Liquid Biopsy in Colorectal Carcinoma: Clinical Applications and Challenges

 , ,
, ,  and
and 
Abstract
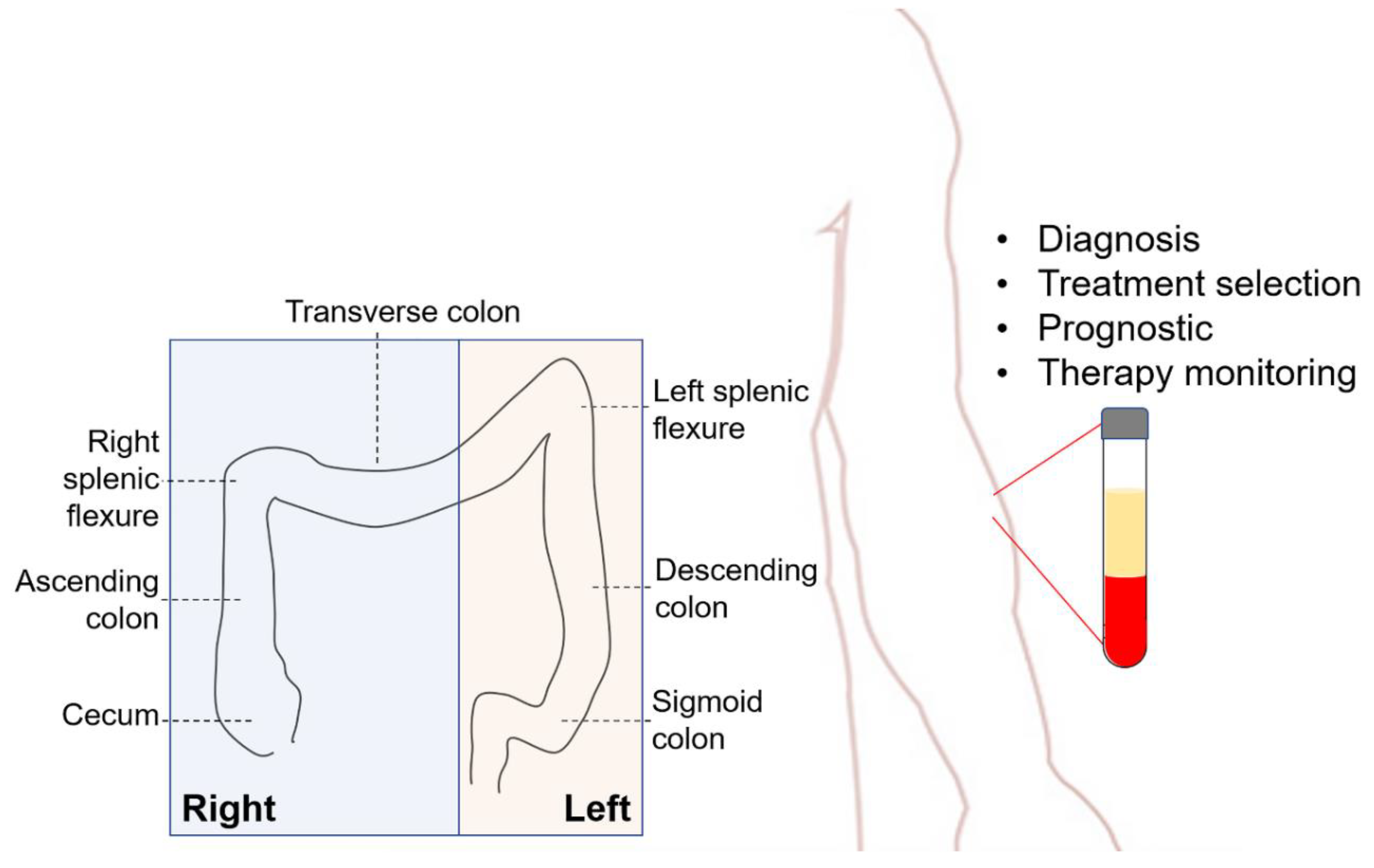
1. Introduction
2. Liquid Biopsy for CRC
2.1. Circulating Tumor Cells
2.2. Cell-Free DNA
3. Liquid Biopsy Platforms
3.1. Detection of Circulating Tumor Cells
3.1.1. CellSearch®
3.1.2. Epic Sciences/High-Definition Single-Cell Assay
3.1.3. The Isolation by Size of Epithelial Tumor Cells®
3.2. cfDNA Analysis
4. Clinical Applications
4.1. Diagnostics
4.2. Treatment Selection
4.3. Prognostics
5. Challenges
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2016, 66, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Araghi, M.; Soerjomataram, I.; Jenkins, M.A.; Brierley, J.; Morris, E.; Bray, F.; Arnold, M. Global trends in colorectal cancer mortality: Projections to the year 2035. Int. J. Cancer 2019, 144, 2992–3000. [Google Scholar] [CrossRef]
- Sharp, L.; O’Leary, E.; O’Ceilleachair, A.; Skally, M.; Hanly, P. Financial Impact of Colorectal Cancer and Its Consequences. Dis. Colon Rectum 2018, 61, 27–35. [Google Scholar] [CrossRef]
- John, S.K.P.; George, S.; Primrose, J.N.; Fozard, J.B.J. Symptoms and signs in patients with colorectal cancer. Color. Dis. 2010, 13, 17–25. [Google Scholar] [CrossRef]
- Uraoka, T.; Hosoe, N.; Yahagi, N. Colonoscopy: Is it as effective as an advanced diagnostic tool for colorectal cancer screening? Expert Rev. Gastroenterol. Hepatol. 2014, 9, 129–132. [Google Scholar] [CrossRef][Green Version]
- Baek, S.K. Laterality: Right-Sided and Left-Sided Colon Cancer. Ann. Coloproctol. 2017, 33, 205–206. [Google Scholar] [CrossRef][Green Version]
- Helvaci, K.; Eraslan, E.; Yildiz, F.; Tufan, G.; Demirci, U.; Berna Oksuzoglu, O.; Yalcintas Arslan, U. Comparison of clinicopathological and survival features of right and left colon cancers. J. BUON Off. J. Balk. Union Oncol. 2019, 24, 1845–1851. [Google Scholar]
- Mik, M.; Dziki, Ł.; Trzciński, R. Risk factors of 30-day mortality following surgery for colorectal cancer. Pol. J. Surg. 2016, 88, 26–31. [Google Scholar] [CrossRef]
- Doubeni, C.A.; Corley, D.A.; Quinn, V.P.; Jensen, C.D.; Zauber, A.G.; Goodman, M.; Johnson, J.R.; Mehta, S.J.; Becerra, T.A.; Zhao, W.K.; et al. Effectiveness of screening colonoscopy in reducing the risk of death from right and left colon cancer: A large community-based study. Gut 2016, 67, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Nawa, T.; Kato, J.; Kawamoto, H.; Okada, H.; Yamamoto, H.; Kohno, H.; Endo, H.; Shiratori, Y. Differences between right- and left-sided colon cancer in patient characteristics, cancer morphology and histology. J. Gastroenterol. Hepatol. 2008, 23, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Richman, S.D.; Chambers, P.; Seymour, M.T.; Daly, C.; Grant, S.; Hemmings, G.; Quirke, P. Intra-tumoral Heterogeneity of KRAS and BRAF Mutation Status in Patients with Advanced Colorectal Cancer (aCRC) and Cost-Effectiveness of Multiple Sample Testing. Anal. Cell. Pathol. 2011, 34, 61–66. [Google Scholar] [CrossRef]
- Smith, G.; Carey, F.A.; Beattie, J.; Wilkie, M.J.V.; Lightfoot, T.J.; Coxhead, J.; Garner, R.C.; Steele, R.J.; Wolf, C.R. Mutations in APC, Kirsten-ras, and p53—Alternative genetic pathways to colorectal cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 9433–9438. [Google Scholar] [CrossRef] [PubMed]
- Ionov, Y.; Peinado, M.A.; Malkhosyan, S.; Shibata, D.; Perucho, M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature 1993, 363, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Samowitz, W.S. Poor Survival Associated with the BRAF V600E Mutation in Microsatellite-Stable Colon Cancers. Cancer Res. 2005, 65, 6063–6069. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Catalano, P.J.; Benson, A.B.; O’Dwyer, P.; Hamilton, S.R.; Issa, J.-P. Association between DNA methylation and shortened survival in patients with advanced colorectal cancer treated with 5-fluorouracil based chemotherapy. Clin. Cancer Res. 2007, 13, 6093–6098. [Google Scholar] [CrossRef]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.; Kang, G.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef]
- Hadjihannas, M.V.; Brückner, M.; Jerchow, B.; Birchmeier, W.; Dietmaier, W.; Behrens, J. Aberrant Wnt/beta-catenin signaling can induce chromosomal instability in colon cancer. Proc. Natl. Acad. Sci. USA 2006, 103, 10747–10752. [Google Scholar] [CrossRef]
- Cisyk, A.; Penner-Goeke, S.; Lichtensztejn, Z.; Nugent, Z.; Wightman, R.; Singh, H.; McManus, K.J. Characterizing the prevalence of chromosome instability in interval colorectal cancer. Neoplasia 2015, 17, 306–316. [Google Scholar] [CrossRef]
- Thomas, D.C.; Umar, A.; Kunkel, T. Microsatellite instability and mismatch repair defects in cancer cells. Mutat. Res. Mol. Mech. Mutagen. 1996, 350, 201–205. [Google Scholar] [CrossRef]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar] [PubMed]
- Alexander, J.; Watanabe, T.; Wu, T.-T.; Rashid, A.; Li, S.; Hamilton, S.R. Histopathological Identification of Colon Cancer with Microsatellite Instability. Am. J. Pathol. 2001, 158, 527–535. [Google Scholar] [CrossRef]
- Lanza, G.; Gafà, R.; Maestri, I.; Santini, A.; Matteuzzi, M.; Cavazzini, L. Immunohistochemical Pattern of MLH1/MSH2 Expression Is Related to Clinical and Pathological Features in Colorectal Adenocarcinomas with Microsatellite Instability. Mod. Pathol. 2002, 15, 741–749. [Google Scholar] [CrossRef]
- Ricciardiello, L.; Ceccarelli, C.; Angiolini, G.; Pariali, M.; Chieco, P.; Paterini, P.; Biasco, G.; Martinelli, G.N.; Roda, E.; Bazzoli, F. High Thymidylate Synthase Expression in Colorectal Cancer with Microsatellite Instability: Implications for Chemotherapeutic Strategies. Clin. Cancer Res. 2005, 11, 4234–4240. [Google Scholar] [CrossRef]
- Trautmann, K.; Terdiman, J.P.; French, A.J.; Roydasgupta, R.; Sein, N.; Kakar, S.; Fridlyand, J.; Snijders, A.M.; Albertson, N.G.; Thibodeau, S.N.; et al. Chromosomal Instability in Microsatellite-Unstable and Stable Colon Cancer. Clin. Cancer Res. 2006, 12, 6379–6385. [Google Scholar] [CrossRef]
- Peltomaki, P. Deficient DNA mismatch repair: A common etiologic factor for colon cancer. Hum. Mol. Genet. 2001, 10, 735–740. [Google Scholar] [CrossRef]
- Hinoue, T.; Weisenberger, D.J.; Lange, C.P.; Shen, H.; Byun, H.-M.; Berg, D.V.D.; Malik, S.; Pan, F.; Noushmehr, H.; Van Dijk, C.M.; et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2011, 22, 271–282. [Google Scholar] [CrossRef]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Meyerhardt, J.A.; Loda, M.; Giovannucci, E.L.; Fuchs, C.S. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut 2008, 58, 90–96. [Google Scholar] [CrossRef]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.-P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef]
- Huang, D.; Sun, W.; Zhou, Y.; Li, P.; Chen, F.; Chen, H.; Xia, D.; Xu, E.; Lai, M.; Wu, Y.; et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. 2018, 37, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell 2018, 33, 125–136.e3. [Google Scholar] [CrossRef] [PubMed]
