Adaptor CAR Platforms—Next Generation of T Cell-Based Cancer Immunotherapy

 ,
, 

Abstract
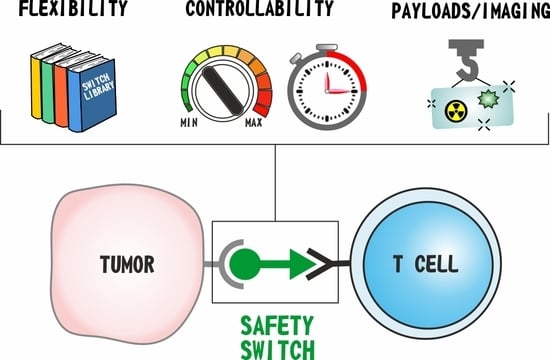
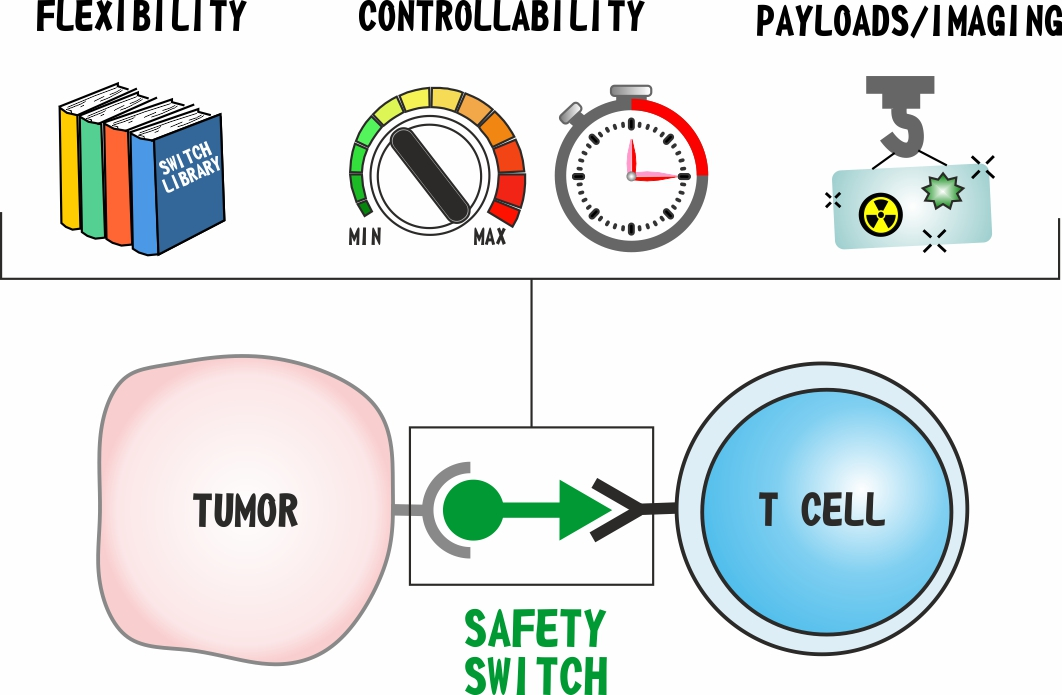
1. Introduction
2. Adaptor CAR Design
- Fc-binding adaptor CARs;
- Tag-specific adaptor CARs;
- Bispecific antibody (bsAb)-binding adaptor CARs.
2.1. Fc-Binding Adaptor CARs
2.2. Tag-Binding Adaptor CARs
2.3. BsAb-Binding Adaptor CARs
3. Controlling Therapy-Related Side Effects with Adaptor CARs
4. Improving Treatment Efficiency and Target Specificity
5. Co-Delivery of Payloads via Adaptor Molecules
6. Clinical Translation of Adaptor CAR Platforms
7. Future Directions of Adaptor CARs
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eshhar, Z. The T-body approach: Redirecting T cells with antibody specificity. Handb. Exp. Pharmacol. 2008, 181, 329–342. [Google Scholar]
- MacKay, M.; Afshinnekoo, E.; Rub, J.; Hassan, C.; Khunte, M.; Baskaran, N.; Owens, B.; Liu, L.; Roboz, G.J.; Guzman, M.L.; et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat. Biotechnol. 2020, 38, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Freyer, C.W. Tisagenlecleucel: The First CAR on the Highway to Remission for Acute Lymphoblastic Leukemia. J. Adv. Pract. Oncol. 2018, 9, 537–544. [Google Scholar] [PubMed]
- Bouchkouj, N.; Kasamon, Y.L.; de Claro, R.A.; George, B.; Lin, X.; Lee, S.; Blumenthal, G.M.; Bryan, W.; McKee, A.E.; Pazdur, R. FDA Approval Summary: Axicabtagene Ciloleucel for Relapsed or Refractory Large B-cell Lymphoma. Clin. Cancer Res. 2019, 25, 1702–1708. [Google Scholar] [CrossRef]
- Ali, S.; Kjeken, R.; Niederlaender, C.; Markey, G.; Saunders, T.S.; Opsata, M.; Moltu, K.; Bremnes, B.; Grønevik, E.; Muusse, M.; et al. The European Medicines Agency Review of Kymriah (Tisagenlecleucel) for the Treatment of Acute Lymphoblastic Leukemia and Diffuse Large B-Cell Lymphoma. Oncologist 2020, 25, e321–e327. [Google Scholar] [CrossRef] [PubMed]
- Fry, T.J.; Shah, N.N.; Orentas, R.J.; Stetler-Stevenson, M.; Yuan, C.M.; Ramakrishna, S.; Wolters, P.; Martin, S.; Delbrook, C.; Yates, B.; et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018, 24, 20–28. [Google Scholar] [CrossRef]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed]
- Shalabi, H.; Kraft, I.L.; Wang, H.W.; Yuan, C.M.; Yates, B.; Delbrook, C.; Zimbelman, J.D.; Giller, R.; Stetler-Stevenson, M.; Jaffe, E.S.; et al. Sequential loss of tumor surface antigens following chimeric antigen receptor T-cell therapies in diffuse large B-cell lymphoma. Haematologica 2018, 103, e215–e218. [Google Scholar] [CrossRef] [PubMed]
- Fesnak, A.D.; June, C.H.; Levine, B.L. Engineered T cells: The promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 2016, 16, 566–581. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow. Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef]
- Gill, S.; Maus, M.V.; Porter, D.L. Chimeric antigen receptor T cell therapy: 25years in the making. Blood Rev. 2016, 30, 157–167. [Google Scholar] [CrossRef]
- Cartellieri, M.; Bachmann, M.; Feldmann, A.; Bippes, C.; Stamova, S.; Wehner, R.; Temme, A.; Schmitz, M. Chimeric antigen receptor-engineered T cells for immunotherapy of cancer. J. Biomed. Biotechnol. 2010, 2010, 956304. [Google Scholar] [CrossRef]
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer. 2019, 120, 26–37. [Google Scholar] [CrossRef]
- Hoyos, V.; Savoldo, B.; Quintarelli, C.; Mahendravada, A.; Zhang, M.; Vera, J.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Dotti, G. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia 2010, 24, 1160–1170. [Google Scholar] [CrossRef]
- Budde, L.E.; Berger, C.; Lin, Y.; Wang, J.; Lin, X.; Frayo, S.E.; Brouns, S.A.; Spencer, D.M.; Till, B.G.; Jensen, M.C.; et al. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS ONE 2013, 8, e82742. [Google Scholar] [CrossRef]
