Regulation of Src Family Kinases during Colorectal Cancer Development and Its Clinical Implications


{kind=link}
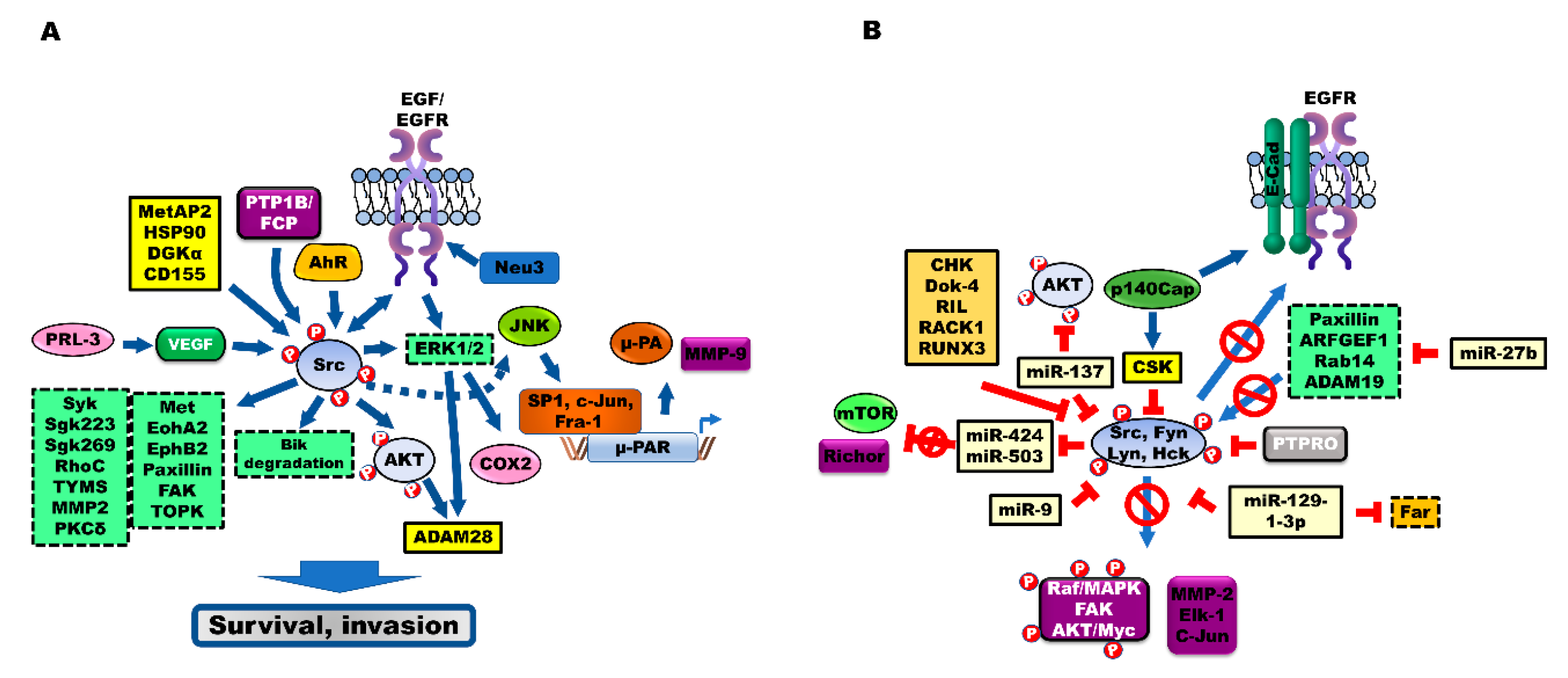{kind=link}
{kind=link}
Abstract
1. Introduction
2. Receptor-Mediated Signaling Pathways Activate Src during the Progression of CRC
2.1. Epidermal Growth Factor Receptor (EGFR)
2.2. Vascular Endothelial Growth Factor Receptor (VEGFR) and Fibroblast Growth Factor Receptor (FGFR)
2.3. Interleukin (IL)-4/IL-13/IL-13Rα2
2.4. IL-6 Signal Transducer (IL6ST), IL-6R and IL-11R
2.5. G-Proteins/GPCR
3. Modulators That Activate Src during the Progression of CRC
3.1. Aryl Hydrocarbon Receptor (AhR) Signaling
3.2. Receptor-Type Protein Tyrosine Phosphatase (PTPR) Positively Regulates Src
3.3. Other Modulators of Src
4. Src Activates Src Substrates during the Progression of CRC
5. Tumor Suppressors Inhibit Src Activation during the Progression of CRC
5.1. C-Terminal Src Kinase (CSK)
5.2. PTPRs
5.3. miRNAs Negatively Regulate Src
5.4. Other Src Repressors
6. Modulation of EMT by Src during CRC Progression
6.1. Correlation between Src and EMT in Colon Cancer
6.2. Activators of Src-Mediated EMT
6.2.1. Caveolin (CAV-1)
6.2.2. SPROUTY2 (SPRY2)
6.2.3. Twist-1/PDGFR
6.2.4. Integrin α5β1
6.2.5. Protein Tyrosine Phosphatase Receptor Type A (RPTPα)
6.2.6. CLDN1
6.2.7. Inositol-Requiring Enzyme 1α (IRE1α)
6.3. Suppressors of Src-Mediated EMT
7. Src Induces and Maintains Cancer Stem Cells (CSCs) in CRC
8. Therapeutic Implications of Src Inhibitors in CRC Progression
8.1. Evaluation of Preclinical and Clinical Src Inhibitors
8.2. HDAC Inhibitors
8.3. MetAP2 Inhibitors
8.4. Andrographolide
8.5. Trametinib
8.6. PKC Inhibitors
8.7. Curcumin
8.8. Folic Acid or Folinic Acid
8.9. Capecitabine
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- White, M.F.; Werth, D.K.; Pastan, I.; Kahn, C.R. Phosphorylation of the solubilized insulin receptor by the gene product of the Rous sarcoma virus, pp60src. J. Cell Biochem. 1984, 26, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Yeatman, T.J. A renaissance for SRC. Nat. Rev. Cancer 2004, 4, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.C.; Song, L.X.; Haura, E.B. Src kinases as therapeutic targets for cancer. Nat. Rev. Clin. Oncol. 2009, 6, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, M.B. Phosphotyrosine-binding domains in signal transduction. Nat. Rev. Mol. Cell Biol. 2002, 3, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Talamonti, M.S.; Roh, M.S.; Curley, S.A.; Gallick, G.E. Increase in activity and level of pp60c-src in progressive stages of human colorectal cancer. J. Clin. Investig. 1993, 91, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Termuhlen, P.M.; Curley, S.A.; Talamonti, M.S.; Saboorian, M.H.; Gallick, G.E. Site-specific differences in pp60c-src activity in human colorectal metastases. J. Surg. Res. 1993, 54, 293–298. [Google Scholar] [CrossRef]
- Iravani, S.; Mao, W.G.; Fu, L.; Karl, R.; Yeatman, T.; Jove, R.; Coppola, D. Elevated c-Src protein expression is an early event in colonic neoplasia. Lab. Investig. 1998, 78, 365–371. [Google Scholar]
- Martinez-Perez, J.; Lopez-Calderero, I.; Saez, C.; Benavent, M.; Limon, M.L.; Gonzalez-Exposito, R.; Soldevilla, B.; Riesco-Martinez, M.C.; Salamanca, J.; Carnero, A.; et al. Prognostic relevance of Src activation in stage II-III colon cancer. Hum. Pathol. 2017, 67, 119–125. [Google Scholar] [CrossRef]
- Irby, R.; Mao, W.G.; Coppola, D.; Jove, R.; Gamero, A.; Cuthbertson, D.; Fujita, D.J.; Yeatman, T.J. Overexpression of normal c-Src in poorly metastatic human colon cancer cells enhances primary tumor growth but not metastatic potential. Cell Growth Differ. 1997, 8, 1287–1295. [Google Scholar]
- Han, N.M.; Curley, S.A.; Gallick, G.E. Differential activation of pp60(c-src) and pp62(c-yes) in human colorectal carcinoma liver metastases. Clin. Cancer Res. 1996, 2, 1397–1404. [Google Scholar]
- Krabbe, G.; Matyash, V.; Pannasch, U.; Mamer, L.; Boddeke, H.W.G.M.; Kettenmann, H. Activation of serotonin receptors promotes microglial injury-induced motility but attenuates phagocytic activity. Brain Behav. Immun. 2012, 26, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Kumar Singh, P.; Kashyap, A.; Silakari, O. Exploration of the therapeutic aspects of Lck: A kinase target in inflammatory mediated pathological conditions. Biomed. Pharmacother. 2018, 108, 1565–1571. [Google Scholar] [CrossRef] [PubMed]
- McCracken, S.; Kim, C.S.; Xu, Y.; Minden, M.; Miyamoto, N.G. An alternative pathway for expression of p56lck from type I promoter transcripts in colon carcinoma. Oncogene 1997, 15, 2929–2937. [Google Scholar] [CrossRef] [PubMed]
- Harashima, N.; Tanaka, K.; Sasatomi, T.; Shimizu, K.; Miyagi, Y.; Yamada, A.; Tamura, M.; Yamana, H.; Itoh, K.; Shichijo, S. Recognition of the Lck tyrosine kinase as a tumor antigen by cytotoxic T lymphocytes of cancer patients with distant metastases. Eur J. Immunol. 2001, 31, 323–332. [Google Scholar] [CrossRef]
- Mao, W.; Irby, R.; Coppola, D.; Fu, L.; Wloch, M.; Turner, J.; Yu, H.; Garcia, R.; Jove, R.; Yeatman, T.J. Activation of c-Src by receptor tyrosine kinases in human colon cancer cells with high metastatic potential. Oncogene 1997, 15, 3083–3090. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Liu, S.; Zhang, J.; Liu, J.; Xu, L.; Liu, Y.; Qu, X. Cetuximab-induced MET activation acts as a novel resistance mechanism in colon cancer cells. Int. J. Mol. Sci. 2014, 15, 5838–5851. [Google Scholar] [CrossRef]
- Empereur, S.; Djelloul, S.; DiGioia, Y.; Bruyneel, E.; Mareel, M.; VanHengel, J.; VanRoy, F.; Comoglio, P.; Courtneidge, S.; Paraskeva, C.; et al. Progression of familial adenomatous polyposis (FAP) colonic cells after transfer of the src or polyoma middle T oncogenes: Cooperation between src and HGF/Met in invasion. Br. J. Cancer 1997, 75, 241–250. [Google Scholar] [CrossRef][Green Version]
- Cursi, S.; Rufini, A.; Stagni, V.; Condo, I.; Matafora, V.; Bachi, A.; Bonifazi, A.P.; Coppola, L.; Superti-Furga, G.; Testi, R.; et al. Src kinase phosphorylates Caspase-8 on Tyr380: A novel mechanism of apoptosis suppression. EMBO J. 2006, 25, 1895–1905. [Google Scholar] [CrossRef]
- Frey, M.R.; Hilliard, V.C.; Mullane, M.T.; Polk, D.B. ErbB4 promotes cyclooxygenase-2 expression and cell survival in colon epithelial cells. Lab. Investig. 2010, 90, 1415–1424. [Google Scholar] [CrossRef]
- Williams, C.S.; Bernard, J.K.; Demory Beckler, M.; Almohazey, D.; Washington, M.K.; Smith, J.J.; Frey, M.R. ERBB4 is over-expressed in human colon cancer and enhances cellular transformation. Carcinogenesis 2015, 36, 710–718. [Google Scholar] [CrossRef]