- Horst, D. Plastizität der WNT-Signalwegaktivität im Kolonkarzinom. Der Pathol. 2012, 33, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Kongkanuntn, R.; Bubb, V.J.; Sansom, O.J.; Wyllie, A.H.; Harrison, D.J.; Clarke, A. Dysregulated expression of β-catenin marks early neoplastic change in Apc mutant mice, but not all lesions arising in Msh2 deficient mice. Oncogene 1999, 18, 7219–7225. [Google Scholar] [CrossRef]
- Jeong, W.-J.; Yoon, J.-B.; Park, J.-C.; Lee, S.-H.; Kaduwal, S.; Kim, H.; Choi, K.-Y. Ras Stabilization Through Aberrant Activation of Wnt/ -Catenin Signaling Promotes Intestinal Tumorigenesis. Sci. Signal. 2012, 5, ra30. [Google Scholar] [CrossRef] [PubMed]
- Lemieux, E.; Cagnol, S.; Beaudry, K.; Carrier, J.; Rivard, N. Oncogenic KRAS signalling promotes the Wnt/β-catenin pathway through LRP6 in colorectal cancer. Oncogene 2014, 34, 4914–4927. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Haling, J.R.; Chen, H.; Song, K.; Price, S.; Heald, R.; Hewitt, J.F.M.; Zak, M.; Peck, A.; Orr, C.; et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature 2013, 501, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M.; Kendall, K.R.; Wang, Y.; Cheung, A.; Haigis, M.C.; Glickman, J.N.; Niwa-Kawakita, M.; Sweet-Cordero, A.; Sebolt-Leopold, J.; Shannon, K.M.; et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat. Genet. 2008, 40, 600–608. [Google Scholar] [CrossRef]
- Pacold, M.E.; Suire, S.; Perisic, O.; Lara-González, S.; Davis, C.T.; Walker, E.H.; Hawkins, P.; Stephens, L.R.; Eccleston, J.F.; Williams, R.L. Crystal Structure and Functional Analysis of Ras Binding to Its Effector Phosphoinositide 3-Kinase γ. Cell 2000, 103, 931–944. [Google Scholar] [CrossRef]
- Murillo, M.M.; Zelenay, S.; Nye, E.; Castellano, E.; Lassailly, F.; Stamp, G.; Downward, J. RAS interaction with PI3K p110α is required for tumor-induced angiogenesis. J. Clin. Investig. 2014, 124, 3601–3611. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-Type BRAF Is Required for Response to Panitumumab or Cetuximab in Metastatic Colorectal Cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef]
- Souglakos, J.; Philips, J.; Wang, R.; Marwah, S.; Silver, M.; Tzardi, M.; Silver, J.; Ogino, S.; Hooshmand, S.; Kwak, E.; et al. Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br. J. Cancer 2009, 101, 465–472. [Google Scholar] [CrossRef]
- Yokota, T.; Ura, T.; Shibata, N.; Takahari, D.; Shitara, K.; Nomura, M.; Kondo, C.; Mizota, A.; Utsunomiya, S.; Muro, K.; et al. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br. J. Cancer 2011, 104, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guo, F.; Shi, X.; Zhang, L.; Zhang, A.; Jin, H.; He, Y. BRAF V600E mutation and KRAS codon 13 mutations predict poor survival in Chinese colorectal cancer patients. BMC Cancer 2014, 14, 802. [Google Scholar] [CrossRef] [PubMed]
- Sahin, I.H.; Kazmi, S.M.; Yorio, J.T.; Bhadkamkar, N.A.; Kee, B.K.; Garrett, C.R. Rare Though Not Mutually Exclusive: A Report of Three Cases of Concomitant KRAS and BRAF Mutation and a Review of the Literature. J. Cancer 2013, 4, 320–322. [Google Scholar] [CrossRef] [PubMed]
- Li, A.-J.; Li, H.-G.; Tang, E.-J.; Wu, W.; Chen, Y.; Jiang, H.-H.; Lin, M.-B.; Yin, L. PIK3CA and TP53 mutations predict overall survival of stage II/III colorectal cancer patients. World J. Gastroenterol. 2018, 24, 631–640. [Google Scholar] [CrossRef]
- Lu, T.; Li, J. Clinical applications of urinary cell-free DNA in cancer: Current insights and promising future. Am. J. Cancer Res. 2017, 7, 2318–2332. [Google Scholar]
- Sidransky, D.; Tokino, T.; Hamilton, S.R.; Kinzler, K.; Levin, B.; Frost, P.; Vogelstein, B. Identification of ras oncogene mutations in the stool of patients with curable colorectal tumors. Science 1992, 256, 102–105. [Google Scholar] [CrossRef]
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martinez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventós, C.; Tang, J.; et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 2015, 6, 8839. [Google Scholar] [CrossRef]
- Stefancu, A.; Badarinza, M.; Moisoiu, V.; Iancu, S.D.; Serban, O.; Leopold, N.; Fodor, D. SERS-based liquid biopsy of saliva and serum from patients with Sjögren’s syndrome. Anal. Bioanal. Chem. 2019, 411, 5877–5883. [Google Scholar] [CrossRef]
- Song, Z.; Cai, Z.; Yan, J.; Shao, Y.W.; Zhang, Y. Liquid biopsies using pleural effusion-derived exosomal DNA in advanced lung adenocarcinoma. Transl. Lung Cancer Res. 2019, 8, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Peterson, V.M.; Castro, C.M.; Chung, J.; Miller, N.C.; Ullal, A.V.; Castano, M.D.; Penson, R.T.; Lee, H.; Birrer, M.J.; Weissleder, R. Ascites analysis by a microfluidic chip allows tumor-cell profiling. Proc. Natl. Acad. Sci. USA 2013, 110, E4978–E4986. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-Y.; Oskarsson, T.; Acharyya, S.; Nguyen, N.X.; Zhang, X.H.-F.; Norton, L.; Massagué, J. Tumor Self-Seeding by Circulating Cancer Cells. Cell 2009, 139, 1315–1326. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, C.; Li, J.; Luttgen, M.S.; Kolatkar, A.; Kendall, J.T.; Flores, E.; Topp, Z.; Samlowski, W.E.; McClay, E.; Bethel, K.; et al. Limited genomic heterogeneity of circulating melanoma cells in advanced stage patients. Phys. Biol. 2015, 12, 016008. [Google Scholar] [CrossRef]
- Che, J.; Yu, V.; Garon, E.B.; Goldman, J.W.; Di Carlo, D. Biophysical isolation and identification of circulating tumor cells. Lab Chip 2017, 17, 1452–1461. [Google Scholar] [CrossRef]
- Harouaka, R.; Nisic, M.; Zheng, S.-Y. Circulating tumor cell enrichment based on physical properties. J. Lab. Autom. 2013, 18, 455–468. [Google Scholar] [CrossRef]
- Marrinucci, D.; Bethel, K.; Kolatkar, A.; Luttgen, M.S.; Malchiodi, M.; Baehring, F.; Voigt, K.; Lazar, D.; Nieva, J.J.; Bazhenova, L.; et al. Fluid biopsy in patients with metastatic prostate, pancreatic and breast cancers. Phys. Biol. 2012, 9, 016003. [Google Scholar] [CrossRef]
- Ashworth, T.R. A case of cancer in which cells similar to those in the tumours were seen in the blood after death. Aust. Med. J. 1869, 14, 146. [Google Scholar]
- Hofman, V.; Ilie, M.; Long, E.; Selva, E.; Bonnetaud, C.; Molina, T.; Venissac, N.; Mouroux, J.; Vielh, P.; Hofman, P. Detection of circulating tumor cells as a prognostic factor in patients undergoing radical surgery for non-small-cell lung carcinoma: Comparison of the efficacy of the CellSearch Assay™ and the isolation by size of epithelial tumor cell method. Int. J. Cancer 2011, 129, 1651–1660. [Google Scholar] [CrossRef] [PubMed]
- Müller, V.; Stahmann, N.; Riethdorf, S.; Rau, T.; Zabel, T.; Goetz, A.; Jänicke, F.; Pantel, K. Circulating Tumor Cells in Breast Cancer: Correlation to Bone Marrow Micrometastases, Heterogeneous Response to Systemic Therapy and Low Proliferative Activity. Clin. Cancer Res. 2005, 11, 3678–3685. [Google Scholar] [CrossRef] [PubMed]