- Diaconu, I.; Ballard, B.; Zhang, M.; Chen, Y.; West, J.; Dotti, G.; Savoldo, B. Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol. Ther. 2017, 25, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Larson, S.M.; Truscott, L.C.; Chiou, T.T.; Patel, A.; Kao, R.; Tu, A.; Tyagi, T.; Lu, X.; Elashoff, D.; De Oliveira, S.N. Pre-clinical development of gene modification of haematopoietic stem cells with chimeric antigen receptors for cancer immunotherapy. Hum. Vaccines Immunother. 2017, 13, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Vogler, I.; Newrzela, S.; Hartmann, S.; Schneider, N.; von Laer, D.; Koehl, U.; Grez, M. An improved bicistronic CD20/tCD34 vector for efficient purification and in vivo depletion of gene-modified T cells for adoptive immunotherapy. Mol. Ther. 2010, 18, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Philip, B.; Kokalaki, E.; Mekkaoui, L.; Thomas, S.; Straathof, K.; Flutter, B.; Marin, V.; Marafioti, T.; Chakraverty, R.; Linch, D.; et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 2014, 124, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chang, W.C.; Wong, C.W.; Colcher, D.; Sherman, M.; Ostberg, J.R.; Forman, S.J.; Riddell, S.R.; Jensen, M.C. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 2011, 118, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Paszkiewicz, P.J.; Fräßle, S.P.; Srivastava, S.; Sommermeyer, D.; Hudecek, M.; Drexler, I.; Sadelain, M.; Liu, L.; Jensen, M.C.; Riddell, S.R.; et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Investig. 2016, 126, 4262–4272. [Google Scholar] [CrossRef]
- Clémenceau, B.; Congy-Jolivet, N.; Gallot, G.; Vivien, R.; Gaschet, J.; Thibault, G.; Vié, H. Antibody-dependent cellular cytotoxicity (ADCC) is mediated by genetically modified antigen-specific human T lymphocytes. Blood 2006, 107, 4669–4677. [Google Scholar] [CrossRef]
- Ochi, F.; Fujiwara, H.; Tanimoto, K.; Asai, H.; Miyazaki, Y.; Okamoto, S.; Mineno, J.; Kuzushima, K.; Shiku, H.; Barrett, J.; et al. Gene-modified human α/β-T cells expressing a chimeric CD16-CD3ζ receptor as adoptively transferable effector cells for anticancer monoclonal antibody therapy. Cancer Immunol. Res. 2014, 2, 249–262. [Google Scholar] [CrossRef]
- Tanaka, H.; Fujiwara, H.; Ochi, F.; Tanimoto, K.; Casey, N.; Okamoto, S.; Mineno, J.; Kuzushima, K.; Shiku, H.; Sugiyama, T.; et al. Development of Engineered T Cells Expressing a Chimeric CD16-CD3ζ Receptor to Improve the Clinical Efficacy of Mogamulizumab Therapy Against Adult T-Cell Leukemia. Clin. Cancer Res. 2016, 22, 4405–4416. [Google Scholar] [CrossRef]
- Kudo, K.; Imai, C.; Lorenzini, P.; Kamiya, T.; Kono, K.; Davidoff, A.M.; Chng, W.J.; Campana, D. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. 2014, 74, 93–103. [Google Scholar] [CrossRef]
- Rataj, F.; Jacobi, S.J.; Stoiber, S.; Asang, F.; Ogonek, J.; Tokarew, N.; Cadilha, B.L.; van Puijenbroek, E.; Heise, C.; Duewell, P.; et al. High-affinity CD16-polymorphism and Fc-engineered antibodies enable activity of CD16-chimeric antigen receptor-modified T cells for cancer therapy. Br. J. Cancer 2019, 120, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Caratelli, S.; Arriga, R.; Sconocchia, T.; Ottaviani, A.; Lanzilli, G.; Pastore, D.; Cenciarelli, C.; Venditti, A.; Del Principe, M.I.; Lauro, D.; et al. In vitro elimination of epidermal growth factor receptor-overexpressing cancer cells by CD32A-chimeric receptor T cells in combination with cetuximab or panitumumab. Int. J. Cancer 2020, 146, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Goulet, D.R.; Atkins, W.M. Considerations for the Design of Antibody-Based Therapeutics. J. Pharm. Sci. 2020, 109, 74–103. [Google Scholar] [CrossRef] [PubMed]
- Strohl, W.R. Current progress in innovative engineered antibodies. Protein Cell. 2018, 9, 86–120. [Google Scholar] [CrossRef]
- Urbanska, K.; Lanitis, E.; Poussin, M.; Lynn, R.; Gavin, B.P.; Kelderman, S.; Yu, J.; Scholler, N.; Powell, D.J., Jr. A universal strategy for adoptive immunotherapy of cancer through use of a novel T cell antigen receptor. Cancer Res. 2012, 72, 1844–1852. [Google Scholar] [CrossRef]