- Lien, G.S.; Wu, M.S.; Bien, M.Y.; Chen, C.H.; Lin, C.H.; Chen, B.C. Epidermal growth factor stimulates nuclear factor-kappaB activation and heme oxygenase-1 expression via c-Src, NADPH oxidase, PI3K, and Akt in human colon cancer cells. PLoS ONE 2014, 9, e104891. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kelber, J.A.; Tran Cao, H.S.; Cantin, G.T.; Lin, R.; Wang, W.; Kaushal, S.; Bristow, J.M.; Edgington, T.S.; Hoffman, R.M.; et al. Pseudopodium-enriched atypical kinase 1 regulates the cytoskeleton and cancer progression [corrected]. Proc. Natl. Acad. Sci. USA 2010, 107, 10920–10925. [Google Scholar] [CrossRef] [PubMed]
- Schoenherr, C.; Serrels, B.; Proby, C.; Cunningham, D.L.; Findlay, J.E.; Baillie, G.S.; Heath, J.K.; Frame, M.C. Eps8 controls Src- and FAK-dependent phenotypes in squamous carcinoma cells. J. Cell Sci. 2014, 127, 5303–5316. [Google Scholar] [CrossRef] [PubMed]
- Maa, M.C.; Lee, J.C.; Chen, Y.J.; Chen, Y.J.; Lee, Y.C.; Wang, S.T.; Huang, C.C.; Chow, N.H.; Leu, T.H. Eps8 facilitates cellular growth and motility of colon cancer cells by increasing the expression and activity of focal adhesion kinase. J. Biol. Chem. 2007, 282, 19399–19409. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M.; Staley, C.A.; Liu, W.B.; Fleming, R.Y.D.; Parikh, N.U.; Bucana, C.D.; Gallick, G.E. Down-regulation of vascular endothelial growth factor in a human colon carcinoma cell line transfected with an antisense expression vector specific for c-src. J. Biol. Chem. 1998, 273, 1052–1057. [Google Scholar] [CrossRef]
- Lesslie, D.P.; Summy, J.M.; Parikh, N.U.; Fan, F.; Trevino, J.G.; Sawyer, T.K.; Metcalf, C.A.; Shakespeare, W.C.; Hicklin, D.J.; Ellis, L.M.; et al. Vascular endothelial growth factor receptor-1 mediates migration of human colorectal carcinoma cells by activation of Src family kinases. Br. J. Cancer 2006, 94, 1710–1717. [Google Scholar] [CrossRef]
- Zeng, M.; Kikuchi, H.; Pino, M.S.; Chung, D.C. Hypoxia activates the K-ras proto-oncogene to stimulate angiogenesis and inhibit apoptosis in colon cancer cells. PLoS ONE 2010, 5, e10966. [Google Scholar] [CrossRef]
- Bange, J.; Prechtl, D.; Cheburkin, Y.; Specht, K.; Harbeck, N.; Schmitt, M.; Knyazeva, T.; Muller, S.; Gartner, S.; Sures, I.; et al. Cancer progression and tumor cell motility are associated with the FGFR4 Arg(388) allele. Cancer Res. 2002, 62, 840–847. [Google Scholar]
- Pelaez-Garcia, A.; Barderas, R.; Torres, S.; Hernandez-Varas, P.; Teixido, J.; Bonilla, F.; de Herreros, A.G.; Casal, J.I. FGFR4 Role in Epithelial–mesenchymal transition and Its Therapeutic Value in Colorectal Cancer. PLoS ONE 2013, 8, e63695. [Google Scholar] [CrossRef]
- Barderas, R.; Bartolome, R.A.; Fernandez-Acenero, M.J.; Torres, S.; Casal, J.I. High expression of IL-13 receptor alpha2 in colorectal cancer is associated with invasion, liver metastasis, and poor prognosis. Cancer Res. 2012, 72, 2780–2790. [Google Scholar] [CrossRef]
- Bartolome, R.A.; Garcia-Palmero, I.; Torres, S.; Lopez-Lucendo, M.; Balyasnikova, I.V.; Casal, J.I. IL13 Receptor alpha2 Signaling Requires a Scaffold Protein, FAM120A, to Activate the FAK and PI3K Pathways in Colon Cancer Metastasis. Cancer Res. 2015, 75, 2434–2444. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. Drugging Wnt signalling in cancer. Embo J. 2012, 31, 2737–2746. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Moroishi, T.; de Jong, P.R.; Krawczyk, M.; Grebbin, B.M.; Luo, H.Y.; Xu, R.H.; Golob-Schwarzl, N.; Schweiger, C.; Wang, K.P.; et al. YAP-IL-6ST autoregulatory loop activated on APC loss controls colonic tumorigenesis. Proc. Natl. Acad. Sci. USA 2017, 114, 1643–1648. [Google Scholar] [CrossRef] [PubMed]
- Duffy, A.G.; Greten, T.F. Liver cancer: Regorafenib as second-line therapy in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 141–142. [Google Scholar] [CrossRef]
- Fogelman, D.; Cubillo, A.; Garcia-Alfonso, P.; Miron, M.L.L.; Nemunaitis, J.; Flora, D.; Borg, C.; Mineur, L.; Vieitez, J.M.; Cohn, A.; et al. Randomized, double-blind, phase two study of ruxolitinib plus regorafenib in patients with relapsed/refractory metastatic colorectal cancer. Cancer Med. 2018, 7, 5382–5393. [Google Scholar] [CrossRef]
- Pai, R.; Soreghan, B.; Szabo, I.L.; Pavelka, M.; Baatar, D.; Tarnawski, A.S. Prostaglandin E2 transactivates EGF receptor: A novel mechanism for promoting colon cancer growth and gastrointestinal hypertrophy. Nat. Med. 2002, 8, 289–293. [Google Scholar] [CrossRef]
- Fukuda, R.; Kelly, B.; Semenza, G.L. Vascular endothelial growth factor gene expression in colon cancer cells exposed to prostaglandin E2 is mediated by hypoxia-inducible factor 1. Cancer Res. 2003, 63, 2330–2334. [Google Scholar]
- Xu, J.W.; Ikeda, K.; Yamori, Y. Upregulation of endothelial nitric oxide synthase by cyanidin-3-glucoside, a typical anthocyanin pigment. Hypertension 2004, 44, 217–222. [Google Scholar] [CrossRef]
- Hu, H.; Han, T.; Zhuo, M.; Wu, L.L.; Yuan, C.; Wu, L.; Lei, W.; Jiao, F.; Wang, L.W. Elevated COX-2 Expression Promotes Angiogenesis Through EGFR/p38-MAPK/Sp1-Dependent Signalling in Pancreatic Cancer. Sci Rep. 2017, 7, 470. [Google Scholar] [CrossRef]
- Buchanan, F.G.; Wang, D.; Bargiacchi, F.; DuBois, R.N. Prostaglandin E2 regulates cell migration via the intracellular activation of the epidermal growth factor receptor. J. Biol. Chem. 2003, 278, 35451–35457. [Google Scholar] [CrossRef]
- Wojtukiewicz, M.Z.; Hempel, D.; Sierko, E.; Tucker, S.C.; Honn, K.V. Protease-activated receptors (PARs)--biology and role in cancer invasion and metastasis. Cancer Metastasis Rev. 2015, 34, 775–796. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Regnauld, K.; Bruyneel, E.; Nguyen, Q.D.; Mareel, M.; Emami, S.; Gespach, C. Suppression of cellular invasion by activated G-protein subunits Galphao, Galphai1, Galphai2, and Galphai3 and sequestration of Gbetagamma. Mol. Pharmacol. 2001, 60, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.D.; Faivre, S.; Bruyneel, E.; Rivat, C.; Seto, M.; Endo, T.; Mareel, M.; Emami, S.; Gespach, C. RhoA- and RhoD-dependent regulatory switch of Galpha subunit signaling by PAR-1 receptors in cellular invasion. FASEB J. 2002, 16, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Darmoul, D.; Gratio, V.; Devaud, H.; Laburthe, M. Protease-activated receptor 2 in colon cancer: Trypsin-induced MAPK phosphorylation and cell proliferation are mediated by epidermal growth factor receptor transactivation. J. Biol. Chem. 2004, 279, 20927–20934. [Google Scholar] [CrossRef]
- Darmoul, D.; Gratio, V.; Devaud, H.; Peiretti, F.; Laburthe, M. Activation of proteinase-activated receptor 1 promotes human colon cancer cell proliferation through epidermal growth factor receptor transactivation. Mol. Cancer Res. 2004, 2, 514–522. [Google Scholar]
- Gratio, V.; Walker, F.; Lehy, T.; Laburthe, M.; Darmoul, D. Aberrant expression of proteinase-activated receptor 4 promotes colon cancer cell proliferation through a persistent signaling that involves Src and ErbB-2 kinase. Int. J. Cancer 2009, 124, 1517–1525. [Google Scholar] [CrossRef]
- Hellmich, M.R.; Rui, X.L.; Hellmich, H.L.; Fleming, R.Y.D.; Evers, B.M.; Townsend, C.M. Human colorectal cancers express a constitutively active cholecystokinin-B/gastrin receptor that stimulates cell growth. J. Biol. Chem. 2000, 275, 32122–32128. [Google Scholar] [CrossRef]
