Feasibility of Targeting Traf2-and-Nck-Interacting Kinase in Synovial Sarcoma

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
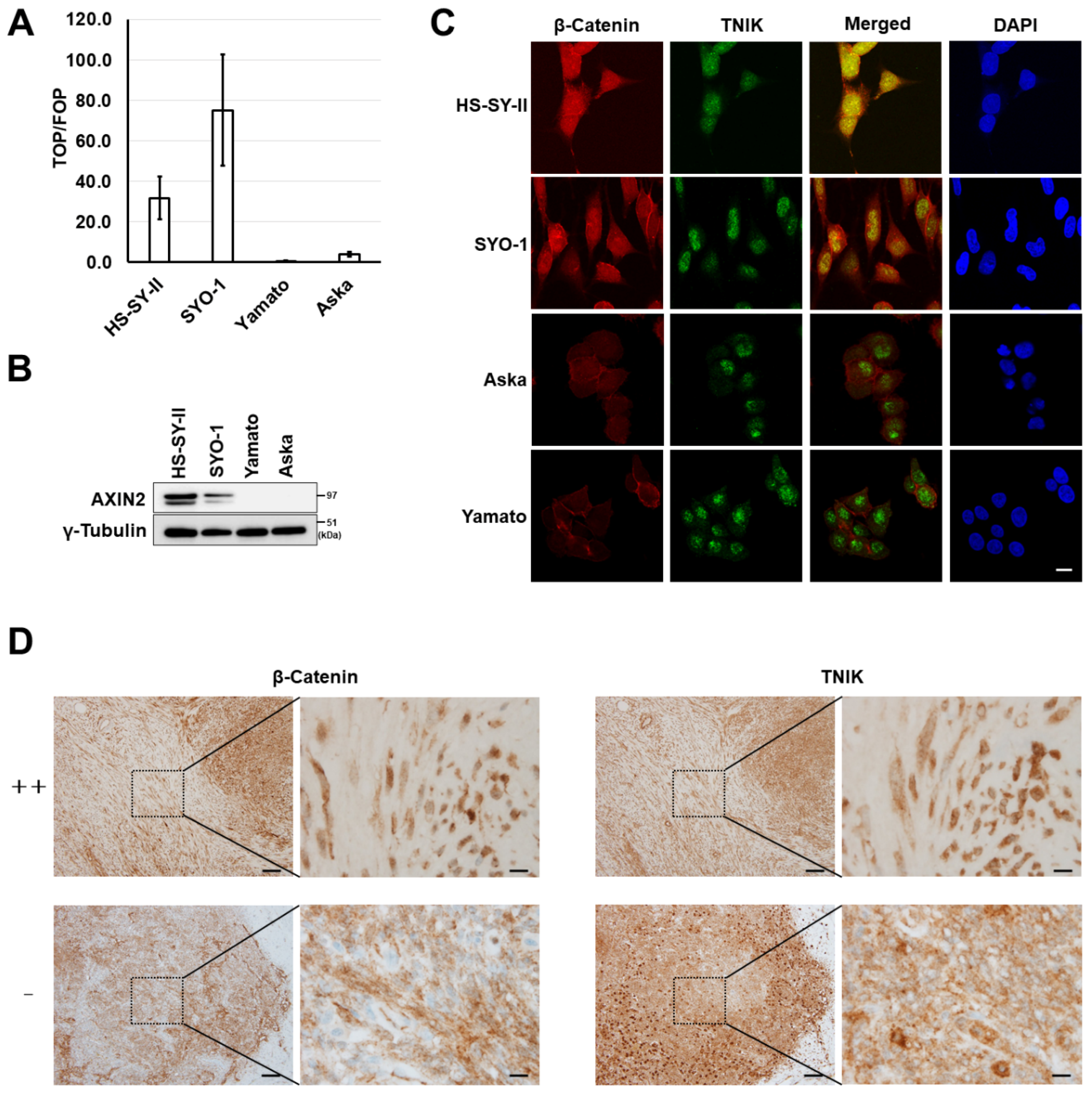
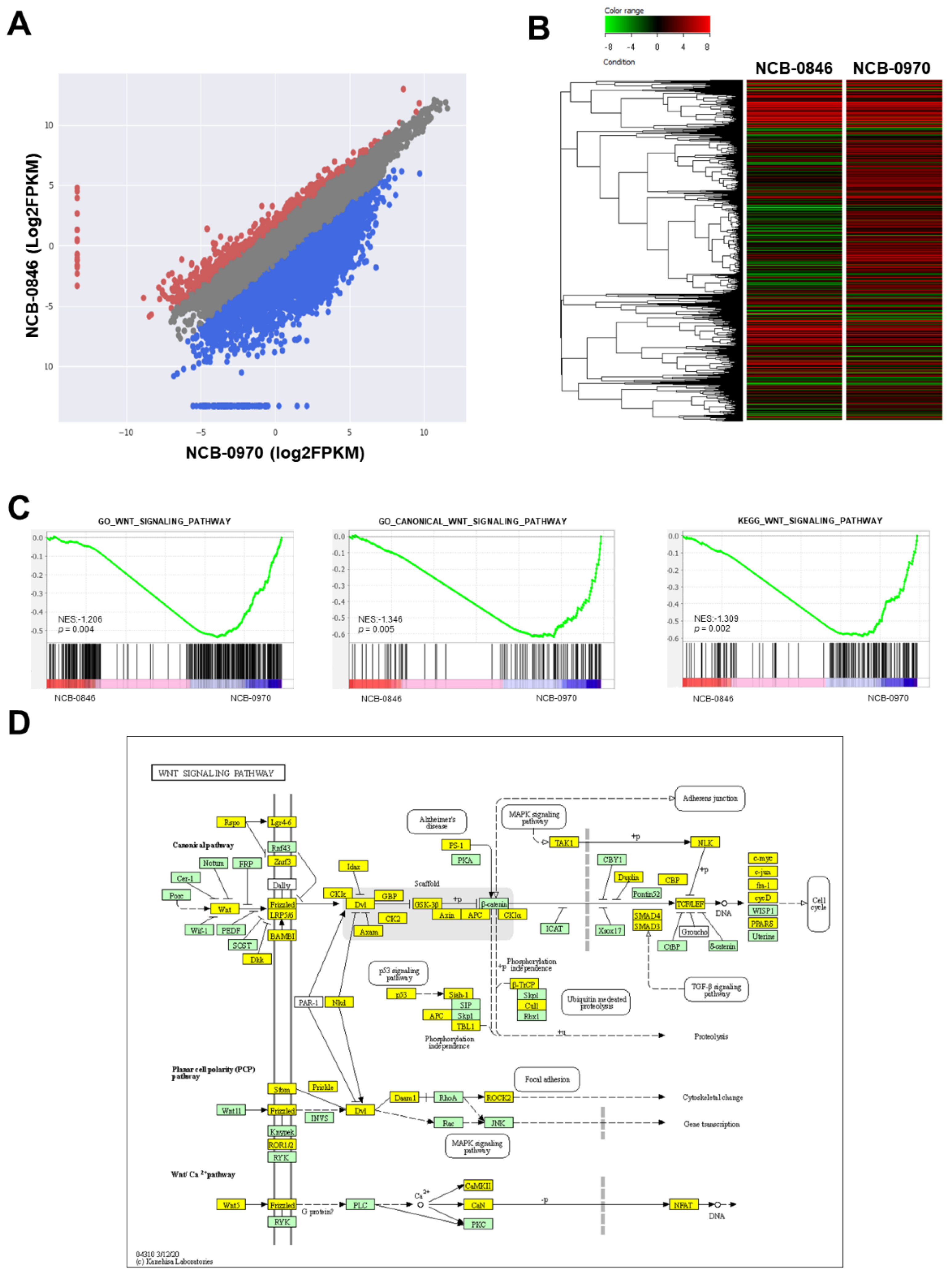
2.1. Activation of Wnt