- Lohmueller, J.J.; Ham, J.D.; Kvorjak, M.; Finn, O.J. mSA2 affinity-enhanced biotin-binding CAR T cells for universal tumor targeting. Oncoimmunology 2017, 7, e1368604. [Google Scholar] [CrossRef]
- Stratton, S.L.; Horvath, T.D.; Bogusiewicz, A.; Matthews, N.I.; Henrich, C.L.; Spencer, H.J.; Moran, J.H.; Mock, D.M. Plasma concentration of 3-hydroxyisovaleryl carnitine is an early and sensitive indicator of marginal biotin deficiency in humans. Am. J. Clin. Nutr. 2010, 92, 1399–1405. [Google Scholar] [CrossRef]
- Dale, G.L.; Gaddy, P.; Pikul, F.J. Antibodies against biotinylated proteins are present in normal human serum. J. Lab. Clin. Med. 1994, 123, 365–371. [Google Scholar]
- Paganelli, G.; Magnani, P.; Zito, F.; Villa, E.; Sudati, F.; Lopalco, L.; Rossetti, C.; Malcovati, M.; Chiolerio, F.; Seccamani, E.; et al. Three-step monoclonal antibody tumor targeting in carcinoembryonic antigen-positive patients. Cancer Res. 1991, 51, 5960–5966. [Google Scholar]
- Paganelli, G.; Chinol, M.; Maggiolo, M.; Sidoli, A.; Corti, A.; Baroni, S.; Siccardi, A.G. The three-step pretargeting approach reduces the human anti-mouse antibody response in patients submitted to radioimmunoscintigraphy and radioimmunotherapy. Eur. J. Nucl. Med. 1997, 24, 350–351. [Google Scholar] [CrossRef]
- Tamada, K.; Geng, D.; Sakoda, Y.; Bansal, N.; Srivastava, R.; Li, Z.; Davila, E. Redirecting gene-modified T cells toward various cancer types using tagged antibodies. Clin. Cancer Res. 2012, 18, 6436–6445. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Ma, J.S.; Yun, H.; Cao, Y.; Kim, J.Y.; Chi, V.; Wang, D.; Woods, A.; Sherwood, L.; Caballero, D.; et al. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J. Am. Chem. Soc. 2015, 137, 2832–2835. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Rodgers, D.T.; Du, J.; Ahmad, I.; Hampton, E.N.; Ma, J.S.; Mazagova, M.; Choi, S.H.; Yun, H.Y.; Xiao, H.; et al. Design of Switchable Chimeric Antigen Receptor T Cells Targeting Breast Cancer. Angew. Chem. Int. Ed. Engl. 2016, 55, 7520–7524. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.S.; Kim, J.Y.; Kazane, S.A.; Choi, S.H.; Yun, H.Y.; Kim, M.S.; Rodgers, D.T.; Pugh, H.M.; Singer, O.; Sun, S.B.; et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E450–E458. [Google Scholar] [CrossRef]
- Zhang, E.; Gu, J.; Xue, J.; Lin, C.; Liu, C.; Li, M.; Hao, J.; Setrerrahmane, S.; Chi, X.; Qi, W.; et al. Accurate control of dual-receptor-engineered T cell activity through a bifunctional anti-angiogenic peptide. J. Hematol. Oncol. 2018, 11, 44. [Google Scholar] [CrossRef]
- Chu, W.; Zhou, Y.; Tang, Q.; Wang, M.; Ji, Y.; Yan, J.; Yin, D.; Zhang, S.; Lu, H.; Shen, J. Bi-specific ligand-controlled chimeric antigen receptor T-cell therapy for non-small cell lung cancer. Biosci. Trends 2018, 12, 298–308. [Google Scholar] [CrossRef]
- Lee, Y.G.; Chu, H.; Lu, Y.; Leamon, C.P.; Srinivasarao, M.; Putt, K.S.; Low, P.S. Regulation of CAR T cell-mediated cytokine release syndrome-like toxicity using low molecular weight adapters. Nat. Commun. 2019, 10, 2681. [Google Scholar] [CrossRef]
- Lee, Y.G.; Marks, I.; Srinivasarao, M.; Kanduluru, A.K.; Mahalingam, S.M.; Liu, X.; Chu, H.; Low, P.S. Use of a Single CAR T Cell and Several Bispecific Adapters Facilitates Eradication of Multiple Antigenically Different Solid Tumors. Cancer Res. 2019, 79, 387–396. [Google Scholar] [CrossRef]
- Lu, Y.J.; Chu, H.; Wheeler, L.W.; Nelson, M.; Westrick, E.; Matthaei, J.F.; Cardle, I.I.; Johnson, A.; Gustafson, J.; Parker, N.; et al. Preclinical Evaluation of Bispecific Adaptor Molecule Controlled Folate Receptor CAR-T Cell Therapy With Special Focus on Pediatric Malignancies. Front. Oncol. 2019, 9, 151. [Google Scholar] [CrossRef]
- Van Dam, G.M.; Themelis, G.; Crane, L.M.; Harlaar, N.J.; Pleijhuis, R.G.; Kelder, W.; Sarantopoulos, A.; de Jong, J.S.; Arts, H.J.; van der Zee, A.G.; et al. Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-α targeting: First in-human results. Nat. Med. 2011, 17, 1315–1319. [Google Scholar] [CrossRef]
