M.; Manda, T.; Matsumoto, S.; Shimomura, K. New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J. Antibiot. 1996, 49, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, H.; Sato, B.; Fujita, T.; Takase, S.; Terano, H.; Okuhara, M. New antitumor substances, FR901463, FR901464 and FR901465. I. Taxonomy, fermentation, isolation, physicochemical properties and biological activities. J. Antibiot. 1996, 49, 1196–1203. [Google Scholar] [CrossRef]
- Uehara, T.; Minoshima, Y.; Sagane, K.; Sugi, N.H.; Mitsuhashi, K.O.; Yamamoto, N.; Kamiyama, H.; Takahashi, K.; Kotake, Y.; Uesugi, M.; et al. Selective degradation of splicing factor CAPERalpha by anticancer sulfonamides. Nat. Chem. Biol. 2017, 13, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Niijhawan, D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356, eaal3755. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Stewart, C.; Abdel-Wahab, O. Altered RNA processing in cancer pathogenesis and therapy. Cancer Discov. 2019, 9, 1493–1510. [Google Scholar] [CrossRef]
- Hatcher, J.M.; Wu, G.; Zeng, C.; Zhu, J.; Meng, F.; Patel, S.; Wang, W.; Ficarro, S.B.; Leggett, A.L.; Powell, C.E.; et al. SRPKIN-1: A covalent SRPK1/2 inhibitor that potently converts VEGF from proangiogenic to anti- angiogenic isoform. Cell Chem. Biol. 2018, 25, 460–470. [Google Scholar] [CrossRef]
- Fukuhara, T.; Hosoya, T.; Shimizu, S.; Sumi, K.; Oshiro, T.; Yoshinaka, Y.; Suzuki, M.; Yamamoto, N.; Herzenberg, L.A.; Herzenberg, L.A.; et al. Utilization of host SR protein kinases and RNA splicing machinery during viral replication. Proc. Natl. Acad. Sci. USA 2006, 103, 11329–11333. [Google Scholar] [CrossRef]
- Desterro, J.; Bak-Gordon, P.; Carmo-Fonseca, M. Targeting mRNA processing as an anticancer strategy. Nat. Rev. Drug Discov. 2020, 19, 112–129. [Google Scholar] [CrossRef] [PubMed]
- Muraki, M.; Ohkawara, B.; Hosoya, T.; Onogi, H.; Koizumi, J.; Koizumi, T.; Sumi, K.; Yomoda, J.; Murray, M.V.; Kimura, H.; et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. J. Biol. Chem. 2004, 279, 24246–24254. [Google Scholar] [CrossRef]
- Iwai, K.; Yaguchi, M.; Nishimura, K.; Yamamoto, Y.; Tamura, T.; Nakata, D.; Dairiki, R.; Kawakita, Y.; Mizojiri, R.; Ito, Y.; et al. Anti-tumor efficacy of a novel CLK inhibitor via targeting RNA splicing and MYC-dependent vulnerability. EMBO Mol. Med. 2018, 10, e8289. [Google Scholar] [CrossRef]
- Araki, S.; Dairiki, R.; Nakayama, Y.; Murai, A.; Miyashita, R.; Iwatani, M.; Nomura, T.; Nakanishi, O. Inhibitors of CLK protein kinases suppress cell growth and induce apoptosis by modulating pre-mRNA splicing. PLoS ONE 2015, 10, e0116929. [Google Scholar] [CrossRef] [PubMed]
- Pilch, B.; Allemand, E.; Facompré, M.; Bailly, C.; Riou, J.F.; Soret, J.; Tazi, J. Splicing inhibition of serine- and arginine-rich splicing factors phosphorylation, spliceosome assembly, and splicing by the antitumor drug NM-506. Cancer Res. 2001, 61, 6876–6884. [Google Scholar] [PubMed]
- Chang, J.G.; Yang, D.M.; Chang, W.H.; Chow, L.P.; Chan, W.L.; Lin, H.H.; Huang, H.D.; Chang, Y.S.; Hung, C.H.; Yang, W.K. Small molecule amiloride modulates oncogenic RNA alternative splicing to devitalize human cancer cells. PLoS ONE 2011, 6, e18643. [Google Scholar] [CrossRef] [PubMed]
- Denichenko, P.; Mogilevsky, M.; Cléry, A.; Welte, T.; Biran, J.; Shimshon, O.; Barnabas, G.D.; Danan-Gotthold, M.; Kumar, S.; Yavin, E.; et al. Specific inhibition of splicing factor activity by decoy RNA oligonucleotides. Nat. Commun. 2019, 10, 1590. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; North, K.; Kim, E.; Jang, E.; Obeng, E.; Lu, S.X.; Liu, B.; Inoue, D.; Yoshimi, A.; Ki, M.; et al. Synthetic lethal and convergent biological effects of cancer-associated spliceosomal gene mutations. Cancer Cell 2018, 34, 225–241.e8. [Google Scholar] [CrossRef]