- Olszewska-Pazdrak, B.; Townsend, C.M.; Hellmich, M.R. Agonist-independent activation of Src tyrosine kinase by a cholecystokinin-2 (CCK2) receptor splice variant. J. Biol. Chem. 2004, 279, 40400–40404. [Google Scholar] [CrossRef]
- Guo, Y.S.; Wen, X.D.; Rui, X.L.; Townsend, C.M. Both glycine-extended gastrin (G-gly) and gastrin (G17) stimulated COX-2 expression via CCK-B receptor in intestinal epithelial cells. Gastroenterology 2001, 120, e507. [Google Scholar] [CrossRef]
- Ferrand, A.; Bertrand, C.; Portolan, G.; Cui, G.L.; Carlson, J.; Pradayrol, L.; Fourmy, D.; Dufresne, M.; Wang, T.C.; Seva, C. Signaling pathways associated with colonic mucosa hyperproliferation in mice overexpressing gastrin precursors. Cancer Res. 2005, 65, 2770–2777. [Google Scholar] [CrossRef]
- Ferrand, A.; Kowalski-Chauvel, A.; Pannequin, J.; Bertrand, C.; Fourmy, D.; Dufresne, M.; Seva, C. Glycine-extended gastrin activates two independent tyrosine-kinases in upstream of p85/p110 phosphatidylinositol 3-kinase in human colonic tumour cells. World J. Gastroenterol. 2006, 12, 1859–1864. [Google Scholar] [CrossRef] [PubMed]
- Servitja, J.M.; Marinissen, M.J.; Sodhi, A.; Bustelo, X.R.; Gutkind, J.S. Rac1 function is required for Src-induced transformation. Evidence of a role for Tiam1 and Vav2 in Rac activation by Src. J. Biol. Chem. 2003, 278, 34339–34346. [Google Scholar] [CrossRef]
- Gianni, D.; Bohl, B.; Courtneidge, S.A.; Bokoch, G.M. The involvement of the tyrosine kinase c-Src in the regulation of reactive oxygen species generation mediated by NADPH oxidase-1. Mol. Biol. Cell 2008, 19, 2984–2994. [Google Scholar] [CrossRef] [PubMed]
- Kajla, S.; Mondol, A.S.; Nagasawa, A.; Zhang, Y.; Kato, M.; Matsuno, K.; Yabe-Nishimura, C.; Kamata, T. A crucial role for Nox 1 in redox-dependent regulation of Wnt-beta-catenin signaling. Faseb J. 2012, 26, 2049–2059. [Google Scholar] [CrossRef] [PubMed]
- Sadok, A.; Bourgarel-Rey, V.; Gattacceca, F.; Penel, C.; Lehmann, M.; Kovacic, H. Nox1-dependent superoxide production controls colon adenocarcinoma cell migration. Biochim. Biophys. Acta 2008, 1783, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Peng, Z.; Raufman, J.P. Src-mediated aryl hydrocarbon and epidermal growth factor receptor cross talk stimulates colon cancer cell proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, 1006–1015. [Google Scholar] [CrossRef]
- Lopez, J.; Hesling, C.; Prudent, J.; Popgeorgiev, N.; Gadet, R.; Mikaelian, I.; Rimokh, R.; Gillet, G.; Gonzalo, P. Src tyrosine kinase inhibits apoptosis through the Erk1/2-dependent degradation of the death accelerator Bik. Cell Death Differ. 2012, 19, 1459–1469. [Google Scholar] [CrossRef]
- Zhu, S.; Bjorge, J.D.; Fujita, D.J. PTP1B contributes to the oncogenic properties of colon cancer cells through src activation. Cancer Res. 2007, 67, 10129–10137. [Google Scholar] [CrossRef]
- Mei, W.H.; Wang, K.M.; Huang, J.; Zheng, X.M. Cell Transformation by PTP1B Truncated Mutants Found in Human Colon and Thyroid Tumors. PLoS ONE 2016, 11, e0166538. [Google Scholar] [CrossRef]
- Huang, J.; Yao, L.; Xu, R.; Wu, H.; Wang, M.; White, B.S.; Shalloway, D.; Zheng, X. Activation of Src and transformation by an RPTPalpha splice mutant found in human tumours. EMBO J. 2011, 30, 3200–3211. [Google Scholar] [CrossRef]
- Saha, S.; Bardelli, A.; Buckhaults, P.; Velculescu, V.E.; Rago, C.; St Croix, B.; Romans, K.E.; Choti, M.A.; Lengauer, C.; Kinzler, K.W.; et al. A phosphatase associated with metastasis of colorectal cancer. Science 2001, 294, 1343–1346. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Li, J.; Wang, H.; Osato, M.; Tang, J.P.; Quah, S.Y.; Gan, B.Q.; Zeng, Q. PRL-3 initiates tumor angiogenesis by recruiting endothelial cells in vitro and in vivo. Cancer Res. 2006, 66, 9625–9635. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.W.; McQueeney, K.E.; Isenberg, J.S.; Pitt, B.R.; Wasserloos, K.A.; Homanics, G.E.; Lazo, J.S. Protein-tyrosine phosphatase 4A3 (PTP4A3) promotes vascular endothelial growth factor signaling and enables endothelial cell motility. J. Biol. Chem. 2014, 289, 5904–5913. [Google Scholar] [CrossRef] [PubMed]
- Fiordalisi, J.J.; Dewar, B.J.; Graves, L.M.; Madigan, J.P.; Cox, A.D. Src-Mediated Phosphorylation of the Tyrosine Phosphatase PRL-3 Is Required for PRL-3 Promotion of Rho Activation, Motility and Invasion. PLoS ONE 2013, 8, e64309. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y. Common therapeutic target for both cancer and obesity. World J. Biol. Chem. 2017, 8, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Selvakumar, P.; Lakshmikuttyamma, A.; Lawman, Z.; Bonham, K.; Dimmock, J.R.; Sharma, R.K. Expression of methionine aminopeptidase 2, N-myristoyltransferase, and N-myristoyltransferase inhibitor protein 71 in HT29. Biochem. Biophys. Res. Commun. 2004, 322, 1012–1017. [Google Scholar] [CrossRef]
- Selvakumar, P.; Lakshmikuttyamma, A.; Kanthan, R.; Kanthan, S.C.; Dimmock, J.R.; Sharma, R.K. High expression of methionine aminopeptidase 2 in human colorectal adenocarcinomas. Clin. Cancer Res. 2004, 10, 2771–2775. [Google Scholar] [CrossRef]
- Tucker, L.A.; Zhang, Q.; Sheppard, G.S.; Lou, P.; Jiang, F.; McKeegan, E.; Lesniewski, R.; Davidsen, S.K.; Bell, R.L.; Wang, J. Ectopic expression of methionine aminopeptidase-2 causes cell transformation and stimulates proliferation. Oncogene 2008, 27, 3967–3976. [Google Scholar] [CrossRef]
- Selvakumar, P.; Lakshmikuttyamma, A.; Das, U.; Pati, H.N.; Dimmock, J.R.; Sharma, R.K. NC2213: A novel methionine aminopeptidase 2 inhibitor in human colon cancer HT29 cells. Mol. Cancer 2009, 8, e65. [Google Scholar] [CrossRef]
- Kakugawa, Y.; Wada, T.; Yamaguchi, K.; Yamanami, H.; Ouchi, K.; Sato, I.; Miyagi, T. Up-regulation of plasma membrane-associated ganglioside sialidase (Neu3) in human colon cancer and its involvement in apoptosis suppression. Proc. Natl. Acad. Sci. USA 2002, 99, 10718–10723. [Google Scholar] [CrossRef]
- Shiozaki, K.; Yamaguchi, K.; Sato, I.; Miyagi, T. Plasma membrane-associated sialidase (NEU3) promotes formation of colonic aberrant crypt foci in azoxymethane-treated transgenic mice. Cancer Sci. 2009, 100, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Takahashi, K.; Shiozaki, K.; Yamaguchi, K.; Moriya, S.; Hosono, M.; Shima, H.; Miyagi, T. Potentiation of epidermal growth factor-mediated oncogenic transformation by sialidase NEU3 leading to Src activation. PLoS ONE 2015, 10, e0120578. [Google Scholar] [CrossRef] [PubMed]
- Yamanami, H.; Shiozaki, K.; Wada, T.; Yamaguchi, K.; Uemura, T.; Kakugawa, Y.; Hujiya, T.; Miyagi, T. Down-regulation of sialidase NEU4 may contribute to invasive properties of human colon cancers. Cancer Sci. 2007, 98, 299–307. [Google Scholar] [CrossRef]