- Koristka, S.; Cartellieri, M.; Feldmann, A.; Arndt, C.; Loff, S.; Michalk, I.; Aliperta, R.; von Bonin, M.; Bornhäuser, M.; Ehninger, A.; et al. Flexible antigen-specific redirection of human regulatory T cells via a novel universal chimeric antigen receptor system. Blood 2014, 124, 3494. [Google Scholar] [CrossRef]
- Cartellieri, M.; Loff, S.; von Bonin, M.; Bejestani, E.P.; Ehninger, A.; Feldmann, A.; Koristka, S.; Arndt, C.; Ehninger, G.; Bachmann, M.P. Unicar: A Novel Modular Retargeting Platform Technology for CAR T Cells. Blood 2015, 126, 5549. [Google Scholar] [CrossRef]
- Carmo-Fonseca, M.; Pfeifer, K.; Schröder, H.C.; Vaz, M.F.; Fonseca, J.E.; Müller, W.E.G.; Bachmann, M. Identification of La ribonucleoproteins as a component of interchromatin granules. Exp. Cell. Res. 1989, 185, 73–85. [Google Scholar] [CrossRef]
- Koristka, S.; Cartellieri, M.; Arndt, C.; Bippes, C.C.; Feldmann, A.; Michalk, I.; Wiefel, K.; Stamova, S.; Schmitz, M.; Ehninger, G.; et al. Retargeting of regulatory T cells to surface-inducible autoantigen La/SS-B. J. Autoimmun. 2013, 42, 105–116. [Google Scholar] [CrossRef]
- Cartellieri, M.; Feldmann, A.; Koristka, S.; Arndt, C.; Loff, S.; Ehninger, A.; von Bonin, M.; Bejestani, E.P.; Ehninger, G.; Bachmann, M.P. Switching CAR T cells on and off: A novel modular platform for retargeting of T cells to AML blasts. Blood Cancer J. 2016, 6, e458. [Google Scholar] [CrossRef]
- Feldmann, A.; Arndt, C.; Bergmann, R.; Loff, S.; Cartellieri, M.; Bachmann, D.; Aliperta, R.; Hetzenecker, M.; Ludwig, F.; Albert, S.; et al. Retargeting of T lymphocytes to PSCA- or PSMA positive prostate cancer cells using the novel modular chimeric antigen receptor platform technology "UniCAR". Oncotarget 2017, 8, 31368–31385. [Google Scholar] [CrossRef]
- Albert, S.; Arndt, C.; Feldmann, A.; Bergmann, R.; Bachmann, D.; Koristka, S.; Ludwig, F.; Ziller-Walter, P.; Kegler, A.; Gärtner, S.; et al. A novel nanobody-based target module for retargeting of T lymphocytes to EGFR-expressing cancer cells via the modular UniCAR platform. Oncoimmunology 2017, 6, e1287246. [Google Scholar] [CrossRef]
- Mitwasi, N.; Feldmann, A.; Bergmann, R.; Berndt, N.; Arndt, C.; Koristka, S.; Kegler, A.; Jureczek, J.; Hoffmann, A.; Ehninger, A.; et al. Development of novel target modules for retargeting of UniCAR T cells to GD2 positive tumor cells. Oncotarget 2017, 8, 108584–108603. [Google Scholar] [CrossRef]
- Bachmann, D.; Aliperta, R.; Bergmann, R.; Feldmann, A.; Koristka, S.; Arndt, C.; Loff, S.; Welzel, P.; Albert, S.; Kegler, A.; et al. Retargeting of UniCAR T cells with an in vivo synthesized target module directed against CD19 positive tumor cells. Oncotarget 2018, 9, 7487–7500. [Google Scholar] [CrossRef]
- Albert, S.; Arndt, C.; Koristka, S.; Berndt, N.; Bergmann, R.; Feldmann, A.; Schmitz, M.; Pietzsch, J.; Steinbach, J.; Bachmann, M. From mono- to bivalent: Improving theranostic properties of target modules for redirection of UniCAR T cells against EGFR-expressing tumor cells in vitro and in vivo. Oncotarget 2018, 9, 25597–25616. [Google Scholar] [CrossRef]
- Loureiro, L.R.; Feldmann, A.; Bergmann, R.; Koristka, S.; Berndt, N.; Arndt, C.; Pietzsch, J.; Novo, C.; Videira, P.; Bachmann, M. Development of a novel target module redirecting UniCAR T cells to Sialyl Tn-expressing tumor cells. Blood Cancer J. 2018, 8, 81. [Google Scholar] [CrossRef]
- Arndt, C.; Feldmann, A.; Koristka, S.; Schäfer, M.; Bergmann, R.; Mitwasi, N.; Berndt, N.; Bachmann, D.; Kegler, A.; Schmitz, M.; et al. A theranostic PSMA ligand for PET imaging and retargeting of T cells expressing the universal chimeric antigen receptor UniCAR. OncoImmunology 2019, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Arndt, C.; Loureiro, L.R.; Feldmann, A.; Jureczek, J.; Bergmann, R.; Máthé, D.; Hegedüs, N.; Berndt, N.; Koristka, S.; Mitwasi, N.; et al. UniCAR T cell immunotherapy enables efficient elimination of radioresistant cancer cells. OncoImmunology 2020, 9, 1743036. [Google Scholar] [CrossRef]