- Lee, S.C.; Dvinge, H.; Kim, E.; Cho, H.; Micol, J.B.; Chung, Y.R.; Durham, B.H.; Yoshimi, A.; Kim, Y.J.; Thomas, M.; et al. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat. Med. 2016, 22, 672–678. [Google Scholar] [CrossRef]
- Shirai, C.L.; Tripathi, M.; Ley, J.N.; Ndonwi, M.; White, B.S.; Tapia, R.; Saez, B.; Bertino, A.; Shao, J.; Kim, S.; et al. Preclinical activity of splicing modulators in U2AF1-mutant MDS–AML. Blood 2015, 126, 1653. [Google Scholar] [CrossRef]
- Assi, R.; Kantarjian, H.M.; Kadia, T.M.; Pemmaraju, N.; Jabbour, E.; Jain, N.; Daver, N.; Estrov, Z.; Uehara, T.; Owa, T.; et al. Final results of a phase 2, open-label study of indisulam, idarubicin, and cytarabine in patients with relapsed or refractory acute myeloid leukemia and high-risk myelodysplastic syndrome. Cancer 2018, 124, 2758–2765. [Google Scholar] [CrossRef]
- Talbot, D.C.; von Pawel, J.; Cattell, E.; Yule, S.M.; Johnston, C.; Zandvliet, A.S.; Huitema, A.D.; Norbury, C.J.; Ellis, P.; Bosquee, L.; et al. A randomized phase II pharmacokinetic and pharmacodynamics study of indisulam as second-line therapy in patients with advanced non-small cell lung cancer. Clin. Cancer Res. 2007, 13, 1816–1822. [Google Scholar] [CrossRef]
- Supuran, C.T. Indisulam: An anticancer sulfonamide in clinical development. Expert Opin. Investig. Drugs 2003, 12, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Lu, S.X.; Pastore, A.; Chen, X.; Imig, J.; Chun-Wei Lee, S.; Hockemeyer, K.; Ghebrechristos, Y.R.; Yoshimi, A.; Inoue, D.; et al. Targeting an RNA-binding protein network in acute myeloid leukemia. Cancer Cell 2019, 35, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Huang, J.; Higgs, B.W.; Hu, Z.; Xiao, Z.; Yao, X.; Conley, S.; Zhong, H.; Liu, Z.; Brohawn, P.; et al. Genomic landscape survey identifies SRSF1 as a key oncodriver in small cell lung cancer. PLoS Genet. 2016, 12, e1005895. [Google Scholar] [CrossRef]
- Shepard, P.J.; Hertel, K.J. The SR protein family. Genome Biol. 2009, 10, 242. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.; Hertel, K.J. Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip. Rev. RNA 2012, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Giannakouros, T.; Nikolakaki, E.; Mylonis, I.; Georgatsou, E. Serine–arginine protein kinases: A small protein kinase family with a large cellular presence. FEBS J. 2011, 278, 570–586. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Fu, X.D. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [CrossRef]
- McClorey, G.; Wood, M.J. An overview of the clinical application of antisense oligonucleotides for RNA-targeting therapies. Curr. Opin. Pharmacol. 2015, 24, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Meister, G.; Eggert, C.; Buhler, D.; Brahms, H.; Kambach, C.; Fischer, U. Methylation of Sm proteins by a complex containing PRMT5 and the putative U snRNP assembly factor pICln. Curr. Biol. 2001, 11, 1990–1994. [Google Scholar] [CrossRef]
- Brahms, H.; Meheus, L.; de Brabandere, V.; Fischer, U.; Lührmann, R. Symmetrical dimethylation of arginine residues in spliceosomal Sm protein B/B’ and the Sm-like protein LSm4, and their interactionwith the SMN protein. RNA 2001, 7, 1531–1542. [Google Scholar] [CrossRef]