- Ahn, J.Y.; Lee, J.S.; Min, H.Y.; Lee, H.Y. Acquired resistance to 5-fluorouracil via HSP90/Src-mediated increase in thymidylate synthase expression in colon cancer. Oncotarget 2015, 6, 32622–32633. [Google Scholar] [CrossRef] [PubMed]
- Torres-Ayuso, P.; Daza-Martin, M.; Martin-Perez, J.; Avila-Flores, A.; Merida, I. Diacylglycerol kinase alpha promotes 3D cancer cell growth and limits drug sensitivity through functional interaction with Src. Oncotarget 2014, 5, 9710–9726. [Google Scholar] [CrossRef] [PubMed]
- Kucan Brlic, P.; Lenac Rovis, T.; Cinamon, G.; Tsukerman, P.; Mandelboim, O.; Jonjic, S. Targeting PVR (CD155) and its receptors in anti-tumor therapy. Cell Mol. Immunol. 2019, 16, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zheng, Q.; Xin, N.; Wang, W.; Zhao, C. CD155, an onco-immunologic molecule in human tumors. Cancer Sci. 2017, 108, 1934–1938. [Google Scholar] [CrossRef]
- Zheng, Q.; Wang, B.; Gao, J.; Xin, N.; Wang, W.; Song, X.; Shao, Y.; Zhao, C. CD155 knockdown promotes apoptosis via AKT/Bcl-2/Bax in colon cancer cells. J. Cell Mol. Med. 2018, 22, 131–140. [Google Scholar] [CrossRef]
- Leroy, C.; Fialin, C.; Sirvent, A.; Simon, V.; Urbach, S.; Poncet, J.; Robert, B.; Jouin, P.; Roche, S. Quantitative phosphoproteomics reveals a cluster of tyrosine kinases that mediates Src invasive activity in advanced colon carcinoma cells. Cancer Res. 2009, 69, 2279–2286. [Google Scholar] [CrossRef]
- Aligayer, H.; Boyd, D.D.; Heiss, M.M.; Abdalla, E.K.; Curley, S.A.; Gallick, G.E. Activation of Src kinase in primary colorectal carcinoma: An indicator of poor clinical prognosis. Cancer 2002, 94, 344–351. [Google Scholar] [CrossRef]
- Allgayer, H.; Wang, H.; Gallick, G.E.; Crabtree, A.; Mazar, A.; Jones, T.; Kraker, A.J.; Boyd, D.D. Transcriptional induction of the urokinase receptor gene by a constitutively active Src-Requirement of an upstream motif (-152/-135) bound with Sp1. J. Biol. Chem. 1999, 274, 18428–18437. [Google Scholar] [CrossRef] [PubMed]
- Leupold, J.H.; Asangani, I.; Maurer, G.D.; Lengyel, E.; Post, S.; Allgayer, H. Src induces urokinase receptor gene expression and invasion/intravasation via activator protein-1/p-c-Jun in colorectal cancer. Mol. Cancer Res. 2007, 5, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Oliva, K.; Wang, Y.; Quinn, M.; Rice, G. Proteomic profiling of proteins associated with urokinase plasminogen activator receptor in a colon cancer cell line using an antisense approach. Proteomics 2003, 3, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhang, Q.; Wang, X.; Li, T.; Wan, Y.; Liu, Y.; Zhu, J. Role of paxillin in colorectal carcinoma and its relationship to clinicopathological features. Chin. Med. J. (Engl) 2014, 127, 423–429. [Google Scholar]
- Wu, D.W.; Huang, C.C.; Chang, S.W.; Chen, T.H.; Lee, H. Bcl-2 stabilization by paxillin confers 5-fluorouracil resistance in colorectal cancer. Cell Death Differ. 2015, 22, 779–789. [Google Scholar] [CrossRef]
- Van Zyp, J.V.; Conway, W.C.; Craig, D.H.; van Zyp, N.V.; Thamilselvan, V.; Basson, M.D. Extracellular pressure stimulates tumor cell adhesion in vitro by paxillin activation. Cancer Biol. Ther. 2006, 5, 1169–1178. [Google Scholar] [CrossRef]
- Thamilselvan, V.; Craig, D.H.; Basson, M.D. FAK association with multiple signal proteins mediates pressure-induced colon cancer cell adhesion via a Src-dependent PI3K/Akt pathway. Faseb J. 2007, 21, 1730–1741. [Google Scholar] [CrossRef]
- Herbert, K.J.; Ashton, T.M.; Prevo, R.; Pirovano, G.; Higgins, G.S. T-LAK cell-originated protein kinase (TOPK): An emerging target for cancer-specific therapeutics. Cell Death Dis. 2018, 9, e1089. [Google Scholar] [CrossRef]
- Xiao, J.J.; Duan, Q.H.; Wang, Z.; Yan, W.; Sun, H.M.; Xue, P.P.; Fan, X.M.; Zeng, X.Y.; Chen, J.; Shao, C.; et al. Phosphorylation of TOPK at Y74, Y272 by Src increases the stability of TOPK and promotes tumorigenesis of colon cancer. Oncotarget 2016, 7, 24483–24494. [Google Scholar] [CrossRef]
- Perletti, G.P.; Marras, E.; Concari, P.; Piccinini, F.; Tashjian, A.H. PKC delta acts as a growth and tumor suppressor in rat colonic epithelial cells. Oncogene 1999, 18, 1251–1256. [Google Scholar] [CrossRef]
- Abe, H.; Mochizuki, S.; Ohara, K.; Ueno, M.; Ochiai, H.; Kitagawa, Y.; Hino, O.; Sato, H.; Okada, Y. Src Plays a Key Role in ADAM28 Expression in v-src-Transformed Epithelial Cells and Human Carcinoma Cells. Am. J. Pathol. 2013, 183, 1667–1678. [Google Scholar] [CrossRef] [PubMed]
- Cam, W.R.; Masaki, T.; Shiratori, Y.; Kato, N.; Ikenoue, T.; Okamoto, M.; Igarashi, K.; Sano, T.; Omata, M. Reduced C-terminal Src kinase activity is correlated inversely with pp60(c-src) activity in colorectal carcinoma. Cancer 2001, 92, 61–70. [Google Scholar] [CrossRef]
- Kunte, D.P.; Wali, R.K.; Koetsier, J.L.; Hart, J.; Kostjukova, M.N.; Kilimnik, A.Y.; Pyatkin, I.G.; Strelnikova, S.R.; Roy, H.K. Down-regulation of the tumor suppressor gene C-terminal Src kinase: An early event during premalignant colonic epithelial hyperproliferation. FEBS Lett. 2005, 579, 3497–3502. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Nada, S.; Yamanashi, Y.; Yamamoto, T.; Nakagawa, H. Csk-a Protein-Tyrosine Kinase Involved in Regulation of Src Family Kinases. J. Biol. Chem. 1991, 266, 24249–24252. [Google Scholar] [PubMed]
- Sirvent, A.; Benistant, C.; Pannequin, J.; Veracini, L.; Simon, V.; Bourgaux, J.F.; Hollande, F.; Cruzalegui, F.; Roche, S. Src family tyrosine kinases-driven colon cancer cell invasion is induced by Csk membrane delocalization. Oncogene 2010, 29, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Tanaka, S.; Suzuki, H.; Takayanagi, H.; Miyazaki, T.; Nakamura, K.; Tsuruo, T. Overexpression of the csk gene suppresses tumor metastasis in vivo. Int. J. Cancer 2000, 88, 384–391. [Google Scholar] [CrossRef]
- Rengifo-Cam, W.; Konishi, A.; Morishita, N.; Matsuoka, H.; Yamori, T.; Nada, S.; Okada, M. Csk defines the ability of integrin-mediated cell adhesion and migration in human colon cancer cells: Implication for a potential role in cancer metastasis. Oncogene 2004, 23, 289–297. [Google Scholar] [CrossRef]
- Di Stefano, P.; Damiano, L.; Cabodi, S.; Aramu, S.; Tordella, L.; Praduroux, A.; Piva, R.; Cavallo, F.; Forni, G.; Silengo, L.; et al. p140Cap protein suppresses tumour cell properties, regulating Csk and Src kinase activity. EMBO J. 2007, 26, 2843–2855. [Google Scholar] [CrossRef]
- Damiano, L.; Di Stefano, P.; Camacho Leal, M.P.; Barba, M.; Mainiero, F.; Cabodi, S.; Tordella, L.; Sapino, A.; Castellano, I.; Canel, M.; et al. p140Cap dual regulation of E-cadherin/EGFR cross-talk and Ras signalling in tumour cell scatter and proliferation. Oncogene 2010, 29, 3677–3690. [Google Scholar] [CrossRef]
- Asbagh, L.A.; Vazquez, I.; Vecchione, L.; Budinska, E.; De Vriendt, V.; Baietti, M.F.; Steklov, M.; Jacobs, B.; Hoe, N.; Singh, S.; et al. The tyrosine phosphatase PTPRO sensitizes colon cancer cells to anti-EGFR therapy through activation of SRC-mediated EGFR signaling. Oncotarget 2014, 5, 10070–10083. [Google Scholar] [CrossRef]