- Mitwasi, N.; Feldmann, A.; Arndt, C.; Koristka, S.; Berndt, N.; Jureczek, J.; Loureiro, L.R.; Bergmann, R.; Máthé, D.; Hegedüs, N.; et al. “UniCAR”-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells. Sci. Rep. 2020, 10, 2141. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, L.R.; Feldmann, A.; Bergmann, R.; Koristka, S.; Berndt, N.; Máthé, D.; Hegedüs, N.; Szigeti, K.; Videira, P.A.; Bachmann, M.; et al. Extended half-life target module for sustainable UniCAR T-cell treatment of STn-expressing cancers. J. Exp. Clin. Cancer Res. 2020, 39, 77. [Google Scholar] [CrossRef]
- Koristka, S.; Ziller-Walter, P.; Bergmann, R.; Arndt, C.; Feldmann, A.; Kegler, A.; Cartellieri, M.; Ehninger, A.; Ehninger, G.; Bornhäuser, M.; et al. Anti-CAR-engineered T cells for epitope-based elimination of autologous CAR T cells. Cancer Immunol. Immunother. 2019, 68, 1401–1415. [Google Scholar] [CrossRef] [PubMed]
- Tröster, H.; Metzger, T.E.; Semsei, I.; Schwemmle, M.; Winterpacht, A.; Zabel, B.; Bachmann, M. One gene, two transcripts: Isolation of an alternative transcript encoding for the autoantigen La/SS-B from a cDNA library of a patient with primary Sjögrens’ syndrome. J. Exp. Med. 1994, 180, 2059–2067. [Google Scholar] [CrossRef]
- Yiannaki, E.E.; Tzioufas, A.G.; Bachmann, M.; Hantoumi, J.; Tsikaris, V.; Sakarellos-Daitsiotis, M.; Sakarellos, C.; Moutsopoulos, H.M. The value of synthetic linear epitope analogues of La/SSB for the detection of autoantibodies to La/SSB; specificity, sensitivity and comparison of methods. Clin. Exp. Immunol. 1998, 112, 152–158. [Google Scholar] [CrossRef]
- Tröster, H.; Bartsch, H.; Klein, R.; Metzger, T.E.; Pollak, G.; Semsei, I.; Schwemmle, M.; Pruijn, G.J.; van Venrooij, W.J.; Bachmann, M. Activation of a murine autoreactive B cell by immunization with human recombinant autoantigen La/SS-B: Characterization of the autoepitope. J. Autoimmun. 1995, 8, 825–842. [Google Scholar] [CrossRef][Green Version]
- Dudek, N.L.; Maier, S.; Chen, Z.J.; Mudd, P.A.; Mannering, S.I.; Jackson, D.C.; Zeng, W.; Keech, C.L.; Hamlin, K.; Pan, Z.J.; et al. T cell epitopes of the La/SSB autoantigen in humanized transgenic mice expressing the HLA class II haplotype DRB1*0301/DQB1*0201. Arthritis. Rheum. 2007, 56, 3387–3398. [Google Scholar] [CrossRef]
- Bachmann, M. The UniCAR system: A modular CAR T cell approach to improve the safety of CAR T cells. Immunol. Lett. 2019, 211, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, D.T.; Mazagova, M.; Hampton, E.N.; Cao, Y.; Ramadoss, N.S.; Hardy, I.R.; Schulman, A.; Du, J.; Wang, F.; Singer, O.; et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc. Natl. Acad. Sci. USA 2016, 113, E459–E468. [Google Scholar] [CrossRef] [PubMed]
- Raj, D.; Yang, M.H.; Rodgers, D.; Hampton, E.N.; Begum, J.; Mustafa, A.; Lorizio, D.; Garces, I.; Propper, D.; Kench, J.G.; et al. Switchable CAR-T cells mediate remission in metastatic pancreatic ductal adenocarcinoma. Gut 2019, 68, 1052–1064. [Google Scholar] [CrossRef] [PubMed]
- Viaud, S.; Ma, J.S.Y.; Hardy, I.R.; Hampton, E.N.; Benish, B.; Sherwood, L.; Nunez, V.; Ackerman, C.J.; Khialeeva, E.; Weglarz, M.; et al. Switchable control over in vivo CAR T expansion, B cell depletion, and induction of memory. Proc. Natl. Acad. Sci. USA 2018, 115, E10898–E10906. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Collins, J.J.; Wong, W.W. Universal Chimeric Antigen Receptors for Multiplexed and Logical Control of T Cell Responses. Cell 2018, 173, 1426–1438. [Google Scholar] [CrossRef]
- Minutolo, N.G.; Sharma, P.; Poussin, M.; Shaw, L.C.; Brown, D.P.; Hollander, E.E.; Smole, A.; Rodriguez-Garcia, A.; Hui, J.Z.; Zappala, F.; et al. Quantitative Control of Gene-Engineered T-Cell Activity through the Covalent Attachment of Targeting Ligands to a Universal Immune Receptor. J. Am. Chem. Soc. 2020, 142, 6554–6568. [Google Scholar] [CrossRef]