- Hsu, T.Y.; Simon, L.M.; Neill, N.J.; Marcotte, R.; Sayad, A.; Bland, C.S.; Echeverria, G.V.; Sun, T.; Kurley, S.J.; Tyagi, S.; et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 2015, 525, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Banasavadi-Siddegowda, Y.K.; Welker, A.M.; An, M.; Yang, X.; Zhou, W.; Shi, G.; Imitola, J.; Li, C.; Hsu, S.; Wang, J.; et al. PRMT5 as a druggable target for glioblastoma therapy. Neuro. Oncol. 2018, 20, 753–763. [Google Scholar] [CrossRef]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tran, N.T.; Su, H.; Wang, R.; Lu, Y.; Tang, H.; Aoyagi, S.; Guo, A.; Khodadadi-Jamayran, A.; Zhou, D.; et al. Cross-talk between PRMT1-mediated methylation and ubiquitylation on RBM15 controls RNA splicing. eLife 2015, 4, e07938. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Zhang, L.; Villarreal, O.D.; He, W.; Su, D.; Bedford, E.; Moh, P.; Shen, J.; Shi, X.; Bedford, M.T.; et al. PRMT1 loss sensitizes cells to PRMT5 inhibition. Nucleic Acids Res. 2019, 47, 5038–5048. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Bezzi, M.; Low, D.H.; Ang, W.X.; Teo, S.X.; Gay, F.P.; Al-Haddawi, M.; Tan, S.Y.; Osato, M.; Sabò, A.; et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 2015, 523, 96–100. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Wilson, F.H.; Ruth, J.R.; Paulk, J.; Tsherniak, A.; Marlow, S.E.; Vazquez, F.; Weir, B.A.; Fitzgerald, M.E.; Tanaka, M.; et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 2016, 351, 1214–1218. [Google Scholar] [CrossRef]
- Mavrakis, K.J.; McDonald, E.R.; Schlabach, M.R.; Billy, E.; Hoffman, G.R.; de Weck, A.; Ruddy, D.A.; Venkatesan, K.; Yu, J.; McAllister, G.; et al. Disordered methionine metabolism in MTAP- and CDKN2A-deleted cancers leads to dependence on PRMT5. Science 2016, 351, 1208–1213. [Google Scholar] [CrossRef]
- Vu, L.P.; Cheng, Y.; Kharas, M.G. The biology of m(6)A RNA methylation in normal and malignant hematopoiesis. Cancer Discov. 2019, 9, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Masuda, T.; Canver, M.C.; Seiler, M.; Semba, Y.; Shboul, M.; Al-Raqad, M.; Maeda, M.; Schoonenberg, V.A.C.; Cole, M.A.; et al. Genome-wide CRISPR-Cas9 screen identifies leukemia-specific dependence on a pre-mRNA metabolic pathway regulated by DCPS. Cancer Cell 2018, 33, 386–400. [Google Scholar] [CrossRef]
- Gogliotti, R.G.; Cardona, H.; Singh, J.; Bail, S.; Emery, C.; Kuntz, N.; Jorgensen, M.; Durens, M.; Xia, B.; Barlow, C.; et al. The DcpS inhibitor RG3039 improves survival, function and motor unit pathologies in two SMA mouse models. Hum. Mol. Genet. 2013, 22, 4084–4101. [Google Scholar] [CrossRef] [PubMed]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Rigo, F.; Hua, Y.; Krainer, A.R.; Bennett, C.F. Antisense-based therapy for the treatment of spinal muscular atrophy. J. Cell Biol. 2012, 199, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F. Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Charleston, J.S.; Schnell, F.J.; Dworzak, J.; Donoghue, C.; Lewis, S.; Chen, L.; Young, G.D.; Milici, A.J.; Voss, J.; DeAlwis, U.; et al. Eteplirsen treatment for Duchenne muscular dystrophy: Exon skipping and Dystrophin production. Neurology 2018, 90, e2146–e2154. [Google Scholar] [CrossRef]
- Hua, Y.; Vickers, T.A.; Okunola, H.L.; Bennett, C.F.; Krainer, A.R. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am. J. Hum. Genet. 2008, 82, 834–848. [Google Scholar] [CrossRef]
- Michelson, D.; Ciafaloni, E.; Ashwal, S.; Lewis, E.; Narayanaswami, P.; Oskoui, M.; Armstrong, M.J. Evidence in focus: Nusinersen use in spinal muscular atrophy: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology 2018, 91, 923–933. [Google Scholar] [CrossRef]
- Wang, Z.; Jeon, H.Y.; Rigo, F.; Bennett, C.F.; Krainer, A.R. Manipulation of PK-M mutually exclusive alternative splicing by antisense oligonucleotides. Open Biol. 2012, 2, 120133. [Google Scholar] [CrossRef] [PubMed]