- Zhao, Y.Q.; Scott, A.; Zhang, P.; Hao, Y.J.; Feng, X.J.; Somasundaram, S.; Khalil, A.M.; Willis, J.; Wang, Z.H. Regulation of paxillin-p130-PI3K-AKT signaling axis by Src and PTPRT impacts colon tumorigenesis. Oncotarget 2017, 8, 48782–48793. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal. Transduct. Target. Ther. 2016, 1, e15004. [Google Scholar] [CrossRef] [PubMed]
- Kokuda, R.; Watanabe, R.; Okuzaki, D.; Akamatsu, H.; Oneyama, C. MicroRNA-137-mediated Src oncogenic signaling promotes cancer progression. Genes Cells 2018. [Google Scholar] [CrossRef] [PubMed]
- Oneyama, C.; Kito, Y.; Asai, R.; Ikeda, J.; Yoshida, T.; Okuzaki, D.; Kokuda, R.; Kakumoto, K.; Takayama, K.; Inoue, S.; et al. MiR-424/503-mediated Rictor upregulation promotes tumor progression. PLoS ONE 2013, 8, e80300. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Ou, M.C.; Fang, C.W.; Lee, T.H.; Tzeng, S.L. High Glucose Concentrations Negatively Regulate the IGF1R/Src/ERK Axis through the MicroRNA-9 in Colorectal Cancer. Cells 2019, 8, 326. [Google Scholar] [CrossRef] [PubMed]
- Oneyama, C.; Morii, E.; Okuzaki, D.; Takahashi, Y.; Ikeda, J.; Wakabayashi, N.; Akamatsu, H.; Tsujimoto, M.; Nishida, T.; Aozasa, K.; et al. MicroRNA-mediated upregulation of integrin-linked kinase promotes Src-induced tumor progression. Oncogene 2012, 31, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, R.; Okuzaki, D.; Okada, M.; Oneyama, C. MicroRNA-27b suppresses tumor progression by regulating ARFGEF1 and focal adhesion signaling. Cancer Sci. 2016, 107, 28–35. [Google Scholar] [CrossRef]
- Okuzaki, D.; Yamauchi, T.; Mitani, F.; Miyata, M.; Ninomiya, Y.; Watanabe, R.; Akamatsu, H.; Oneyama, C. c-Src promotes tumor progression through downregulation of microRNA-129-1-3p. Cancer Sci. 2020, 111, 418–428. [Google Scholar] [CrossRef]
- Chong, Y.P.; Mulhern, T.D.; Zhu, H.J.; Fujita, D.J.; Bjorge, J.D.; Tantiongco, J.P.; Sotirellis, N.; Lio, D.S.S.; Scholz, G.; Cheng, H.C. A novel non-catalytic mechanism employed by the C-terminal Src-homologous kinase to inhibit Src-family kinase activity. J. Biol. Chem. 2004, 279, 20752–20766. [Google Scholar] [CrossRef]
- Zhu, S.; Bjorge, J.D.; Cheng, H.C.; Fujita, D.J. Decreased CHK protein levels are associated with Src activation in colon cancer cells. Oncogene 2008, 27, 2027–2034. [Google Scholar] [CrossRef][Green Version]
- Bedirian, A.; Baldwin, C.; Abe, J.; Takano, T.; Lemay, S. Pleckstrin homology and phosphotyrosine-binding domain-dependent membrane association and tyrosine phosphorylation of Dok-4, an inhibitory adapter molecule expressed in epithelial cells. J. Biol. Chem. 2004, 279, 19335–19349. [Google Scholar] [CrossRef] [PubMed]
- Boumber, Y.A.; Kondo, Y.; Chen, X.Q.; Shen, L.L.; Gharibyan, V.; Konishi, K.; Estey, E.; Kantarjian, H.; Garcia-Manero, G.; Issa, J.P.J. RIL, a LIM gene on 5q31, is silenced by methylation in cancer and sensitizes cancer cells to apoptosis. Cancer Res. 2007, 67, 1997–2005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Tu, Y.Z.; Zhao, J.P.; Chen, K.; Wu, C.Y. Reversion-induced LIM interaction with Src reveals a novel Src inactivation cycle. J. Cell Biol. 2009, 184, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Mamidipudi, V.; Zhang, J.; Lee, K.C.; Cartwright, C.A. RACK1 regulates G1/S progression by suppressing Src kinase activity. Mol. Cell Biol. 2004, 24, 6788–6798. [Google Scholar] [CrossRef] [PubMed]
- Mamidipudi, V.; Cartwright, C.A. A novel pro-apoptotic function of RACK1: Suppression of Src activity in the intrinsic and Akt pathways. Oncogene 2009, 28, 4421–4433. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mamidipudi, V.; Dhillon, N.K.; Parman, T.; Miller, L.D.; Lee, K.C.; Cartwright, C.A. RACK1 inhibits colonic cell growth by regulating Src activity at cell cycle checkpoints. Oncogene 2007, 26, 2914–2924. [Google Scholar] [CrossRef]
- Swaminathan, G.; Cartwright, C.A. Rack1 promotes epithelial cell–cell adhesion by regulating E-cadherin endocytosis. Oncogene 2012, 31, 376–389. [Google Scholar] [CrossRef]
- Ito, Y.; Bae, S.C.; Chuang, L.S.H. The RUNX family: Developmental regulators in cancer. Nat. Rev. Cancer 2015, 15, 81–95. [Google Scholar] [CrossRef]
- Kang, K.A.; Piao, M.J.; Ryu, Y.S.; Maeng, Y.H.; Hyun, J.W. Cytoplasmic Localization of RUNX3 via Histone Deacetylase-Mediated SRC Expression in Oxidative-Stressed Colon Cancer Cells. J. Cell Physiol. 2017, 232, 1914–1921. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Bio. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.; Shintani, S.; Weber, A.; Kirchmair, R.; Wood, M.; Cravens, A.; McSharry, H.; Iwakura, A.; Yoon, Y.S.; Himes, N.; et al. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J. Clin. Invest. 2004, 113, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Irby, R.B.; Yeatman, T.J. Increased Src activity disrupts cadherin/catenin-mediated homotypic adhesion in human colon cancer and transformed rodent cells. Cancer Res. 2002, 62, 2669–2674. [Google Scholar] [PubMed]
- Calcagno, A.M.; Fostel, J.N.; Orchekowski, R.P.; Alston, J.T.; Mattes, W.B.; Siahaan, T.J.; Ware, J.A. Modulation of cell adhesion molecules in various epithelial cell lines after treatment with PP2. Mol. Pharmaceut. 2005, 2, 170–184. [Google Scholar] [CrossRef]
- Nam, J.S.; Ino, Y.; Sakamoto, M.; Hirohashi, S. Src family kinase inhibitor PP2 restores the E-cadherin/catenin cell adhesion system in human cancer cells and reduces cancer metastasis. Clin. Cancer Res. 2002, 8, 2430–2436. [Google Scholar]
- Shi, S.; Garcia, J.G.N.; Roy, S.; Parinandi, N.L.; Natarajan, V. Involvement of c-Src in diperoxovanadate-induced endothelial cell barrier dysfunction. Am. J. Physiol-Lung C 2000, 279, L441–L451. [Google Scholar] [CrossRef]
- Laird, A.D.; Li, G.M.; Moss, K.G.; Blake, R.A.; Broome, M.A.; Cherrington, J.M.; Mendel, D.B. Src family kinase activity is required for signal tranducer and activator of transcription 3 and focal adhesion kinase phosphorylation and vascular endothelial growth factor signaling in vivo and for anchorage-dependent and -independent growth of human tumor cells. Mol. Cancer Ther. 2003, 2, 461–469. [Google Scholar]
- Minard, M.E.; Ellis, L.M.; Gallick, G.E. Tiam1 regulates cell adhesion, migration and apoptosis in colon tumor cells. Clin. Exp. Metastasis 2006, 23, 301–313. [Google Scholar] [CrossRef]
- Windham, T.C.; Parikh, N.U.; Siwak, D.R.; Summy, J.M.; McConkey, D.J.; Kraker, A.J.; Gallick, G.E. Src activation regulates anoikis in human colon tumor cell lines. Oncogene 2002, 21, 7797–7807. [Google Scholar] [CrossRef]
- Fianco, G.; Cenci, C.; Barila, D. Caspase-8 expression and its Src-dependent phosphorylation on Tyr380 promote cancer cell neoplastic transformation and resistance to anoikis. Exp. Cell Res. 2016, 347, 114–122. [Google Scholar] [CrossRef]