Signaling and TNIK in Synovial Sarcoma
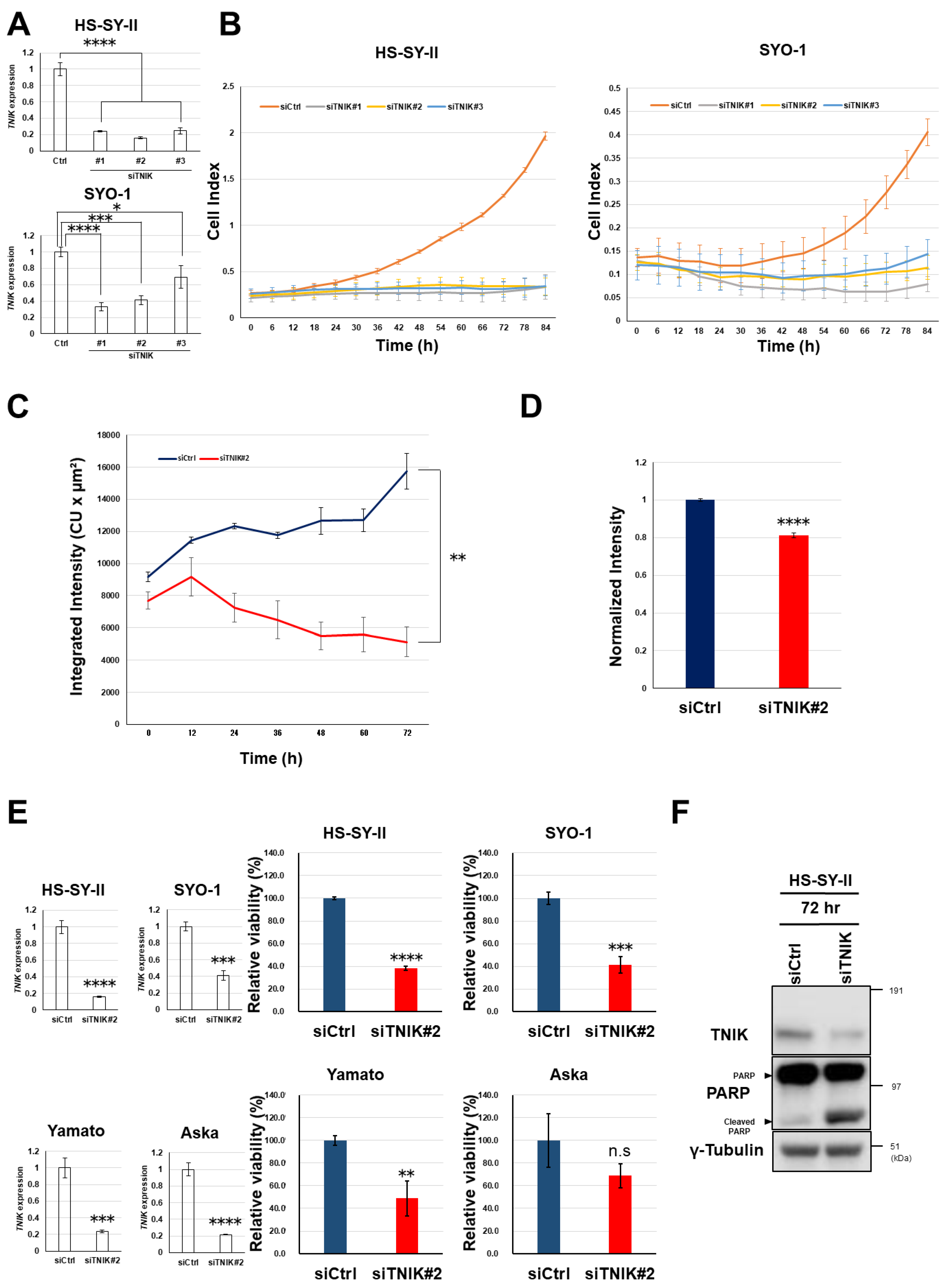
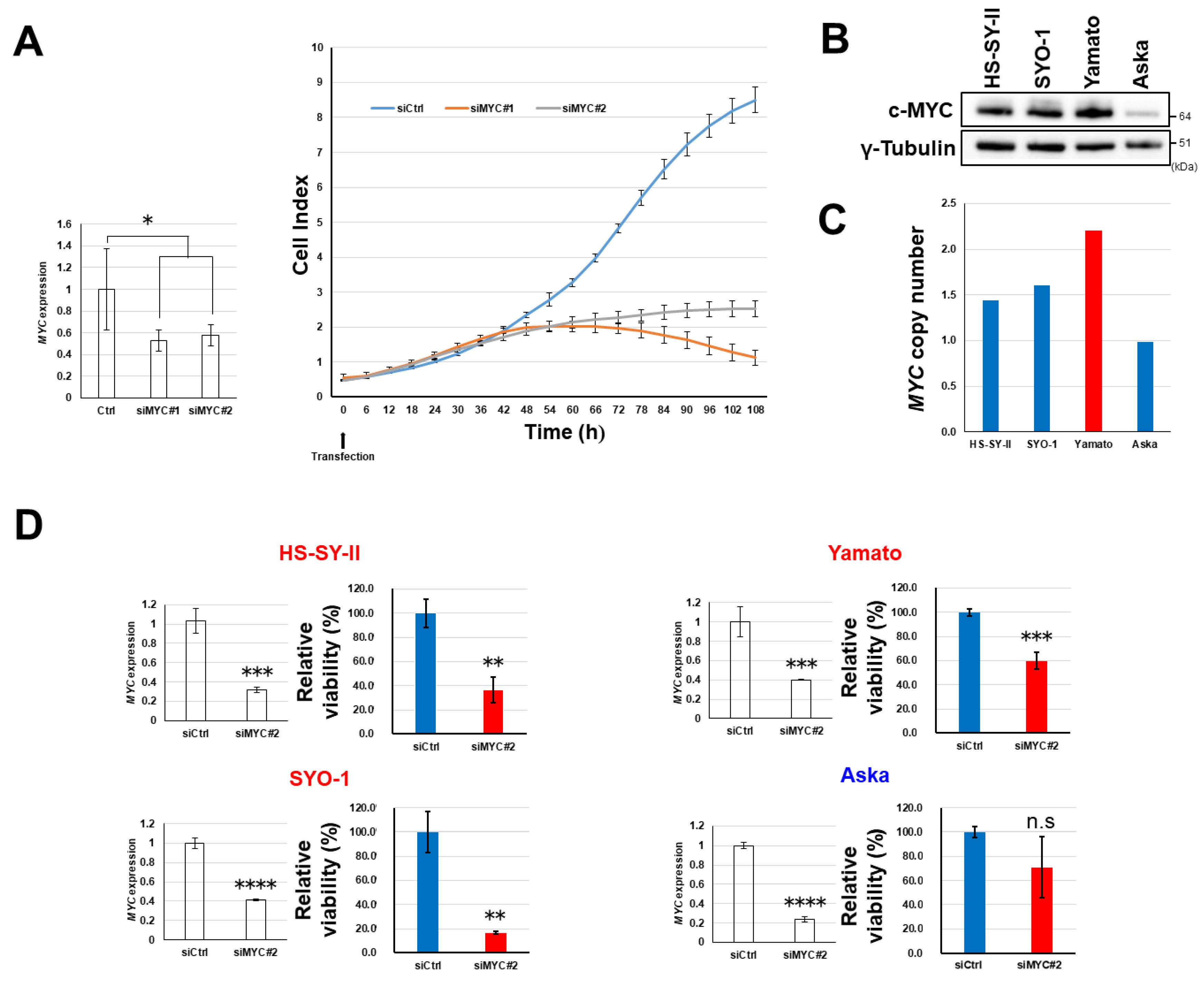