- Hastings, M.L.; Berniac, J.; Liu, Y.H.; Abato, P.; Jodelka, F.M.; Barthel, L.; Kumar, S.; Dudley, C.; Nelson, M.; Larson, K.; et al. Tetracyclines that promote SMN2 exon 7 splicing as therapeutics for spinal muscular atrophy. Sci. Transl. Med. 2009, 1, 5ra12. [Google Scholar] [CrossRef]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef]
- Yoshida, M.; Kataoka, N.; Miyauchi, K.; Ohe, K.; Iida, K.; Yoshida, S.; Nojima, T.; Okuno, Y.; Onogi, H.; Usui, T.; et al. Rectifier of aberrant mRNA splicing recovers tRNA modification in familial dysautonomia. Proc. Natl. Acad. Sci. USA 2015, 112, 2764–2769. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Nandi, S. Choices have consequences: The nexus between DNA repair pathways and genomic instability in cancer. Clin. Transl. Med. 2016, 5, 45. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Nandi, S. Synthetic lethality in DNA repair network: A novel avenue in targeted cancer therapy and combination therapeutics. IUBMB Life 2017, 69, 929–937. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Nandi, S. DNA damage response and cancer therapeutics through the lens of the Fanconi Anemia DNA repair pathway. Cell Commun. Signal. 2017, 15, 41. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Nandi, S. Rare Genetic Diseases with Defects in DNA Repair: Opportunities and Challenges in Orphan Drug Development for Targeted Cancer Therapy. Cancers 2018, 10, 298. [Google Scholar] [CrossRef]
- Ghosh, D.; Venkatramani, P.; Nandi, S.; Bhattacharjee, S. CRISPR–Cas9 a boon or bane: The bumpy road ahead to cancer therapeutics. Cancer Cell Int. 2019, 19, 12. [Google Scholar] [CrossRef]
- Huilgol, D.; Venkataramani, P.; Nandi, S.; Bhattacharjee, S. Targeting transcription factors that govern development and disease: An achilles heel for cancer therapeutics. Genes 2019, 10, 794. [Google Scholar] [CrossRef]
- Kim, Y.J.; Rahman, M.A. NMD in diseases and potential therapies. J. Investig. Genom. 2018, 5, 50–54. [Google Scholar] [CrossRef]
- Nazim, M.; Nasrin, F.; Rahman, M.A. Coordinated regulation of alternative splicing and alternative polyadenylation. J. Genet. Genet. Eng. 2018, 2, 26–34. [Google Scholar]



{kind=link}
{kind=link}
| Gene | Alteration | Tumor Type | Reference |
|---|---|---|---|
| SRSF2 | Mutation | MDS, AML, CMML, RARS, RCMD-RS, MPN, UVM | [4,11,18,19] |
| Hot spot: P95 | |||
| Overexpression | Ovary | ||
| SF3B1 | Mutation | MDS, AML, CMML, CLL, RARS, RCMD-RS, MPN, UVM, MM, Breast, PDAC | [4,11,18,19] |
| Hotspot: K700, E622, R625, H662, K666 | |||
| U2AF1 | Mutation | MDS, AML, CMML, RARS, RCMD-RS, MPN, Lung | [4,11,18,19] |
| Hot spot: S34, Q157 | |||
| ZRSR2 | Mutation | MDS, AML, CMML, RARS, RCMD-RS, MPN | [4,11,18,19] |
| No hot spot | |||
| SRSF1 | Amplification, overexpression | Breast, lung, colon, ovary, thyroid kidney, small intestine | [11,23,24] |
| SRSF3 | Downregulation | Liver | [11,25,26,27] |
| Amplification, overexpression | Ovary, cervix, breast, skin, stomach, bladder, thyroid, kidney, colon, liver | ||
| SRSF5 | Overexpression | Breast | [28] |
| SRSF6 | Amplification, overexpression | Breast, Lung, colon, skin | [23,29,30] |
| SRSF10 | Overexpression | Colorectal | [31] |
| TRA2B | Overexpression | Breast, colon, glioblastoma, ovary | [11,24,32,33] |
| RBM5 | Downregulation | Lung, prostate | [34,35,36] |
| Overexpression | Breast | ||
| RBM10 | Mutation | Lung | [34,37] |
| Overexpression | Breast | ||
| ESRP1, ESRP2 | Overexpression/downregulation | Breast, oral | [38,39] |
| HNRNPA1 | Overexpression | Glioblastoma, breast, colon, lung | [23,40,41,42] |
| HNRNPA2/B1 | Amplification, overexpression | Glioblastoma | [40] |
| HNRNPM | Overexpression | Breast | [43] |
| HNRNPK | Downregulation | AML | [44,45] |
| Overexpression | Breast, pancreatic, skin, esophageal, colorectal, oral | ||
| HNRNPH | Overexpression | Glioblastoma | [46] |