- Li, L.; Fierer, J.O.; Rapoport, T.A.; Howarth, M. Structural analysis and optimization of the covalent association between SpyCatcher and a peptide Tag. J. Mol. Biol. 2014, 426, 309–317. [Google Scholar] [CrossRef]
- Stamova, S.; Koristka, S.; Keil, J.; Arndt, C.; Feldmann, A.; Michalk, I.; Bartsch, H.; Bippes, C.C.; Schmitz, M.; Cartellieri, M.; et al. Cancer immunotherapy by retargeting of immune effector cells via recombinant bispecific antibody constructs. Antibodies 2012, 1, 172–198. [Google Scholar] [CrossRef]
- Urbanska, K.; Lynn, R.C.; Stashwick, C.; Thakur, A.; Lum, L.G.; Powell, D.J., Jr. Targeted cancer immunotherapy via combination of designer bispecific antibody and novel gene-engineered T cells. J. Transl. Med. 2014, 12, 347. [Google Scholar] [CrossRef]
- Karches, C.H.; Benmebarek, M.R.; Schmidbauer, M.L.; Kurzay, M.; Klaus, R.; Geiger, M.; Rataj, F.; Cadilha, B.L.; Lesch, S.; Heise, C.; et al. Bispecific Antibodies Enable Synthetic Agonistic Receptor-Transduced T Cells for Tumor Immunotherapy. Clin. Cancer Res. 2019, 25, 5890–5900. [Google Scholar] [CrossRef]
- Ambrose, C.; Su, L.; Wu, L.; Lobb, R.R.; Rennert, P.D. Abstract 3768: CAR T cells specific for CD19 can be redirected to kill CD19 negative tumors. Cancer Res. 2017, 77, 3768. [Google Scholar] [CrossRef]
- Klesmith, J.R.; Su, L.; Wu, L.; Schrack, I.A.; Dufort, F.J.; Birt, A.; Ambrose, C.; Hackel, B.J.; Lobb, R.R.; Rennert, P.D. Retargeting CD19 Chimeric Antigen Receptor T Cells via Engineered CD19-Fusion Proteins. Mol. Pharm. 2019, 16, 3544–3558. [Google Scholar] [CrossRef] [PubMed]
- Rennert, P.; Su, L.; Dufort, F.; Birt, A.; Sanford, T.; Wu, L.; Ambrose, C.; Lobb, R. A Novel CD19-Anti-CD20 Bridging Protein Prevents and Reverses CD19-Negative Relapse from CAR19 T Cell Treatment In Vivo. Blood 2019, 134, 252. [Google Scholar] [CrossRef]
- Jureczek, J.; Bergmann, R.; Berndt, N.; Koristka, S.; Kegler, A.; Puentes-Cala, E.; Soto, J.A.; Arndt, C.; Bachmann, M.; Feldmann, A. An oligo-His-tag of a targeting module does not influence its biodistribution and the retargeting capabilities of UniCAR T cells. Sci. Rep. 2019, 9, 10547. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, R.J.; Jones, A.L. Fab antibody fragments: Some applications in clinical toxicology. Drug. Saf. 2004, 27, 1115–1133. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, E.Q.; Balthasar, J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2008, 84, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xu, L.C.; Parker, N.; Westrick, E.; Reddy, J.A.; Vetzel, M.; Low, P.S.; Leamon, C.P. Preclinical pharmacokinetics, tissue distribution, and antitumor activity of a folate-hapten conjugate-targeted immunotherapy in hapten-immunized mice. Mol. Cancer Ther. 2006, 5, 3258–3267. [Google Scholar] [CrossRef]
- Fasslrinner, F.; Arndt, C.; Koristka, S.; Feldmann, A.; Altmann, H.; von Bonin, M.; Schmitz, M.; Bornhäuser, M.; Bachmann, M. Midostaurin abrogates CD33-directed UniCAR and CD33-CD3 bispecific antibody therapy in acute myeloid leukaemia. Br. J. Haematol. 2019, 186, 735–740. [Google Scholar] [CrossRef]
- Ghosh, A.; Heston, W.D. Tumor target prostate specific membrane antigen (PSMA) and its regulation in prostate cancer. J. Cell. Biochem. 2004, 91, 528–539. [Google Scholar] [CrossRef]
- Silver, D.A.; Pellicer, I.; Fair, W.R.; Heston, W.D.; Cordon-Cardo, C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997, 3, 81–85. [Google Scholar]
- Lal, A.; Peters, H.; St Croix, B.; Haroon, Z.A.; Dewhirst, M.W.; Strausberg, R.L.; Kaanders, J.H.; van der Kogel, A.J.; Riggins, G.J. Transcriptional response to hypoxia in human tumors. J. Natl. Cancer Inst. 2001, 93, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Puig-Kröger, A.; Sierra-Filardi, E.; Domínguez-Soto, A.; Samaniego, R.; Corcuera, M.T.; Gómez-Aguado, F.; Ratnam, M.; Sánchez-Mateos, P.; Corbí, A.L. Folate receptor beta is expressed by tumor-associated macrophages and constitutes a marker for M2 anti-inflammatory/regulatory macrophages. Cancer Res. 2009, 69, 9395–9403. [Google Scholar] [CrossRef] [PubMed]