- Avizienyte, E.; Fincham, V.J.; Brunton, V.G.; Frame, M.C. Src SH3/2 domain-mediated peripheral accumulation of Src and phospho-myosin is linked to deregulation of E-cadherin and the epithelial–mesenchymal transition. Mol. Biol. Cell 2004, 15, 2794–2803. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, P.L.; Auger, F.A.; Huot, J. Regulation of transendothelial migration of colon cancer cells by E-selectin-mediated activation of p38 and ERK MAP kinases. Oncogene 2006, 25, 6563–6573. [Google Scholar] [CrossRef] [PubMed]
- Porquet, N.; Poirier, A.; Houle, F.; Pin, A.L.; Gout, S.; Tremblay, P.L.; Paquet, E.R.; Klinck, R.; Auger, F.A.; Huot, J. Survival advantages conferred to colon cancer cells by E-selectin-induced activation of the PI3K-NFkappaB survival axis downstream of Death receptor-3. BMC Cancer 2011, 11, e285. [Google Scholar] [CrossRef] [PubMed]
- Sancier, F.; Dumont, A.; Sirvent, A.; Paquay de Plater, L.; Edmonds, T.; David, G.; Jan, M.; de Montrion, C.; Coge, F.; Leonce, S.; et al. Specific oncogenic activity of the Src-family tyrosine kinase c-Yes in colon carcinoma cells. PLoS ONE 2011, 6, e17237. [Google Scholar] [CrossRef] [PubMed]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Thamilselvan, V.; Basson, M.D. Pressure activates colon cancer cell adhesion by inside-out focal adhesion complex and actin cytoskeletal signaling. Gastroenterology 2004, 126, 8–18. [Google Scholar] [CrossRef]
- Van Zyp, J.V.; Thamilselvan, V.; Walsh, M.; Polin, L.; Basson, M.D. Extracellular pressure stimulates colon cancer cell adhesion in vitro and to surgical wounds by Src (sarcoma protein) activation. Am. J. Surg. 2004, 188, 467–473. [Google Scholar] [CrossRef]
- Craig, D.H.; Gayer, C.P.; Schaubert, K.L.; Wei, Y.; Li, J.; Laouar, Y.; Basson, M.D. Increased extracellular pressure enhances cancer cell integrin-binding affinity through phosphorylation of beta1-integrin at threonine 788/789. Am. J. Physiol. Cell Physiol. 2009, 296, 193–204. [Google Scholar] [CrossRef]
- Del Pozo, M.A.; Balasubramanian, N.; Alderson, N.B.; Kiosses, W.B.; Grande-Garcia, A.; Anderson, R.G.; Schwartz, M.A. Phospho-caveolin-1 mediates integrin-regulated membrane domain internalization. Nat. Cell Biol. 2005, 7, 901–908. [Google Scholar] [CrossRef]
- Mi, L.; Zhu, F.; Yang, X.; Lu, J.; Zheng, Y.; Zhao, Q.; Wen, X.; Lu, A.; Wang, M.; Zheng, M.; et al. The metastatic suppressor NDRG1 inhibits EMT, migration and invasion through interaction and promotion of caveolin-1 ubiquitylation in human colorectal cancer cells. Oncogene 2017, 36, 4323–4335. [Google Scholar] [CrossRef]
- Joshi, B.; Strugnell, S.S.; Goetz, J.G.; Kojic, L.D.; Cox, M.E.; Griffith, O.L.; Chan, S.K.; Jones, S.J.; Leung, S.P.; Masoudi, H.; et al. Phosphorylated Caveolin-1 Regulates Rho/ROCK-Dependent Focal Adhesion Dynamics and Tumor Cell Migration and Invasion. Cancer Res. 2008, 68, 8210–8220. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Utsunomiya, T.; Mimori, K.; Tanaka, F.; Inoue, H.; Nagahara, H.; Murayama, S.; Mori, M. Clinical significance of human kallikrein gene 6 messenger RNA expression in colorectal cancer. Clin. Cancer Res. 2005, 11, 2889–2893. [Google Scholar] [CrossRef]
- Henkhaus, R.S.; Roy, U.K.; Cavallo-Medved, D.; Sloane, B.F.; Gerner, E.W.; Ignatenko, N.A. Caveolin-1-mediated expression and secretion of kallikrein 6 in colon cancer cells. Neoplasia 2008, 10, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Sells, E.; Pandey, R.; Abril, E.R.; Hsu, C.H.; Krouse, R.S.; Nagle, R.B.; Pampalakis, G.; Sotiropoulou, G.; Ignatenko, N.A. Kallikrein 6 protease advances colon tumorigenesis via induction of the high mobility group A2 protein. Oncotarget 2019, 10, 6062–6078. [Google Scholar] [CrossRef] [PubMed]
- Holgren, C.; Dougherty, U.; Edwin, F.; Cerasi, D.; Taylor, I.; Fichera, A.; Joseph, L.; Bissonnette, M.; Khare, S. Sprouty-2 controls c-Met expression and metastatic potential of colon cancer cells: Sprouty/c-Met upregulation in human colonic adenocarcinomas. Oncogene 2010, 29, 5241–5253. [Google Scholar] [CrossRef]
- Egan, J.E.; Hall, A.B.; Yatsula, B.A.; Bar-Sagi, D. The bimodal regulation of epidermal growth factor signaling by human Sprouty proteins. Proc. Natl. Acad. Sci. USA 2002, 99, 6041–6046. [Google Scholar] [CrossRef]
- Barbachano, A.; Fernandez-Barral, A.; Pereira, F.; Segura, M.F.; Ordonez-Moran, P.; Carrillo-de Santa Pau, E.; Gonzalez-Sancho, J.M.; Hanniford, D.; Martinez, N.; Costales-Carrera, A.; et al. SPROUTY-2 represses the epithelial phenotype of colon carcinoma cells via upregulation of ZEB1 mediated by ETS1 and miR-200/miR-150. Oncogene 2016, 35, 2991–3003. [Google Scholar] [CrossRef]
- Eddy, R.J.; Weidmann, M.D.; Sharma, V.P.; Condeelis, J.S. Tumor Cell Invadopodia: Invasive Protrusions that Orchestrate Metastasis. Trends Cell Biol. 2017, 27, 595–607. [Google Scholar] [CrossRef]
- Eckert, M.A.; Lwin, T.M.; Chang, A.T.; Kim, J.; Danis, E.; Ohno-Machado, L.; Yang, J. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 2011, 19, 372–386. [Google Scholar] [CrossRef]
- Vitale, S.; Avizienyte, E.; Brunton, V.G.; Frame, M.C. Focal adhesion kinase is not required for Src-induced formation of invadopodia in KM12C colon cancer cells and can interfere with their assembly. Eur. J. Cell Biol. 2008, 87, 569–579. [Google Scholar] [CrossRef]
- Geiszt, M.; Lekstrom, K.; Witta, J.; Leto, T.L. Proteins homologous to p47phox and p67phox support superoxide production by NAD(P)H oxidase 1 in colon epithelial cells. J. Biol. Chem. 2003, 278, 20006–20012. [Google Scholar] [CrossRef] [PubMed]
- Gianni, D.; Taulet, N.; DerMardirossian, C.; Bokoch, G.M. c-Src-mediated phosphorylation of NoxA1 and Tks4 induces the reactive oxygen species (ROS)-dependent formation of functional invadopodia in human colon cancer cells. Mol. Biol. Cell 2010, 21, 4287–4298. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Streit, M.; Kaiser, R.; Herzberg, F.; Schirner, M.; Schramm, K.; Kaufmann, C.; Henneken, M.; Schafer-Korting, M.; Thiel, E.; et al. De novo expression of the alpha5beta1-fibronectin receptor in HT29 colon-cancer cells reduces activity of C-SRC. Increase of C-SRC activity by attachment on fibronectin. Int. J. Cancer 1998, 76, 91–98. [Google Scholar] [CrossRef]
- Avizienyte, E.; Wyke, A.W.; Jones, R.J.; McLean, G.W.; Westhoff, M.A.; Brunton, V.G.; Frame, M.C. Src-induced de-regulation of E-cadherin in colon cancer cells requires integrin signalling. Nat. Cell Biol 2002, 4, 632–638. [Google Scholar] [CrossRef]
- Jones, R.J.; Avizienyte, E.; Wyke, A.W.; Owens, D.W.; Brunton, V.G.; Frame, M.C. Elevated c-Src is linked to altered cell-matrix adhesion rather than proliferation in KM12C human colorectal cancer cells. Br. J. Cancer 2002, 87, 1128–1135. [Google Scholar] [CrossRef]
- Brunton, V.G.; Avizienyte, E.; Fincham, V.J.; Serrels, B.; Metcalf, C.A.; Sawyer, T.K.; Frame, M.C. Identification of Src-specific phosphorylation site on focal adhesion kinase: Dissection of the role of Src SH2 and catalytic functions and their consequences for tumor cell behavior. Cancer Res. 2005, 65, 1335–1342. [Google Scholar] [CrossRef]