| PTB | Overexpression | Breast, ovary, oral | [27,47,48] |
| QKI | Downregulation | Lung adenocarcinoma, prostate, oral | [49,50] |
| Compound | Target of Inhibition | Mechanism(s) of Action | Reference | |
|---|---|---|---|---|
| Pladienolide: | SF3B complex | Interact with SF3B1 and promote conformational alteration | [10,116,117,118] | |
| Pladienolides A–G | Antitumor activities reported in human BSY-1, PC-3, OVCAR-3, DU-145, WiDr, HCT-116 cells, primary human colon cancer cells, xenograft mouse model, etc. | |||
| Pladienolide analog: | SF3B complex | Interacts with SF3B1 and promotes splicing alteration | [10,118,119,120] | |
| E7107 | Inhibition of tumor growth in human breast, ovary, non-small cell lung cancer, xenograft mouse model, etc. | |||
| Spliceostatin: | SF3B complex | Interface interaction of U2 snRNP with pre- | [10,118,121,122] | |
| FR901463 | ||||
| FR901464 | mRNA | |||
| FR901465 | ||||
| Meayamycin B | Antitumor activities reported in murine solid tumors (colon 38 and Meth A), human lung adenocarcinoma solid tumor (A549), human colorectal carcinoma (HCT-116), human prostate tumor (PC-3), human rhabdomyosarcoma (rh18), xenograft mouse model, etc. | |||
| Spliceostatin A | ||||
| Sudemycins | ||||
| Sufonamides: | RBM39 | Bind a receptor of the CRL4 E3 ubiquitin ligase complex (known as DCAF15), and direct ubiquitin-mediated degradation of RBM39 | [123,124,125] | |
| Indisulam | ||||
| Tasisulam | ||||
| E7820 | ||||
| SRPK inhibitor: | SRPK1 and SRPK2 | Competitively inhibit SRPK1 and SRPK2, subsequently affect SR protein phosphorylation | [126,127,128] | |
| SRPIN340 | ||||
| SRPKIN-1 | SRPIN340 is reported to block angiogenesis and related tumor growth in nude mice | |||
| SRPKIN-1 is reported to convert the proangiogenic VEGF isoform to the angiogenic isoform | ||||
| CLK inhibitor: | CLK1, CLK2, CLK4 | Competitively inhibit CLK1, CLK2, and CLK4, and subsequently affect SR protein phosphorylation | [10,129,130] | |
| T-025 | ||||
| TG003 | T-025 is reported to affect global alternative splicing, inducing cancer cell death, and inhibiting cancer cell growth | |||
| SRPK and CLK | SRPK1, SRPK2, CLK1, CLK2 | Inhibit SRPK1, SRPK2, CLK1, and CLK2, and subsequently affect SR protein phosphorylation | [10,118,131] | |
| inhibitor: | ||||
| Cpd-1 | ||||
| Cpd-2 | ||||
| Cpd-3 | ||||
| Topoisomerase I inhibitor: | Topoisomerase I | Inhibitory effect prior to step one of splicing | [132] | |
| NB-506 | Inhibits SRSF1 phosphorylation and splicing of Bcl-X, CD44, SRSF2, and Sty in P388 cultured cells | |||
| Amiloride | Unknown | Inhibits SRSF1 phosphorylation and splicing of Bcl-X, HIPK3, and RON/MISTR1 in Huh-7 cultured cells | [133] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.A.; Nasrin, F.; Bhattacharjee, S.; Nandi, S. Hallmarks of Splicing Defects in Cancer: Clinical Applications in the Era of Personalized Medicine. Cancers 2020, 12, 1381. https://doi.org/10.3390/cancers12061381
Rahman MA, Nasrin F, Bhattacharjee S, Nandi S. Hallmarks of Splicing Defects in Cancer: Clinical Applications in the Era of Personalized Medicine. Cancers. 2020; 12(6):1381. https://doi.org/10.3390/cancers12061381
Chicago/Turabian StyleRahman, Mohammad Alinoor, Farhana Nasrin, Sonali Bhattacharjee, and Saikat Nandi. 2020. "Hallmarks of Splicing Defects in Cancer: Clinical Applications in the Era of Personalized Medicine" Cancers 12, no. 6: 1381. https://doi.org/10.3390/cancers12061381
APA StyleRahman, M. A., Nasrin, F., Bhattacharjee, S., & Nandi, S. (2020). Hallmarks of Splicing Defects in Cancer: Clinical Applications in the Era of Personalized Medicine. Cancers, 12(6), 1381. https://doi.org/10.3390/cancers12061381