- Kegler, A.; Koristka, S.; Bergmann, R.; Berndt, N.; Arndt, C.; Feldmann, A.; Hoffmann, A.; Bornhäuser, M.; Schmitz, M.; Bachmann, M.P. T cells engrafted with a UniCAR 28/z outperform UniCAR BB/z-transduced T cells in the face of regulatory T cell-mediated immunosuppression. Oncoimmunology 2019, 8, e1621676. [Google Scholar] [CrossRef] [PubMed]
- Loff, S.; Meyer, J.-E.; Dietrich, J.; Spehr, J.; Riewaldt, J.; von Bonin, M.; Gründer, C.; Franke, K.; Feldmann, A.; Bachmann, M.; et al. Late-Stage Preclinical Characterization of Switchable CD123-Specific CAR-T for Treatment of Acute Leukemia. Blood 2018, 132, 964. [Google Scholar] [CrossRef]
- Minutolo, N.G.; Hollander, E.E.; Powell, D.J., Jr. The Emergence of Universal Immune Receptor T Cell Therapy for Cancer. Front. Oncol. 2019, 9, 176. [Google Scholar] [CrossRef]
- FDA Places Clinical Hold on Unum Therapeutics Phase 1 Trial Evaluating ACTR087 for Relapsed/Refractory CD20+ B Cell Non-Hodgkin Lymphoma. Available online: https://www.trialsitenews.com/fda-places-clinical-hold-on-unum-therapeutics-phase-1-trial-evaluating-actr087-for-relapsed-refractory-cd20-b-cell-non-hodgkin-lymphoma/ (accessed on 27 April 2020).
- Unum Therapeutics Provides Updates to its Phase 1 Trial of ACTR707 for HER2+ Solid Tumor Cancers. Available online: https://www.globenewswire.com/news-release/2020/01/29/1976692/0/en/Unum-Therapeutics-Provides-Updates-to-its-Phase-1-Trial-of-ACTR707-for-HER2-Solid-Tumor-Cancers.html (accessed on 27 April 2020).
- AbbVie & Scripps-based Calibr Moves Novel ‘Switchable’ CAR-T Technology to Phase I Clinical Trial. Available online: https://www.trialsitenews.com/abbvie-scripps-based-calibr-moves-novel-switchable-car-t-technology-to-phase-i-clinical-trial/ (accessed on 27 April 2020).
- Endocyte Provides Third Quarter 2018 Financial Results and Operational Update. Available online: https://www.globenewswire.com/news-release/2018/11/07/1647347/0/en/Endocyte-Provides-Third-Quarter-2018-Financial-Results-and-Operational-Update.html (accessed on 27 April 2020).
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Clémenceau, B.; Valsesia-Wittmann, S.; Jallas, A.C.; Vivien, R.; Rousseau, R.; Marabelle, A.; Caux, C.; Vié, H. In Vitro and In Vivo Comparison of Lymphocytes Transduced with a Human CD16 or with a Chimeric Antigen Receptor Reveals Potential Off-Target Interactions due to the IgG2 CH2-CH3 CAR-Spacer. J. Immunol. Res. 2015, 2015, 482089. [Google Scholar] [CrossRef]
- Tonn, T.; Schwabe, D.; Klingemann, H.G.; Becker, S.; Esser, R.; Koehl, U.; Suttorp, M.; Seifried, E.; Ottmann, O.G.; Bug, G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy 2013, 15, 1563–1570. [Google Scholar] [CrossRef]
- Arai, S.; Meagher, R.; Swearingen, M.; Myint, H.; Rich, E.; Martinson, J.; Klingemann, H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: A phase I trial. Cytotherapy 2008, 10, 625–632. [Google Scholar] [CrossRef]
- Williams, B.A.; Law, A.D.; Routy, B.; denHollander, N.; Gupta, V.; Wang, X.H.; Chaboureau, A.; Viswanathan, S.; Keating, A. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget 2017, 8, 89256–89268. [Google Scholar] [CrossRef]
- Boyiadzis, M.; Agha, M.; Redner, R.L.; Sehgal, A.; Im, A.; Hou, J.Z.; Farah, R.; Dorritie, K.A.; Raptis, A.; Lim, S.H.; et al. Phase 1 clinical trial of adoptive immunotherapy using “off-the-shelf” activated natural killer cells in patients with refractory and relapsed acute myeloid leukemia. Cytotherapy 2017, 19, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Andreesen, R.; Hennemann, B.; Krause, S.W. Adoptive immunotherapy of cancer using monocyte-derived macrophages: Rationale, current status, and perspectives. J. Leukoc. Biol. 1998, 64, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Burger, M.; Thiounn, N.; Denzinger, S.; Kondas, J.; Benoit, G.; Chapado, M.S.; Jimenz-Cruz, F.J.; Kisbenedek, L.; Szabo, Z.; Zsolt, D.; et al. The application of adjuvant autologous antravesical macrophage cell therapy vs. BCG in non-muscle invasive bladder cancer: A multicenter, randomized trial. J. Transl. Med. 2010, 8, 54. [Google Scholar] [CrossRef]