- Onidani, K.; Shoji, H.; Kakizaki, T.; Yoshimoto, S.; Okaya, S.; Miura, N.; Sekikawa, S.; Furuta, K.; Lim, C.T.; Shibahara, T.; et al. Monitoring of cancer patients via next-generation sequencing of patient-derived circulating tumor cells and tumor DNA. Cancer Sci. 2019, 110, 2590–2599. [Google Scholar] [CrossRef]
- Scher, H.I.; Lu, D.; Schreiber, N.A.; Louw, J.; Graf, R.P.; Vargas, H.A.; Johnson, A.; Jendrisak, A.; Bambury, R.; Danila, D.; et al. Association of AR-V7 on Circulating Tumor Cells as a Treatment-Specific Biomarker With Outcomes and Survival in Castration-Resistant Prostate Cancer. JAMA Oncol. 2016, 2, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Gasch, C.; Bauernhofer, T.; Pichler, M.; Langer-Freitag, S.; Reeh, M.; Seifert, A.M.; Mauermann, O.; Izbicki, J.; Pantel, K.; Riethdorf, S. Heterogeneity of Epidermal Growth Factor Receptor Status and Mutations of KRAS/PIK3CA in Circulating Tumor Cells of Patients with Colorectal Cancer. Clin. Chem. 2013, 59, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Malihi, P.D.; Morikado, M.; Welter, L.; Liu, S.T.; Miller, E.T.; Cadaneanu, R.M.; Knudsen, B.S.; Lewis, M.S.; Carlsson, A.; Velasco, C.R.; et al. Clonal diversity revealed by morphoproteomic and copy number profiles of single prostate cancer cells at diagnosis. Converg. Sci. Phys. Oncol. 2018, 4, 015003. [Google Scholar] [CrossRef]
- Thiele, J.-A.; Pitule, P.; Hicks, J.; Kuhn, P. Single-Cell Analysis of Circulating Tumor Cells. Adv. Struct. Saf. Stud. 2019, 1908, 243–264. [Google Scholar] [CrossRef]
- Carlsson, A.; Nair, V.S.; Luttgen, M.S.; Keu, K.V.; Horng, G.; Vasanawala, M.; Kolatkar, A.; Jamali, M.; Iagaru, A.H.; Kuschner, W.; et al. Circulating tumor microemboli diagnostics for patients with non-small-cell lung cancer. J. Thorac. Oncol. 2014, 9, 1111–1119. [Google Scholar] [CrossRef]
- Steeg, P.S. Tumor metastasis: Mechanistic insights and clinical challenges. Nat. Med. 2006, 12, 895–904. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Guibert, N.; Delaunay, M.; Lusque, A.; Boubekeur, N.; Rouquette, I.; Clermont, E.; Gouin, S.; Dormoy, I.; Favre, G.; Mazieres, J.; et al. PD-L1 expression in circulating tumor cells of advanced non-small cell lung cancer patients treated with nivolumab. Lung Cancer 2018, 120, 108–112. [Google Scholar] [CrossRef]
- Boffa, D.J.; Graf, R.P.; Salazar, M.C.; Hoag, J.; Lu, D.; Krupa, R.; Louw, J.; Dugan, L.; Wang, Y.; Landers, M.; et al. Cellular Expression of PD-L1 in the Peripheral Blood of Lung Cancer Patients is Associated with Worse Survival. Cancer Epidemiol. Biomark. Prev. 2017, 26, 1139–1145. [Google Scholar] [CrossRef]
- Mandel, P.; Metais, P. Les acides nucleiques du plasma sanguine chez l’homme. Comptes Rendus Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar] [PubMed]
- Anker, P.; Stroun, M.; Maurice, P.A. Spontaneous release of DNA by human blood lymphocytes as shown in an in vitro system. Cancer Res. 1975, 35, 2375–2382. [Google Scholar]
- Wang, W.; Kong, P.; Ma, G.; Li, L.; Zhu, J.; Xia, T.; Xie, H.; Zhou, W.; Wang, S. Characterization of the release and biological significance of cell-free DNA from breast cancer cell lines. Oncotarget 2017, 8, 43180–43191. [Google Scholar] [CrossRef]
- Stroun, M.; Lyautey, J.; Lederrey, C.; Olson-Sand, A.; Anker, P. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin. Chim. Acta 2001, 313, 139–142. [Google Scholar] [CrossRef]
- Mouliere, F.; Thierry, A. The importance of examining the proportion of circulating DNA originating from tumor, microenvironment and normal cells in colorectal cancer patients. Expert Opin. Biol. Ther. 2012, 12, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.M.D.; Chan, K.C.A.; Sun, H.; Chen, E.Z.; Jiang, P.; Lun, F.M.F.; Zheng, Y.W.; Leung, T.Y.; Lau, T.K.; Cantor, C.; et al. Maternal Plasma DNA Sequencing Reveals the Genome-Wide Genetic and Mutational Profile of the Fetus. Sci. Transl. Med. 2010, 2, 61ra91. [Google Scholar] [CrossRef]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Analysis of the Size Distributions of Fetal and Maternal Cell-Free DNA by Paired-End Sequencing. Clin. Chem. 2010, 56, 1279–1286. [Google Scholar] [CrossRef]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar]
- Hao, T.B.; Shi, W.; Shen, X.J.; Qi, J.; Wu, X.H.; Wu, Y.; Tang, Y.Y.; Ju, S.Q. Circulating cell-free DNA in serum as a biomarker for diagnosis and prognostic prediction of colorectal cancer. Br. J. Cancer 2014, 111, 1482–1489. [Google Scholar] [CrossRef]
- Mohan, S.; Ayub, M.; Rothwell, D.G.; Gulati, S.; Kilerci, B.; Hollebecque, A.; Leong, H.S.; Smith, N.K.; Sahoo, S.; Descamps, T.; et al. Analysis of circulating cell-free DNA identifies KRAS copy number gain and mutation as a novel prognostic marker in Pancreatic cancer. Sci. Rep. 2019, 9, 11610–11616. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Huang, H.J.; Claes, B.; Falchook, G.S.; Fu, S.; Hong, D.; Ramzanali, N.M.; Nitti, G.; Cabrilo, G.; Tsimberidou, A.M.; et al. BRAF Mutation Testing in Cell-Free DNA from the Plasma of Patients with Advanced Cancers Using a Rapid, Automated Molecular Diagnostics System. Mol. Cancer Ther. 2016, 15, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Morris, V.K.; El Osta, B.; Sorokin, A.V.; Janku, F.; Fu, S.; Overman, M.J.; Piha-Paul, S.A.; Subbiah, V.; Kee, B.; et al. Phase IB Study of Vemurafenib in Combination with Irinotecan and Cetuximab in Patients with Metastatic Colorectal Cancer with BRAFV600E Mutation. Cancer Discov. 2016, 6, 1352–1365. [Google Scholar] [CrossRef] [PubMed]
- Allard, W.J. Tumor Cells Circulate in the Peripheral Blood of All Major Carcinomas but not in Healthy Subjects or Patients With Nonmalignant Diseases. Clin. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef]
- Cohen, S.J.; Terstappen, L.W.; Punt, C.J.; Mitchell, E.P.; Fynan, T.M.; Li, T.; Matera, J.; Doyle, G.V.; Meropol, N.J. Circulating endothelial cells (CEC) and circulating tumor cells (CTC) in patients (pts) with metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2006, 24, 3531. [Google Scholar] [CrossRef]
- De Bono, J.; Scher, H.I.; Montgomery, R.B.; Parker, C.; Miller, M.C.; Tissing, H.; Doyle, G.; Terstappen, L.W.; Pienta, K.J.; Raghavan, D. Circulating Tumor Cells Predict Survival Benefit from Treatment in Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2008, 14, 6302–6309. [Google Scholar] [CrossRef]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.M.M.; et al. Circulating Tumor Cells, Disease Progression, and Survival in Metastatic Breast Cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef]
- Negin, B.P.; Cohen, S.J. Circulating Tumor Cells in Colorectal Cancer: Past, Present, and Future Challenges. Curr. Treat. Options Oncol. 2010, 11, 1–13. [Google Scholar] [CrossRef]
- Folkersma, L.R.; Gómez, C.O.; Manso, L.S.J.; De Castro, S.V.; Romo, I.G.; Lázaro, M.V.; De La Orden, G.V.; Fernández, M.A.; Rubio, E.D.; Moyano, A.S.; et al. Immunomagnetic quantification of circulating tumoral cells in patients with prostate cancer: Clinical and pathological correlation. Arch. Espanoles de Urol. 2010, 63, 23–31. [Google Scholar]