- Craig, D.H.; Haimovich, B.; Basson, M.D. alpha-Actinin-1 phosphorylation modulates pressure-induced colon cancer cell adhesion through regulation of focal adhesion kinase-Src interaction. Am. J. Physiol. Cell Physiol. 2007, 293, C1862–C1874. [Google Scholar] [CrossRef]
- Zheng, X.M.; Wang, Y.; Pallen, C.J. Cell transformation and activation of pp60c-src by overexpression of a protein tyrosine phosphatase. Nature 1992, 359, 336–339. [Google Scholar] [CrossRef]
- Sun, G.; Cheng, S.Y.; Chen, M.; Lim, C.J.; Pallen, C.J. Protein tyrosine phosphatase alpha phosphotyrosyl-789 binds BCAR3 to position Cas for activation at integrin-mediated focal adhesions. Mol. Cell Biol. 2012, 32, 3776–3789. [Google Scholar] [CrossRef]
- Krndija, D.; Schmid, H.; Eismann, J.L.; Lother, U.; Adler, G.; Oswald, F.; Seufferlein, T.; von Wichert, G. Substrate stiffness and the receptor-type tyrosine-protein phosphatase alpha regulate spreading of colon cancer cells through cytoskeletal contractility. Oncogene 2010, 29, 2724–2738. [Google Scholar] [CrossRef]
- Lai, X.; Chen, Q.; Zhu, C.; Deng, R.; Zhao, X.; Chen, C.; Wang, Y.; Yu, J.; Huang, J. Regulation of RPTPalpha-c-Src signalling pathway by miR-218. FEBS J. 2015, 282, 2722–2734. [Google Scholar] [CrossRef]
- Dhawan, P.; Singh, A.B.; Deane, N.G.; No, Y.; Shiou, S.R.; Schmidt, C.; Neff, J.; Washington, M.K.; Beauchamp, R.D. Claudin-1 regulates cellular transformation and metastatic behavior in colon cancer. J. Clin. Invest. 2005, 115, 1765–1776. [Google Scholar] [CrossRef]
- Singh, A.B.; Sharma, A.; Dhawan, P. Claudin-1 expression confers resistance to anoikis in colon cancer cells in a Src-dependent manner. Carcinogenesis 2012, 33, 2538–2547. [Google Scholar] [CrossRef]
- Bhat, A.A.; Ahmad, R.; Uppada, S.B.; Singh, A.B.; Dhawan, P. Claudin-1 promotes TNF-alpha-induced epithelial–mesenchymal transition and migration in colorectal adenocarcinoma cells. Exp. Cell Res. 2016, 349, 119–127. [Google Scholar] [CrossRef]
- Sheng, X.; Nenseth, H.Z.; Qu, S.; Kuzu, O.F.; Frahnow, T.; Simon, L.; Greene, S.; Zeng, Q.; Fazli, L.; Rennie, P.S.; et al. IRE1alpha-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat. Commun. 2019, 10, e323. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, C.; Qin, Y.; Chen, J.; Fang, J. Knockdown of IRE1a suppresses metastatic potential of colon cancer cells through inhibiting FN1-Src/FAK-GTPases signaling. Int. J. Biochem. Cell Biol. 2019, 114, e105572. [Google Scholar] [CrossRef]
- Liau, N.P.D.; Laktyushin, A.; Lucet, I.S.; Murphy, J.M.; Yao, S.; Whitlock, E.; Callaghan, K.; Nicola, N.A.; Kershaw, N.J.; Babon, J.J. The molecular basis of JAK/STAT inhibition by SOCS1. Nat. Commun. 2018, 9, e1558. [Google Scholar] [CrossRef]
- Jung, S.H.; Kim, S.M.; Lee, C.E. Mechanism of suppressors of cytokine signaling 1 inhibition of epithelial–mesenchymal transition signaling through ROS regulation in colon cancer cells: Suppression of Src leading to thioredoxin up-regulation. Oncotarget 2016, 7, 62559–62571. [Google Scholar] [CrossRef]
- Bhat, A.A.; Pope, J.L.; Smith, J.J.; Ahmad, R.; Chen, X.; Washington, M.K.; Beauchamp, R.D.; Singh, A.B.; Dhawan, P. Claudin-7 expression induces mesenchymal to epithelial transformation (MET) to inhibit colon tumorigenesis. Oncogene 2015, 34, 4570–4580. [Google Scholar] [CrossRef]
- Zeller, C.; Hinzmann, B.; Seitz, S.; Prokoph, H.; Burkhard-Goettges, E.; Fischer, J.; Jandrig, B.; Schwarz, L.E.; Rosenthal, A.; Scherneck, S. SASH1: A candidate tumor suppressor gene on chromosome 6q24.3 is downregulated in breast cancer. Oncogene 2003, 22, 2972–2983. [Google Scholar] [CrossRef]
- Burgess, J.T.; Bolderson, E.; Saunus, J.M.; Zhang, S.D.; Reid, L.E.; McNicol, A.M.; Lakhani, S.R.; Cuff, K.; Richard, K.; Richard, D.J.; et al. SASH1 mediates sensitivity of breast cancer cells to chloropyramine and is associated with prognosis in breast cancer. Oncotarget 2016, 7, 72807–72818. [Google Scholar] [CrossRef]
- Franke, F.C.; Muller, J.; Abal, M.; Medina, E.D.; Nitsche, U.; Weidmann, H.; Chardonnet, S.; Ninio, E.; Janssen, K.P. The Tumor Suppressor SASH1 Interacts with the Signal Adaptor CRKL to Inhibit Epithelial–mesenchymal transition and Metastasis in Colorectal Cancer. Cell Mol Gastroenter 2019, 7, 33–53. [Google Scholar] [CrossRef]
- Tsunekuni, K.; Konno, M.; Haraguchi, N.; Koseki, J.; Asai, A.; Matsuoka, K.; Kobunai, T.; Takechi, T.; Doki, Y.; Mori, M.; et al. CD44/CD133-Positive Colorectal Cancer Stem Cells are Sensitive to Trifluridine Exposure. Sci. Rep. 2019, 9, 14861. [Google Scholar] [CrossRef]
- Finke, L.H.; Terpe, H.J.; Zorb, C.; Haensch, W.; Schlag, P.M. Colorectal cancer prognosis and expression of exon-v6-containing CD44 proteins. Lancet 1995, 345, e583. [Google Scholar] [CrossRef]
- Matsumura, Y.; Tarin, D. Significance of CD44 gene products for cancer diagnosis and disease evaluation. Lancet 1992, 340, 1053–1058. [Google Scholar] [CrossRef]
- Ju, S.Y.; Chiou, S.H.; Su, Y. Maintenance of the stemness in CD44(+) HCT-15 and HCT-116 human colon cancer cells requires miR-203 suppression. Stem Cell Res. 2014, 12, 86–100. [Google Scholar] [CrossRef]
- Su, Y.J.; Lai, H.M.; Chang, Y.W.; Chen, G.Y.; Lee, J.L. Direct reprogramming of stem cell properties in colon cancer cells by CD44. EMBO J. 2011, 30, 3186–3199. [Google Scholar] [CrossRef]
- Bates, R.C.; Edwards, N.S.; Burns, G.F.; Fisher, D.E. A CD44 survival pathway triggers chemoresistance via lyn kinase and phosphoinositide 3-kinase/Akt in colon carcinoma cells. Cancer Res. 2001, 61, 5275–5283. [Google Scholar]
- Su, Y.J.; Lin, W.H.; Chang, Y.W.; Wei, K.C.; Liang, C.L.; Chen, S.C.; Lee, J.L. Polarized cell migration induces cancer type-specific CD133/integrin/Src/Akt/GSK3beta/beta-catenin signaling required for maintenance of cancer stem cell properties. Oncotarget 2015, 6, 38029–38045. [Google Scholar] [CrossRef]
- Jung, J.; Choi, S.C.; Lee, H.N.; Han, G.Y.; Kim, C.W. Inhibition of c-Yes Induces Differentiation of HT-29 Human Colon Cancer Stem Cells through Midbody Elongation. Tissue Eng. Regen. Med. 2016, 13, 261–269. [Google Scholar] [CrossRef]
- Watanabe, K.; Meyer, M.J.; Strizzi, L.; Lee, J.M.; Gonzales, M.; Bianco, C.; Nagaoka, T.; Farid, S.S.; Margaryan, N.; Hendrix, M.J.; et al. Cripto-1 is a cell surface marker for a tumorigenic, undifferentiated subpopulation in human embryonal carcinoma cells. Stem Cells 2010, 28, 1303–1314. [Google Scholar] [CrossRef]
- Francescangeli, F.; Contavalli, P.; De Angelis, M.L.; Baiocchi, M.; Gambara, G.; Pagliuca, A.; Fiorenzano, A.; Prezioso, C.; Boe, A.; Todaro, M.; et al. Dynamic regulation of the cancer stem cell compartment by Cripto-1 in colorectal cancer. Cell Death Differ. 2015, 22, 1700–1713. [Google Scholar] [CrossRef]
- Chen, T.H.; Chen, C.Y.; Wen, H.C.; Chang, C.C.; Wang, H.D.; Chuu, C.P.; Chang, C.H. YAP promotes myogenic differentiation via the MEK5-ERK5 pathway. Faseb J. 2017, 31, 2963–2972. [Google Scholar] [CrossRef]