- Brunstein, C.G.; Miller, J.S.; Cao, Q.; McKenna, D.H.; Hippen, K.L.; Curtsinger, J.; Defor, T.; Levine, B.L.; June, C.H.; Rubinstein, P.; et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: Safety profile and detection kinetics. Blood 2011, 117, 1061e1070. [Google Scholar] [CrossRef]
- Di Ianni, M.; Falzetti, F.; Carotti, A.; Terenzi, A.; Castellino, F.; Bonifacio, E.; Del Papa, B.; Zei, T.; Ostini, R.I.; Cecchini, D.; et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 2011, 117, 3921e3928. [Google Scholar] [CrossRef]
- Marek-Trzonkowska, N.; Myśliwiec, M.; Dobyszuk, A.; Grabowska, M.; Derkowska, I.; Juścińska, J.; Owczuk, R.; Szadkowska, A.; Witkowski, P.; Młynarski, W.; et al. Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets - results of one year follow-up. Clin. Immunol. 2014, 153, 23–30. [Google Scholar] [CrossRef]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef]
- Sicard, A.; Boardman, D.A.; Levings, M.K. Taking regulatory T-cell therapy one step further. Curr. Opin. Organ. Transplant. 2018, 23, 509–515. [Google Scholar] [CrossRef]
- Ferreira, L.M.R.; Muller, Y.D.; Bluestone, J.A.; Tang, Q. Next-generation regulatory T cell therapy. Nat. Rev. Drug Discov. 2019, 18, 749–769. [Google Scholar] [CrossRef]
- Koristka, S.; Kegler, A.; Bergmann, R.; Arndt, C.; Feldmann, A.; Albert, S.; Cartellieri, M.; Ehninger, A.; Ehninger, G.; Middeke, J.M.; et al. Engrafting human regulatory T cells with a flexible modular chimeric antigen receptor technology. J. Autoimmun. 2018, 90, 116–131. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Age Group (years) | Adaptor CAR | Adaptor Molecule | Location Countries | ClinicalTrial.Gov Reference Number |
|---|---|---|---|---|---|
| refractory or relapsed CD20pos B cell lymphoma | 18–75 | CD16-BB/ζ (ACTR087) | rituximab | United States | NCT02776813 (completed) 08/2016-02/2020 |
| refractory or relapsed CD20pos B cell lymphoma | 18–80 | CD16-28 (ACTR707) | rituximab | United States | NCT03189836 (active) start: 06/2017 |
| relapsed or refractory multiple myeloma | 18–80 | ACTR087 | SEA-BCMA | United States | NCT03266692 (terminated) 02/2018-10/2019 |
| HER2pos advanced malignancies | 18–75 | ACTR087 ACTR707 | trastuzumab | United States | NCT03680560 (terminated) 03/2019-03/2020 |
| B cell lymphomas multiple myeloma HER2pos solid tumors | ≥18 | ACTR087 ACTR707 | Trastuzumab rituximab SEA-BCMA | United States | NCT02840110 (enrolling by invitation) start: 10/2016 |
| CD123pos hematologic and lymphatic malignancies | ≥18 | UniCAR-28/ζ (UniCAR02-T) | CD123 TM (TM123) | Germany | NCT04230265 (recruiting) start: 01/2020 |
| hematologic malignancies | TBA | α-PNE CAR (CLBR001) | CD19 Fab switch (SWI019) | TBA | TBA start: 2020 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arndt, C.; Fasslrinner, F.; Loureiro, L.R.; Koristka, S.; Feldmann, A.; Bachmann, M. Adaptor CAR Platforms—Next Generation of T Cell-Based Cancer Immunotherapy. Cancers 2020, 12, 1302. https://doi.org/10.3390/cancers12051302
Arndt C, Fasslrinner F, Loureiro LR, Koristka S, Feldmann A, Bachmann M. Adaptor CAR Platforms—Next Generation of T Cell-Based Cancer Immunotherapy. Cancers. 2020; 12(5):1302. https://doi.org/10.3390/cancers12051302
Chicago/Turabian StyleArndt, Claudia, Frederick Fasslrinner, Liliana R. Loureiro, Stefanie Koristka, Anja Feldmann, and Michael Bachmann. 2020. "Adaptor CAR Platforms—Next Generation of T Cell-Based Cancer Immunotherapy" Cancers 12, no. 5: 1302. https://doi.org/10.3390/cancers12051302
APA StyleArndt, C., Fasslrinner, F., Loureiro, L. R., Koristka, S., Feldmann, A., & Bachmann, M. (2020). Adaptor CAR Platforms—Next Generation of T Cell-Based Cancer Immunotherapy. Cancers, 12(5), 1302. https://doi.org/10.3390/cancers12051302