- Arrazubi, V.; Mata, E.; Antelo, M.L.; Tarifa, A.; Herrera, J.; Zazpe, C.; Teijeira, L.; Viudez, A.; Suárez, J.; Hernández, I.; et al. Circulating Tumor Cells in Patients Undergoing Resection of Colorectal Cancer Liver Metastases. Clinical Utility for Long-Term Outcome: A Prospective Trial. Ann. Surg. Oncol. 2019, 26, 2805–2811. [Google Scholar] [CrossRef]
- Keomanee-Dizon, K.; Shishido, S.N.; Kuhn, P. Circulating Tumor Cells: High-Throughput Imaging of CTCs and Bioinformatic Analysis. In Methods in Molecular Biology; Springer Science and Business Media LLC: Cham, Switzerland, 2020; Volume 215, pp. 89–104. [Google Scholar]
- Gerdtsson, A.S.; Thiele, J.-A.; Shishido, S.N.; Zheng, S.; Schaffer, R.; Bethel, K.; Curley, S.; Lenz, H.-J.; Hanna, D.L.; Nieva, J.; et al. Single cell correlation analysis of liquid and solid biopsies in metastatic colorectal cancer. Oncotarget 2019, 10, 7016–7030. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rodriguez-Lee, M.; Kolatkar, A.; McCormick, M.; Dago, A.D.; Kendall, J.; Carlsson, N.A.; Bethel, K.; Greenspan, E.J.; Hwang, S.E.; Waitman, K.R.; et al. Effect of Blood Collection Tube Type and Time to Processing on the Enumeration and High-Content Characterization of Circulating Tumor Cells Using the High-Definition Single-Cell Assay. Arch. Pathol. Lab. Med. 2018, 142, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.-A.; Bethel, K.; Kralickova, M.; Kuhn, P. Circulating Tumor Cells: Fluid Surrogates of Solid Tumors. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 419–447. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; Jayaram, A.; Winquist, E.; McLaughlin, B.; Lu, D.; Fleisher, M.; Orr, S.; Lowes, L.; et al. Assessment of the Validity of Nuclear-Localized Androgen Receptor Splice Variant 7 in Circulating Tumor Cells as a Predictive Biomarker for Castration-Resistant Prostate Cancer. JAMA Oncol. 2018, 4, 1179–1186. [Google Scholar] [CrossRef]
- Vona, G.; Sabile, A.; Louha, M.; Sitruk, V.; Romana, S.P.; Schütze, K.; Capron, F.; Franco, M.; Pazzagli, M.; Vekemans, M.; et al. Isolation by Size of Epithelial Tumor Cells. Am. J. Pathol. 2000, 156, 57–63. [Google Scholar] [CrossRef]
- Chinen, L.; De Carvalho, F.M.; Rocha, B.M.M.; Aguiar, C.M.; Abdallah, E.A.; Campanha, D.; Mingues, N.B.; De Oliveira, T.B.; Maciel, M.S.; Cervantes, G.M.; et al. Cytokeratin-based CTC counting unrelated to clinical follow up. J. Thorac. Dis. 2013, 5, 593–599. [Google Scholar]
- Vona, G.; Beroud, C.; Benachi, A.; Quenette, A.; Bonnefont, J.; Romana, S.P.; Dumez, Y.; Lacour, B.; Paterlini-Bréchot, P. Enrichment, Immunomorphological, and Genetic Characterization of Fetal Cells Circulating in Maternal Blood. Am. J. Pathol. 2002, 160, 51–58. [Google Scholar] [CrossRef]
- e Silva, V.S.; Chinen, L.; Abdallah, E.A.; Damascena, A.; Paludo, J.; Chojniak, R.; Dettino, A.; De Mello, C.A.L.; Alves, V.S.; Fanelli, M.F. Early detection of poor outcome in patients with metastatic colorectal cancer: Tumor kinetics evaluated by circulating tumor cells. OncoTargets Ther. 2016, 9, 7503–7513. [Google Scholar] [CrossRef] [PubMed]
- Danila, D.C.; Samoila, A.; Patel, C.; Schreiber, N.; Herkal, A.; Anand, A.; Bastos, D.; Heller, G.; Fleisher, M.; Scher, H.I. Clinical Validity of Detecting Circulating Tumor Cells by AdnaTest Assay Compared With Direct Detection of Tumor mRNA in Stabilized Whole Blood, as a Biomarker Predicting Overall Survival for Metastatic Castration-Resistant Prostate Cancer Patients. Cancer J. 2016, 22, 315–320. [Google Scholar] [CrossRef]
- Todenhöfer, T.; Hennenlotter, J.; Feyerabend, S.; Aufderklamm, S.; Mischinger, J.; Kühs, U.; Gerber, V.; Fetisch, J.; Schilling, D.; Hauch, S.; et al. Preliminary experience on the use of the Adnatest® system for detection of circulating tumor cells in prostate cancer patients. Anticancer Res. 2012, 32, 3507–3513. [Google Scholar]
- Wu, S.; Liu, S.; Liu, Z.; Huang, J.; Pu, X.; Li, J.; Yang, D.; Deng, H.; Yang, N.; Xu, J. Classification of Circulating Tumor Cells by Epithelial-Mesenchymal Transition Markers. PLoS ONE 2015, 10, e0123976. [Google Scholar] [CrossRef]
- Zhao, R.; Cai, Z.; Li, S.; Cheng, Y.; Gao, H.; Liu, F.; Wu, S.; Liu, S.; Dong, Y.; Zheng, L.; et al. Expression and clinical relevance of epithelial and mesenchymal markers in circulating tumor cells from colorectal cancer. Oncotarget 2016, 8, 9293–9302. [Google Scholar] [CrossRef] [PubMed]
- Gasiorowski, L.; Dyszkiewicz, W.; Zielinski, P. In-vivo isolation of circulating tumor cells in non-small cell lung cancer patients by CellCollector. Neoplasma 2017, 64, 938–944. [Google Scholar] [CrossRef]
- He, Y.; Shi, J.; Shi, G.; Xu, X.; Liu, Q.; Liu, C.; Gao, Z.; Bai, J.; Shan, B. Using the New CellCollector to Capture Circulating Tumor Cells from Blood in Different Groups of Pulmonary Disease: A Cohort Study. Sci. Rep. 2017, 7, 9542. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.-S.; You, J.-F.; Hung, H.-Y.; Hsieh, P.-S.; Hsieh, B.; Lenz, H.-J.; Idos, G.; Friedland, S.; Pan, J.Y.-J.; Shao, H.-J.; et al. Novel Circulating Tumor Cell Assay for Detection of Colorectal Adenomas and Cancer. Clin. Transl. Gastroenterol. 2019, 10, e00088. [Google Scholar] [CrossRef]
- Gupta, P.; Gulzar, Z.; Hsieh, B.; Lim, A.; Watson, D.; Mei, R. Analytical validation of the CellMax platform for early detection of cancer by enumeration of rare circulating tumor cells. J. Circ. Biomark. 2019, 8, 1849454419899214. [Google Scholar] [CrossRef]
- Jaeger, B.A.S.; Jueckstock, J.; Andergassen, U.; Salmen, J.; Schochter, F.; Fink, V.; Alunni-Fabbroni, M.; Rezai, M.; Beck, T.; Beckmann, M.W.; et al. Evaluation of Two Different Analytical Methods for Circulating Tumor Cell Detection in Peripheral Blood of Patients with Primary Breast Cancer. BioMed Res. Int. 2014, 2014, 491459. [Google Scholar] [CrossRef]
- Wang, L.; Balasubramanian, P.; Chen, A.P.; Kummar, S.; Evrard, Y.A.; Kinders, R.J. Promise and limits of the CellSearch platform for evaluating pharmacodynamics in circulating tumor cells. Semin. Oncol. 2016, 43, 464–475. [Google Scholar] [CrossRef]
- Bin Lim, S.; Yeo, T.; Di Lee, W.; Bhagat, A.A.S.; Tan, S.J.; Tan, D.S.W.; Lim, W.-T.; Lim, C.T. Addressing cellular heterogeneity in tumor and circulation for refined prognostication. Proc. Natl. Acad. Sci. USA 2019, 116, 17957–17962. [Google Scholar] [CrossRef]
- Lee, Y.; Guan, G.; Bhagat, A.A. ClearCell® FX, a label-free microfluidics technology for enrichment of viable circulating tumor cells. Cytom. Part A 2018, 93, 1251–1254. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Zhang, Z.; Gao, X.H.; Shen, Z.; Jing, Y.; Lu, H.; Li, H.; Yang, X.; Cui, X.; Li, Y.; et al. Clinical significance of detecting circulating tumor cells in colorectal cancer using subtraction enrichment and immunostaining-fluorescence in situ hybridization (SE-iFISH). Oncotarget 2017, 8, 21639–21649. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Jia, S.; Li, H.; Yu, Y.; Liu, G.; Wu, Y.; Liu, X.; Liu, C.; Zhou, Y.; Zhang, Z.; et al. Characterization of circulating tumor cells in newly diagnosed breast cancer. Oncol. Lett. 2017, 15, 2522–2528. [Google Scholar] [CrossRef] [PubMed]