- Abe, J.; Takahashi, M.; Ishida, M.; Lee, J.D.; Berk, B.C. c-Src is required for oxidative stress-mediated activation of big mitogen-activated protein kinase 1 (BMK1). J. Biol. Chem. 1997, 272, 20389–20394. [Google Scholar] [CrossRef]
- Pereira, D.M.; Gomes, S.E.; Borralho, P.M.; Rodrigues, C.M.P. MEK5/ERK5 activation regulates colon cancer stem-like cell properties. Cell Death Discov. 2019, 5, e68. [Google Scholar] [CrossRef]
- Boyd, D.D.; Wang, H.; Avila, H.; Parikh, N.U.; Kessler, H.; Magdolen, V.; Gallick, G.E. Combination of an SRC kinase inhibitor with a novel pharmacological antagonist of the urokinase receptor diminishes in vitro colon cancer invasiveness. Clin. Cancer Res. 2004, 10, 1545–1555. [Google Scholar] [CrossRef]
- Kopetz, S.; Lesslie, D.P.; Dallas, N.A.; Park, S.I.; Johnson, M.; Parikh, N.U.; Kim, M.P.; Abbruzzese, J.L.; Ellis, L.M.; Chandra, J.; et al. Synergistic Activity of the Src Family Kinase Inhibitor Dasatinib and Oxaliplatin in Colon Carcinoma Cells Is Mediated by Oxidative Stress. Cancer Res. 2009, 69, 3842–3849. [Google Scholar] [CrossRef]
- Jia, J.; Starodub, A.; Cushman, I.; Liu, Y.; Marshall, D.J.; Hurwitz, H.I.; Nixon, A.B. Dual inhibition of alphaV integrins and Src kinase activity as a combination therapy strategy for colorectal cancer. Anticancer Drugs 2013, 24, 237–250. [Google Scholar] [CrossRef]
- Golas, J.M.; Lucas, J.; Etienne, C.; Golas, J.; Discafani, C.; Sridharan, L.; Boghaert, E.; Arndt, K.; Ye, F.; Boschelli, D.H.; et al. SKI-606, a Src/Abl inhibitor with in vivo activity in colon tumor xenograft models. Cancer Res. 2005, 65, 5358–5364. [Google Scholar] [CrossRef]
- Foss, F.M.; Veillette, A.; Sartor, O.; Rosen, N.; Bolen, J.B. Alterations in the expression of pp60c-src and p56lck associated with butyrate-induced differentiation of human colon carcinoma cells. Oncogene Res. 1989, 5, 13–23. [Google Scholar]
- Lee, J.C.; Maa, M.C.; Yu, H.S.; Wang, J.H.; Yen, C.K.; Wang, S.T.; Chen, Y.J.; Liu, Y.; Jin, Y.T.; Leu, T.H. Butyrate regulates the expression of c-Src and focal adhesion kinase and inhibits cell invasion of human colon cancer cells. Mol. Carcinog. 2005, 43, 207–214. [Google Scholar] [CrossRef]
- Kostyniuk, C.L.; Dehm, S.M.; Batten, D.; Bonham, K. The ubiquitous and tissue specific promoters of the human SRC gene are repressed by inhibitors of histone deacetylases. Oncogene 2002, 21, 6340–6347. [Google Scholar] [CrossRef] [PubMed]
- Leu, T.H.; Yeh, H.H.; Huang, C.C.; Chuang, Y.C.; Su, S.L.; Maa, M.C. Participation of p97Eps8 in Src-mediated transformation. J. Biol. Chem. 2004, 279, 9875–9881. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.H.; Sang, S.M.; Ho, C.T.; Lin, J.K. Garcinol modulates tyrosine phosphorylation of FAK and subsequently induces apoptosis through down-regulation of Src, ERK, and Akt survival signaling in human colon cancer cells. J. Cell Biochem. 2005, 96, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, C.L.; Smith-Windsor, E.L.; Bonham, K. Src family kinase members have a common response to histone deacetylase inhibitors in human colon cancer cells. Int. J. Cancer 2006, 118, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Mologni, L.; Cleris, L.; Magistroni, V.; Piazza, R.; Boschelli, F.; Formelli, F.; Gambacorti-Passerini, C. Valproic acid enhances bosutinib cytotoxicity in colon cancer cells. Int. J. Cancer 2009, 124, 1990–1996. [Google Scholar] [CrossRef]
- Wang, J.; Tucker, L.A.; Stavropoulos, J.; Zhang, Q.; Wang, Y.C.; Bukofzer, G.; Niquette, A.; Meulbroek, J.A.; Barnes, D.M.; Shen, J.; et al. Correlation of tumor growth suppression and methionine aminopetidase-2 activity blockade using an orally active inhibitor. Proc. Natl. Acad. Sci. USA 2008, 105, 1838–1843. [Google Scholar] [CrossRef]
- Liang, F.P.; Lin, C.H.; Kuo, C.D.; Chao, H.P.; Fu, S.L. Suppression of v-Src transformation by andrographolide via degradation of the v-Src protein and attenuation of the Erk signaling pathway. J. Biol. Chem. 2008, 283, 5023–5033. [Google Scholar] [CrossRef]
- Kraker, A.J.; Hartl, B.G.; Amar, A.M.; Barvian, M.R.; Showalter, H.D.H.; Moore, C.W. Biochemical and cellular effects of c-Src kinase-selective pyrido[2,3-d]pyrimidine tyrosine kinase inhibitors. Biochem. Pharmacol. 2000, 60, 885–898. [Google Scholar] [CrossRef]
- Walsh, M.F.; Woo, R.K.Y.; Gomez, R.; Basson, M.D. Extracellular pressure stimulates colon cancer cell proliferation via a mechanism requiring PKC and tyrosine kinase signals. Cell Proliferat. 2004, 37, 427–441. [Google Scholar] [CrossRef]
- Thamilselvan, V.; Basson, M.D. The role of the cytoskeleton in differentially regulating pressure-mediated effects on malignant colonocyte focal adhesion signaling and cell adhesion. Carcinogenesis 2005, 26, 1687–1697. [Google Scholar] [CrossRef] [PubMed]
- Downing, K.H. Structural basis for the interaction of tubulin with proteins and drugs that affect microtubule dynamics. Annu. Rev. Cell Dev. Biol. 2000, 16, 89–111. [Google Scholar] [CrossRef] [PubMed]
- Craig, D.H.; Owen, C.R.; Conway, W.C.; Walsh, M.F.; Downey, C.; Basson, M.D. Colchicine inhibits pressure-induced tumor cell implantation within surgical wounds and enhances tumor-free survival in mice. J. Clin. Invest. 2008, 118, 3170–3180. [Google Scholar] [CrossRef] [PubMed]
- Nautiyal, J.; Banerjee, S.; Kanwar, S.S.; Yu, Y.; Patel, B.B.; Sarkar, F.H.; Majumdar, A.P. Curcumin enhances dasatinib-induced inhibition of growth and transformation of colon cancer cells. Int. J. Cancer 2011, 128, 951–961. [Google Scholar] [CrossRef]
- Kuo, C.T.; Chang, C.; Lee, W.S. Folic acid inhibits COLO-205 colon cancer cell proliferation through activating the FRalpha/c-SRC/ERK1/2/NFkappaB/TP53 pathway: In vitro and in vivo studies. Sci. Rep. 2015, 5, 11187. [Google Scholar] [CrossRef]
- Sim, J.J.; Park, M.H.; Baek, J.H.; Lee, H.; Jeong, K.Y.; Kim, H.M. Investigation into Enhancing Capecitabine Efficacy in Colorectal Cancer by Inhibiting Focal Adhesion Kinase Signaling. Anticancer Res. 2018, 38, 4667–4676. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef]



© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, W. Regulation of Src Family Kinases during Colorectal Cancer Development and Its Clinical Implications. Cancers 2020, 12, 1339. https://doi.org/10.3390/cancers12051339
Jin W. Regulation of Src Family Kinases during Colorectal Cancer Development and Its Clinical Implications. Cancers. 2020; 12(5):1339. https://doi.org/10.3390/cancers12051339
Chicago/Turabian StyleJin, Wook. 2020. "Regulation of Src Family Kinases during Colorectal Cancer Development and Its Clinical Implications" Cancers 12, no. 5: 1339. https://doi.org/10.3390/cancers12051339
APA StyleJin, W. (2020). Regulation of Src Family Kinases during Colorectal Cancer Development and Its Clinical Implications. Cancers, 12(5), 1339. https://doi.org/10.3390/cancers12051339