- D’Oronzo, S.; Lovero, D.; Palmirotta, R.; Stucci, L.S.; Tucci, M.; Felici, C.; Cascardi, E.; Giardina, C.; Cafforio, P.; Silvestris, F. Dissection of major cancer gene variants in subsets of circulating tumor cells in advanced breast cancer. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Hayashi, K.; Kawakami, K.; Miwa, Y.; Hayashi, H.; Yamamoto, M. KRAS mutation analysis of single circulating tumor cells from patients with metastatic colorectal cancer. BMC Cancer 2017, 17, 311. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Fusi, A.; Klopocki, E.; Schmittel, A.; Tinhofer, I.; Nonnemacher, A.; Keilholz, U. Negative enrichment by immunomagnetic nanobeads for unbiased characterization of circulating tumor cells from peripheral blood of cancer patients. J. Transl. Med. 2011, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, N.P.; Kumari, S.; Neyaz, A.; Gupta, S.; Agarwal, A.; Singhal, A.; Husain, N. EpCAM-based Flow Cytometric Detection of Circulating Tumor Cells in Gallbladder Carcinoma Cases. Asian Pac. J. Cancer Prev. 2017, 18, 3429–3437. [Google Scholar] [PubMed]
- Stott, S.L.; Hsu, C.-H.; Tsukrov, D.I.; Yu, M.; Miyamoto, D.T.; Waltman, B.A.; Rothenberg, S.M.; Shah, A.M.; Smas, M.E.; Korir, G.K.; et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. USA 2010, 107, 18392–18397. [Google Scholar] [CrossRef]
- Xue, P.; Ye, K.; Gao, J.; Wu, Y.; Guo, J.; Hui, K.M.; Kang, Y. Isolation and elution of Hep3B circulating tumor cells using a dual-functional herringbone chip. Microfluid. Nanofluid. 2013, 16, 605–612. [Google Scholar] [CrossRef]
- Castle, J.; Morris, K.; Pritchard, S.; Kirwan, C.C. Challenges in enumeration of CTCs in breast cancer using techniques independent of cytokeratin expression. PLoS ONE 2017, 12, e0175647. [Google Scholar] [CrossRef]
- Farace, F.; Massard, C.; Vimond, N.; Drusch, F.; Jacques, N.; Billiot, F.; Laplanche, A.; Chauchereau, A.; Lacroix, L.; Planchard, D.; et al. A direct comparison of CellSearch and ISET for circulating tumour-cell detection in patients with metastatic carcinomas. Br. J. Cancer 2011, 105, 847–853. [Google Scholar] [CrossRef]
- Cann, G.M.; Gulzar, Z.G.; Cooper, S.; Li, R.; Luo, S.; Tat, M.; Stuart, S.; Schroth, G.; Srinivas, S.; Ronaghi, M.; et al. mRNA-Seq of Single Prostate Cancer Circulating Tumor Cells Reveals Recapitulation of Gene Expression and Pathways Found in Prostate Cancer. PLoS ONE 2012, 7, e49144. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Krishnakumar, S.; Powell, A.A.; Zhang, H.; Mindrinos, M.; Telli, M.L.; Davis, R.W.; Jeffrey, S.S. Single cell mutational analysis of PIK3CA in circulating tumor cells and metastases in breast cancer reveals heterogeneity, discordance, and mutation persistence in cultured disseminated tumor cells from bone marrow. BMC Cancer 2014, 14, 456. [Google Scholar] [CrossRef] [PubMed]
- Bobek, V.; Matkowski, R.; Gürlich, R.; Grabowski, K.; Szelachowska, J.; Lischke, R.; Schutzner, J.; Harustiak, T.; Pazdro, A.; Rzechonek, A.; et al. Cultivation of circulating tumor cells in esophageal cancer. Folia Histochem. Cytobiol. 2014, 52, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Kolostova, K.; Matkowski, R.; Jędryka, M.; Soter, K.; Cegan, M.; Pinkas, M.; Jakabova, A.; Pavlasek, J.; Spicka, J.; Bobek, V. The added value of circulating tumor cells examination in ovarian cancer staging. Am. J. Cancer Res. 2015, 5, 3363–3375. [Google Scholar] [PubMed]
- Gertler, R.; Rosenberg, R.; Fuehrer, K.; Dahm, M.; Nekarda, H.; Siewert, J.R. Detection of circulating tumor cells in blood using an optimized density gradient centrifugation. Methods Mol. Biol. 2003, 162, 149–155. [Google Scholar] [CrossRef]
- Kaifi, J.T.; Kunkel, M.; Das, A.; Harouaka, R.; Dicker, D.T.; Li, G.; Zhu, J.; Clawson, G.A.; Yang, Z.; Reed, M.F.; et al. Circulating tumor cell isolation during resection of colorectal cancer lung and liver metastases: A prospective trial with different detection techniques. Cancer Biol. Ther. 2015, 16, 699–708. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Stoehlmacher, J.; Pantel, K.; Goekkurt, E. Detection and Monitoring of Cell-Free DNA in Blood of Patients with Colorectal Cancer. Ann. N. Y. Acad. Sci. 2008, 1137, 190–196. [Google Scholar] [CrossRef]
- Czeiger, D.; Shaked, G.; Eini, H.; Vered, I.; Belochitski, O.; Avriel, A.; Ariad, S.; Douvdevani, A. Measurement of Circulating Cell-Free DNA Levels by a New Simple Fluorescent Test in Patients With Primary Colorectal Cancer. Am. J. Clin. Pathol. 2011, 135, 264–270. [Google Scholar] [CrossRef]
- Beaver, J.A.; Jelovac, D.; Balukrishna, S.; Cochran, R.L.; Croessmann, S.; Zabransky, D.J.; Wong, H.Y.; Toro, P.V.; Cidado, J.; Blair, B.G.; et al. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin. Cancer Res. 2014, 20, 2643–2650. [Google Scholar] [CrossRef]
- Baslan, T.; Kendall, J.; Ward, B.; Cox, H.; Leotta, A.; Rodgers, L.; Riggs, M.; D’Italia, S.; Sun, G.; Yong, M.; et al. Optimizing sparse sequencing of single cells for highly multiplex copy number profiling. Genome Res. 2015, 25, 714–724. [Google Scholar] [CrossRef]
- Glenn, T.C. Field guide to next?generation DNA sequencers. Mol. Ecol. Resour. 2011, 11, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Molparia, B.; Oliveira, G.; Wagner, J.L.; Spencer, E.G.; Torkamani, A. A feasibility study of colorectal cancer diagnosis via circulating tumor DNA derived CNV detection. PLoS ONE 2018, 13, e0196826. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dittmar, R.; Xia, S.; Zhang, H.; Du, M.; Huang, C.; Druliner, B.R.; Boardman, L.; Wang, L. Cell-free DNA copy number variations in plasma from colorectal cancer patients. Mol. Oncol. 2017, 11, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Birkenkamp-Demtröder, K.; Nordentoft, I.K.; Christensen, E.; Høyer, S.; Reinert, T.; Vang, S.; Borre, M.; Agerbæk, M.; Jensen, J.B.; Ørntoft, T.F.; et al. Genomic Alterations in Liquid Biopsies from Patients with Bladder Cancer. Eur. Urol. 2016, 70, 75–82. [Google Scholar] [CrossRef]
- Zonta, E.; Garlan, F.; Pécuchet, N.; Perez-Toralla, K.; Caen, O.; Milbury, C.; Didelot, A.; Fabre, E.; Blons, H.; Laurent-Puig, P.; et al. Multiplex Detection of Rare Mutations by Picoliter Droplet Based Digital PCR: Sensitivity and Specificity Considerations. PLoS ONE 2016, 11, e0159094. [Google Scholar] [CrossRef] [PubMed]
- Dressman, D.; Yan, H.; Traverso, G.; Kinzler, K.W.; Vogelstein, B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Natl. Acad. Sci. USA 2003, 100, 8817–8822. [Google Scholar] [CrossRef]
- Schmiegel, W.H.; Scott, R.J.; Dooley, S.; Lewis, W.; Meldrum, C.J.; Pockney, P.G.; Draganic, B.; Smith, S.; Hewitt, C.; Philimore, H.; et al. Blood-based detection ofRASmutations to guide anti-EGFR therapy in colorectal cancer patients: Concordance of results from circulating tumor DNA and tissue-basedRAStesting. Mol. Oncol. 2017, 11, 208–219. [Google Scholar] [CrossRef]
- Taly, V.; Pekin, D.; Benhaim, L.; Kotsopoulos, S.K.; Le Corre, D.; Li, X.; Atochin, I.; Link, D.R.; Griffiths, A.D.; Pallier, K.; et al. Multiplex Picodroplet Digital PCR to Detect KRAS Mutations in Circulating DNA from the Plasma of Colorectal Cancer Patients. Clin. Chem. 2013, 59, 1722–1731. [Google Scholar] [CrossRef]
- Zhu, G.; Ye, X.; Dong, Z.; Lu, Y.C.; Sun, Y.; Liu, Y.; McCormack, R.; Gu, Y.; Liu, X. Highly Sensitive Droplet Digital PCR Method for Detection of EGFR-Activating Mutations in Plasma Cell–Free DNA from Patients with Advanced Non–Small Cell Lung Cancer. J. Mol. Diagn. 2015, 17, 265–272. [Google Scholar] [CrossRef]
- Hughesman, C.B.; Lu, X.J.D.; Liu, K.Y.P.; Zhu, Y.; Towle, R.M.; Haynes, C.; Poh, C.F. Detection of clinically relevant copy number alterations in oral cancer progression using multiplexed droplet digital PCR. Sci. Rep. 2017, 7, 11855. [Google Scholar] [CrossRef]
- Gale, D.; Lawson, A.R.J.; Howarth, K.; Madi, M.; Durham, B.; Smalley, S.; Calaway, J.; Blais, S.; Jones, G.; Clark, J.; et al. Development of a highly sensitive liquid biopsy platform to detect clinically-relevant cancer mutations at low allele fractions in cell-free DNA. PLoS ONE 2018, 13, e0194630. [Google Scholar] [CrossRef] [PubMed]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.Y.; Kaper, F.; Dawson, S.-J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive Identification and Monitoring of Cancer Mutations by Targeted Deep Sequencing of Plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.R.; Schmitt, M.W.; Fox, E.; Kohrn, B.F.; Salk, J.J.; Ahn, E.H.; Prindle, M.J.; Kuong, K.J.; Shen, J.-C.; Risques, R.-A.; et al. Detecting ultralow-frequency mutations by Duplex Sequencing. Nat. Protoc. 2014, 9, 2586–2606. [Google Scholar] [CrossRef]
- Iwahashi, N.; Sakai, K.; Noguchi, T.; Yahata, T.; Matsukawa, H.; Toujima, S.; Nishio, K.; Ino, K. Liquid biopsy-based comprehensive gene mutation profiling for gynecological cancer using CAncer Personalized Profiling by deep Sequencing. Sci. Rep. 2019, 9, 10426. [Google Scholar] [CrossRef] [PubMed]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Fredebohm, J.; Mehnert, D.H.; Löber, A.-K.; Holtrup, F.; Van Rahden, V.; Angenendt, P.; Diehl, F. Detection and Quantification of KIT Mutations in ctDNA by Plasma Safe-SeqS; Springer Science and Business Media LLC: Cham, Switzerland, 2016; Volume 924, pp. 187–189. [Google Scholar]
- Zheng, H.; Ladouceur, M.; Greenwood, C.M.T.; Richards, J.B. Effect of Genome-Wide Genotyping and Reference Panels on Rare Variants Imputation. J. Genet. Genome 2012, 39, 545–550. [Google Scholar] [CrossRef]
- Devos, T.; Tetzner, R.; Model, F.; Weiss, G.; Schuster, M.; Distler, J.; Steiger, K.V.; Grützmann, R.; Pilarsky, C.; Habermann, J.K.; et al. Circulating Methylated SEPT9 DNA in Plasma Is a Biomarker for Colorectal Cancer. Clin. Chem. 2009, 55, 1337–1346. [Google Scholar] [CrossRef]
- Warren, J.D.; Xiong, W.; Bunker, A.M.; Vaughn, C.P.; Furtado, L.V.; Owen, W.E.; Fang, J.; Samowitz, W.S.; Heichman, K.A. Septin 9 methylated DNA is a sensitive and specific blood test for colorectal cancer. BMC Med. 2011, 9, 133. [Google Scholar] [CrossRef]
- Solassol, J.; Vendrell, J.; Märkl, B.; Haas, C.; Bellosillo, B.; Montagut, C.; Smith, M.; O’Sullivan, B.; D’Haene, N.; Le Mercier, M.; et al. Multi-Center Evaluation of the Fully Automated PCR-Based Idylla™ KRAS Mutation Assay for Rapid KRAS Mutation Status Determination on Formalin-Fixed Paraffin-Embedded Tissue of Human Colorectal Cancer. PLoS ONE 2016, 11, e0163444. [Google Scholar] [CrossRef]
- Zwaenepoel, K.; Duelund, J.H.; De Winne, K.; Maes, V.; Weyn, C.; Lambin, S.; Dendooven, R.; Broeckx, G.; Steiniche, T.; Pauwels, P. Clinical Performance of the Idylla MSI Test for a Rapid Assessment of the DNA Microsatellite Status in Human Colorectal Cancer. J. Mol. Diagn. 2020, 22, 386–395. [Google Scholar] [CrossRef]
- García-Foncillas, J.; Tabernero, J.; Élez, E.; Aranda, E.; Benavides, M.; Camps, C.; Jantus-Lewintre, E.; López, R.; Muinelo-Romay, L.; Montagut, C.; et al. Prospective multicenter real-world RAS mutation comparison between OncoBEAM-based liquid biopsy and tissue analysis in metastatic colorectal cancer. Br. J. Cancer 2018, 119, 1464–1470. [Google Scholar] [CrossRef]
- Wan, N.; Weinberg, D.; Liu, T.-Y.; Niehaus, K.; Delubac, D.; Kannan, A.; White, B.; Ariazi, E.A.; Bailey, M.; Bertin, M.; et al. Su1658–Machine Learning Enables Detection of Early-Stage Colorectal Cancer by Whole-Genome Sequencing of Plasma Cell-Free Dna. Gastroenterology 2019, 156, 832. [Google Scholar] [CrossRef]
- Russo, M.; Siravegna, G.; Blaszkowsky, L.S.; Corti, G.; Crisafulli, G.; Ahronian, L.G.; Mussolin, B.; Kwak, E.L.; Buscarino, M.; Lazzari, L.; et al. Abstract 878: Tumor heterogeneity and lesion-specific response to targeted therapy in colorectal cancer. Exp. Mol. Ther. 2016, 76, 878. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2007, 14, 985–990. [Google Scholar] [CrossRef]
- Iwanicki-Caron, I.; Di Fiore, F.; Roque, I.; Astruc, E.; Stetiu, M.; Duclos, A.; Tougeron, D.; Saillard, S.; Thureau, S.; Benichou, J.; et al. Usefulness of the Serum Carcinoembryonic Antigen Kinetic for Chemotherapy Monitoring in Patients With Unresectable Metastasis of Colorectal Cancer. J. Clin. Oncol. 2008, 26, 3681–3686. [Google Scholar] [CrossRef]
- Li, M.; Li, J.-Y.; Zhao, A.-L.; He, J.-S.; Zhou, L.-X.; Li, Y.-A.; Gu, J. Comparison of carcinoembryonic antigen prognostic value in serum and tumour tissue of patients with colorectal cancer. Color. Dis. 2009, 11, 276–281. [Google Scholar] [CrossRef]
- Yang, K.M.; Park, I.J.; Kim, C.W.; Roh, S.A.; Cho, D.-H.; Kim, J.C. The prognostic significance and treatment modality for elevated pre- and postoperative serum CEA in colorectal cancer patients. Ann. Surg. Treat. Res. 2016, 91, 165–171. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, F.; Zhou, Q.; Yang, S.; Sun, X.; Wang, G.; Li, Z.; Zhang, Z.; Song, J.; Liu, J.; et al. Pre-operative to post-operative serum carcinoembryonic antigen ratio is a prognostic indicator in colorectal cancer. Oncotarget 2017, 8, 54672–54682. [Google Scholar] [CrossRef]
- Imperiale, T.F.; Ransohoff, D.F.; Itzkowitz, S.H.; Brenner, H.; Werner, S.; Chen, H.; Senore, C.; Segnan, N.; Lee, J.K.; Terdiman, J.P.; et al. Multitarget stool DNA testing for colorectal-cancer screening. N. Engl. J. Med. 2014, 371, 187–188. [Google Scholar] [CrossRef]
- Song, L.; Jia, J.; Peng, X.; Xiao, W.; Li, Y. The performance of the SEPT9 gene methylation assay and a comparison with other CRC screening tests: A meta-analysis. Sci. Rep. 2017, 7, 3032. [Google Scholar] [CrossRef]
- Church, T.R.; Wandell, M.; Lofton-Day, C.; Mongin, S.J.; Burger, M.; Payne, S.R.; Castanos-Velez, E.; Blumenstein, B.A.; Rösch, T.; Osborn, N.; et al. Prospective evaluation of methylated SEPT9 in plasma for detection of asymptomatic colorectal cancer. Gut 2013, 63, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Kim, J.S.; Lee, C.S.; Kim, J.-Y. KRAS discordance between primary and recurrent tumors after radical resection of colorectal cancers. J. Surg. Oncol. 2015, 111, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, F.; Carloni, S.; Zoli, W.; Ulivi, P.; Gallerani, G.; Fici, P.; Chiadini, E.; Passardi, A.; Frassineti, G.L.; Ragazzini, A.; et al. Detection and recovery of circulating colon cancer cells using a dielectrophoresis-based device: KRAS mutation status in pure CTCs. Cancer Lett. 2013, 335, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Crisafulli, G.; Sogari, A.; Reilly, N.M.; Arena, S.; Lamba, S.; Bartolini, A.; Amodio, V.; Magrì, A.; Novara, L.; et al. Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019, 366, 1473–1480. [Google Scholar] [CrossRef]
- Bardelli, A.; Siena, S. Molecular Mechanisms of Resistance to Cetuximab and Panitumumab in Colorectal Cancer. J. Clin. Oncol. 2010, 28, 1254–1261. [Google Scholar] [CrossRef]
- Luo, H.; Zhao, Q.; Wei, W.; Zheng, L.; Yi, S.; Li, G.; Wang, W.; Sheng, H.; Pu, H.; Mo, H.; et al. Circulating tumor DNA methylation profiles enable early diagnosis, prognosis prediction, and screening for colorectal cancer. Sci. Transl. Med. 2020, 12, eaax7533. [Google Scholar] [CrossRef]
- Aravanis, A.M.; Lee, M.; Klausner, R.D. Next-Generation Sequencing of Circulating Tumor DNA for Early Cancer Detection. Cell 2017, 168, 571–574. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.V.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef]
- Kolostova, K.; Matkowski, R.; Gürlich, R.; Grabowski, K.; Soter, K.; Lischke, R.; Schutzner, J.; Bobek, V. Detection and cultivation of circulating tumor cells in gastric cancer. Cytotechnology 2015, 68, 1095–1102. [Google Scholar] [CrossRef]
- Eliášová, P.; Pinkas, M.; Kolostova, K.; Gürlich, R.; Bobek, V. Circulating tumor cells in different stages of colorectal cancer. Folia Histochem. Cytobiol. 2017, 55, 1–5. [Google Scholar] [CrossRef]
- De Macedo, M.P.; De Melo, F.M.; Ribeiro, J.D.S.S.; De Mello, C.A.L.; Begnami, M.D.F.D.S.; Soares, F.A.; Carraro, D.M.; Cunha, I.W. RAS mutations vary between lesions in synchronous primary Colorectal Cancer: Testing only one lesion is not sufficient to guide anti-EGFR treatment decisions. Oncoscience 2015, 2, 125. [Google Scholar] [CrossRef][Green Version]
- Morelli, M.P.; Overman, M.J.; Dasari, A.; Kazmi, S.M.A.; Mazard, T.; Vilar, E.; Morris, V.K.; Lee, M.S.; Herron, D.; Eng, C.; et al. Characterizing the patterns of clonal selection in circulating tumor DNA from patients with colorectal cancer refractory to anti-EGFR treatment. Ann. Oncol. 2015, 26, 731–736. [Google Scholar] [CrossRef]
- Vidal, J.; Muinelo, L.; Dalmases, A.; Jones, F.; Edelstein, D.; Iglesias, M.; Orrillo, M.; Abalo, A.; Rodríguez, C.; Brozos, E.; et al. Plasma ctDNA RAS mutation analysis for the diagnosis and treatment monitoring of metastatic colorectal cancer patients. Ann. Oncol. 2017, 28, 1325–1332. [Google Scholar] [CrossRef]
- Thierry, A.; Pastor, B.; Jiang, Z.-Q.; Katsiampoura, A.D.; Parseghian, C.; Loree, J.; Overman, M.J.; Sanchez, C.; El Messaoudi, S.; Ychou, M.; et al. Circulating DNA Demonstrates Convergent Evolution and Common Resistance Mechanisms during Treatment of Colorectal Cancer. Clin. Cancer Res. 2017, 23, 4578–4591. [Google Scholar] [CrossRef]
- Khan, K.; Cunningham, D.; Werner, B.; Vlachogiannis, G.; Spiteri, I.; Heide, T.; Mateos, J.F.; Vatsiou, A.; Lampis, A.; Damavandi, M.D.; et al. Longitudinal Liquid Biopsy and Mathematical Modeling of Clonal Evolution Forecast Time to Treatment Failure in the PROSPECT-C Phase II Colorectal Cancer Clinical Trial. Cancer Discov. 2018, 8, 1270–1285. [Google Scholar] [CrossRef]
- Klein-Scory, S.; Maslova, M.; Pohl, M.; Eilert-Micus, C.; Schroers, R.; Schmiegel, W.; Baraniskin, A. Significance of Liquid Biopsy for Monitoring and Therapy Decision of Colorectal Cancer. Transl. Oncol. 2018, 11, 213–220. [Google Scholar] [CrossRef]
- Bin Kuo, Y.; Chen, J.-S.; Fan, C.-W.; Li, Y.-S.; Chan, E.-C. Comparison of KRAS mutation analysis of primary tumors and matched circulating cell-free DNA in plasmas of patients with colorectal cancer. Clin. Chim. Acta 2014, 433, 284–289. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of Circulating Tumor DNA in Early- and Late-Stage Human Malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef]
- Iwai, T.; Yamada, T.; Takahashi, G.; Takeda, K.; Koizumi, M.; Shinji, S.; Matsuda, A.; Yokoyama, Y.; Hara, K.; Ueda, K.; et al. Circulating cell-free long DNA fragments predict post-hepatectomy recurrence of colorectal liver metastases. Eur. J. Surg. Oncol. (EJSO) 2020, 46, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Rahbari, N.N.; Aigner, M.; Thorlund, K.; Mollberg, N.; Motschall, E.; Jensen, K.; Diener, M.K.; Büchler, M.W.; Koch, M.; Weitz, J. Meta-analysis Shows That Detection of Circulating Tumor Cells Indicates Poor Prognosis in Patients With Colorectal Cancer. Gastroenterology 2010, 138, 1714–1726.e13. [Google Scholar] [CrossRef] [PubMed]
- Spindler, K.-L.G.; Appelt, A.L.; Pallisgaard, N.; Andersen, R.F.; Brandslund, I.; Jakobsen, A. Cell-free DNA in healthy individuals, noncancerous disease and strong prognostic value in colorectal cancer. Int. J. Cancer 2014, 135, 2984–2991. [Google Scholar] [CrossRef]
- Tan, Y.; Wu, H. The significant prognostic value of circulating tumor cells in colorectal cancer: A systematic review and meta-analysis. Curr. Probl. Cancer 2018, 42, 95–106. [Google Scholar] [CrossRef]
- Spindler, K.-L.G.; Pallisgaard, N.; Andersen, R.F.; Brandslund, I.; Jakobsen, A. Circulating Free DNA as Biomarker and Source for Mutation Detection in Metastatic Colorectal Cancer. PLoS ONE 2015, 10, e0108247. [Google Scholar] [CrossRef]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.K.; Wu, H.-T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124. [Google Scholar] [CrossRef]
- Cohen, S.J.; Punt, C.J.; Iannotti, N.; Saidman, B.H.; Sabbath, K.D.; Gabrail, N.Y.; Picus, J.; Morse, M.; Mitchell, E.; Miller, M.C.; et al. Relationship of Circulating Tumor Cells to Tumor Response, Progression-Free Survival, and Overall Survival in Patients With Metastatic Colorectal Cancer. J. Clin. Oncol. 2008, 26, 3213–3221. [Google Scholar] [CrossRef]
- Connor, A.A.; McNamara, K.; Al-Sukhni, E.; Diskin, J.; Chan, D.; Ash, C.; Lowes, L.E.; Allan, A.; Zogopoulos, G.; Moulton, C.-A.; et al. Central, But Not Peripheral, Circulating Tumor Cells are Prognostic in Patients Undergoing Resection of Colorectal Cancer Liver Metastases. Ann. Surg. Oncol. 2015, 23, 2168–2175. [Google Scholar] [CrossRef]
- Dizdar, L.; Flügen, G.; Van Dalum, G.; Honisch, E.; Neves, R.P.; Niederacher, D.; Neubauer, H.; Fehm, T.; Rehders, A.; Krieg, A.; et al. Detection of circulating tumor cells in colorectal cancer patients using the GILUPI CellCollector: Results from a prospective, single-center study. Mol. Oncol. 2019, 13, 1548–1558. [Google Scholar] [CrossRef]
- Wong, D.; Moturi, S.; Angkachatchai, V.; Mueller, R.; DeSantis, G.; Boom, D.V.D.; Ehrich, M. Optimizing blood collection, transport and storage conditions for cell free DNA increases access to prenatal testing. Clin. Biochem. 2013, 46, 1099–1104. [Google Scholar] [CrossRef]
- Grölz, D.; Hauch, S.; Schlumpberger, M.; Guenther, K.; Voss, T.; Sprenger-Haussels, M.; Oelmüller, U. Liquid Biopsy Preservation Solutions for Standardized Pre-Analytical Workflows—Venous Whole Blood and Plasma. Curr. Pathobiol. Rep. 2018, 6, 275–286. [Google Scholar] [CrossRef]
- Neumann, M.H.; Bender, S.; Krahn, T.; Schlange, T. ctDNA and CTCs in Liquid Biopsy – Current Status and Where We Need to Progress. Comput. Struct. Biotechnol. J. 2018, 16, 190–195. [Google Scholar] [CrossRef]
- Witzig, T.E.; Bossy, B.; Kimlinger, T.; Roche, P.C.; Ingle, J.N.; Grant, C.; Donohue, J.; Suman, V.J.; Harrington, D.; Torre-Bueno, J.; et al. Detection of circulating cytokeratin-positive cells in the blood of breast cancer patients using immunomagnetic enrichment and digital microscopy. Clin. Cancer Res. 2002, 8, 1085–1091. [Google Scholar]
- Hardingham, J.; Grover, P.; Winter, M.; Hewett, P.J.; Price, T.J.; Thierry, B. Detection and Clinical Significance of Circulating Tumor Cells in Colorectal Cancer—20 Years of Progress. Mol. Med. 2015, 21, S25–S31. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a Master Regulator of Morphogenesis, Plays an Essential Role in Tumor Metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef]
- Torga, G.; Pienta, K.J. Patient-Paired Sample Congruence between 2 Commercial Liquid Biopsy Tests. JAMA Oncol. 2018, 4, 868–870. [Google Scholar] [CrossRef]
- Vivancos, A.; Aranda, E.; Benavides, M.; Élez, E.; Gómez-España, M.A.; Toledano, M.; Alvarez, M.; Parrado, M.R.C.; García-Barberán, V.; Diaz-Rubio, E. Comparison of the Clinical Sensitivity of the Idylla Platform and the OncoBEAM RAS CRC Assay for KRAS Mutation Detection in Liquid Biopsy Samples. Sci. Rep. 2019, 9, 8976. [Google Scholar] [CrossRef]
- Scher, H.I.; Morris, M.J.; Larson, S.; Heller, G. Validation and clinical utility of prostate cancer biomarkers. Nat. Rev. Clin. Oncol. 2013, 10, 225–234. [Google Scholar] [CrossRef][Green Version]
- Pantel, K.; Hille, C.; Scher, H.I. Circulating Tumor Cells in Prostate Cancer: From Discovery to Clinical Utility. Clin. Chem. 2019, 65, 87–99. [Google Scholar] [CrossRef]
- Parkinson, D.R.; McCormack, R.T.; Keating, S.M.; Gutman, S.I.; Hamilton, S.R.; Mansfield, E.A.; Piper, M.A.; Deverka, P.; Frueh, F.W.; Jessup, J.M.; et al. Evidence of Clinical Utility: An Unmet Need in Molecular Diagnostics for Patients with Cancer. Clin. Cancer Res. 2014, 20, 1428–1444. [Google Scholar] [CrossRef]
- Toledo, R.A.; Cubillo, A.; Vega, E.; Garralda, E.; Alvarez, R.; De La Varga, L.U.; Rodriguez-Pascual, J.; Sanchez, G.; Sarno, F.; Prieto, S.H.; et al. Clinical validation of prospective liquid biopsy monitoring in patients with wild-type RAS metastatic colorectal cancer treated with FOLFIRI-cetuximab. Oncotarget 2016, 8, 35289–35300. [Google Scholar] [CrossRef]
- Palmirotta, R.; Lovero, D.; Silvestris, E.; Felici, C.; Quaresmini, D.; Cafforio, P.; Silvestris, F. Next-generation Sequencing (NGS) Analysis on Single Circulating Tumor Cells (CTCs) with No Need of Whole-genome Amplification (WGA). Cancer Genom.-Proteom. 2017, 14, 173–179. [Google Scholar] [CrossRef]
- Ulz, P.; Heitzer, E.; Geigl, J.B.; Speicher, M.R. Patient monitoring through liquid biopsies using circulating tumor DNA. Int. J. Cancer 2017, 141, 887–896. [Google Scholar] [CrossRef]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to Anti-EGFR Therapy in Colorectal Cancer: From Heterogeneity to Convergent Evolution. Cancer Discov. 2014, 4, 1269–1280. [Google Scholar] [CrossRef]


{kind=link}
| Platform | Company/Institution Details | Description | References |
|---|---|---|---|
| AdnaTest | Qiagen GmbH, Hilden, Germany | Antibody targeting EpCAM conjugated to magnetic beads for labeling tumor cells in sample | [100,101] |
| CanPatrol™ CTC | SurExam, Guangzhou, China | Filtration (and CD45+ depletion) 1 | [102,103] |
| CellCollector® | Gilupi GmbH, Postdam, Germany | Nano guidewire inserted into patient cubital vein collecting cell expressing EpCAM | [104,105] |
| CellMax CMx | CellMax Life Inc., Sunnyvale, CA, USA | Blood passing through antibody-coated microfluidic chip targeting EpCAM | [106,107] |
| CellSearch® * | Menarini Silicon Biosystems Spa, Castel Maggiore, Italy | Antibody targeting EpCAM conjugated to magnetic beads for labeling tumor cells in sample | [108,109] |
| ClearCell® FX | Genomax Technologies, Bangkok, Thailand | Blood passing through microfluidic biochip with larger cells along the inner wall | [110,111] |
| Cytelligen® | Cytelligen Inc., San Diego, CA, USA | Antibody targeting CD45 conjugated to magnetic beads for labeling tumor cells in the blood sample | [112,113] |
| DEPArray™ | Menarini Silicon Biosystems Spa, Castel Maggiore, Italy | Cell suspension loaded into microchip-based sorter using dielectrophoresis to trap cells | [114,115] |
| Easysep™ | Stemcell Technologies Inc., Vancouver, BC, Canada | Antibody targeting EpCAM or CD45 conjugated to magnetic beads for labeling tumor cells in the blood sample | [116,117] |
| Epic Sciences/HDSCA * | Epic Sciences Inc., San Diego, CA, USA | After processing, cells are plated on the slide and subsequently characterized based on surface markers | [57,92] |
| Herringbone Chip | Massachusetts General Hospital, Boston, MA, USA | Blood processed through antibody-coated microfluidic chip targeting EpCAM | [118,119] |
| ISET® * | Rarecells Diagnostics SAS, Paris, France | Filtration on pressure-controlled system | [120,121] |
| MagSweeper™ | Stanford University, Stanford, CA, USA | Antibody targeting EpCAM or CD133 conjugated to magnetic beads for labeling tumor cells in blood | [122,123] |
| MetaCell® | MetaCell s.r.o., Ostrava, Czech Republic | Capillary-action driven size-based separation | [124,125] |
| Oncoquick® | Greiner Bio-One International GmbH, Kremsmünster, Austria | The denser blood compartment migrates through the porous barrier of the polypropylene centrifugation tube | [126,127] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kolenčík, D.; Shishido, S.N.; Pitule, P.; Mason, J.; Hicks, J.; Kuhn, P. Liquid Biopsy in Colorectal Carcinoma: Clinical Applications and Challenges. Cancers 2020, 12, 1376. https://doi.org/10.3390/cancers12061376
Kolenčík D, Shishido SN, Pitule P, Mason J, Hicks J, Kuhn P. Liquid Biopsy in Colorectal Carcinoma: Clinical Applications and Challenges. Cancers. 2020; 12(6):1376. https://doi.org/10.3390/cancers12061376
Chicago/Turabian StyleKolenčík, Drahomír, Stephanie N. Shishido, Pavel Pitule, Jeremy Mason, James Hicks, and Peter Kuhn. 2020. "Liquid Biopsy in Colorectal Carcinoma: Clinical Applications and Challenges" Cancers 12, no. 6: 1376. https://doi.org/10.3390/cancers12061376
APA StyleKolenčík, D., Shishido, S. N., Pitule, P., Mason, J., Hicks, J., & Kuhn, P. (2020). Liquid Biopsy in Colorectal Carcinoma: Clinical Applications and Challenges. Cancers, 12(6), 1376. https://doi.org/10.3390/cancers12061376