2.2. Growth Suppression of Synovial Sarcoma Cells Through Silencing of TNIK
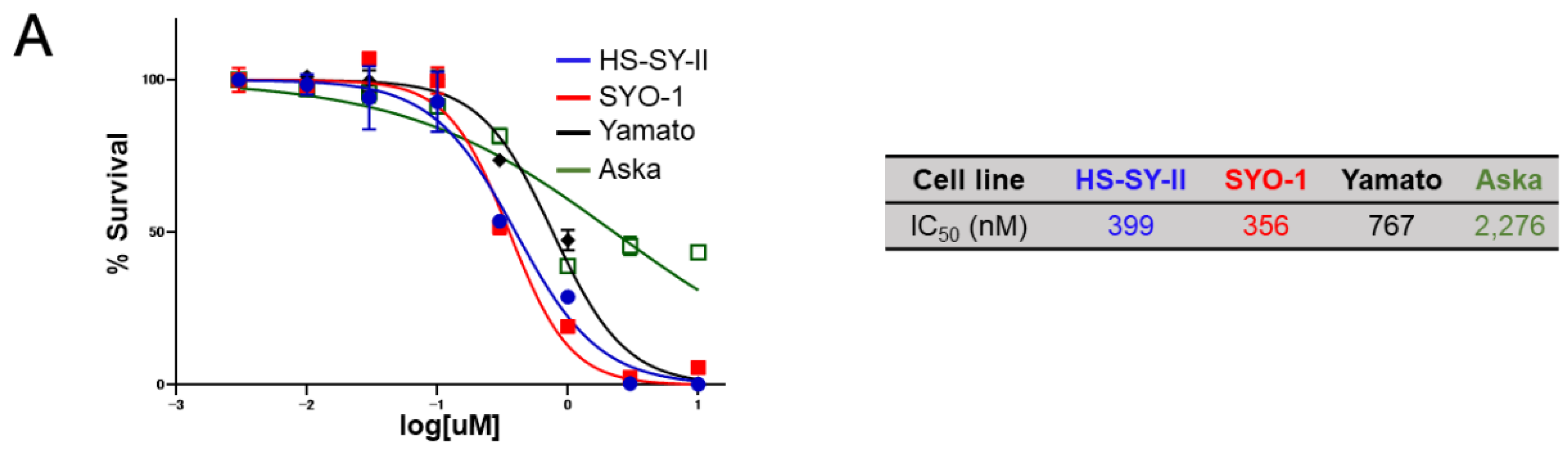
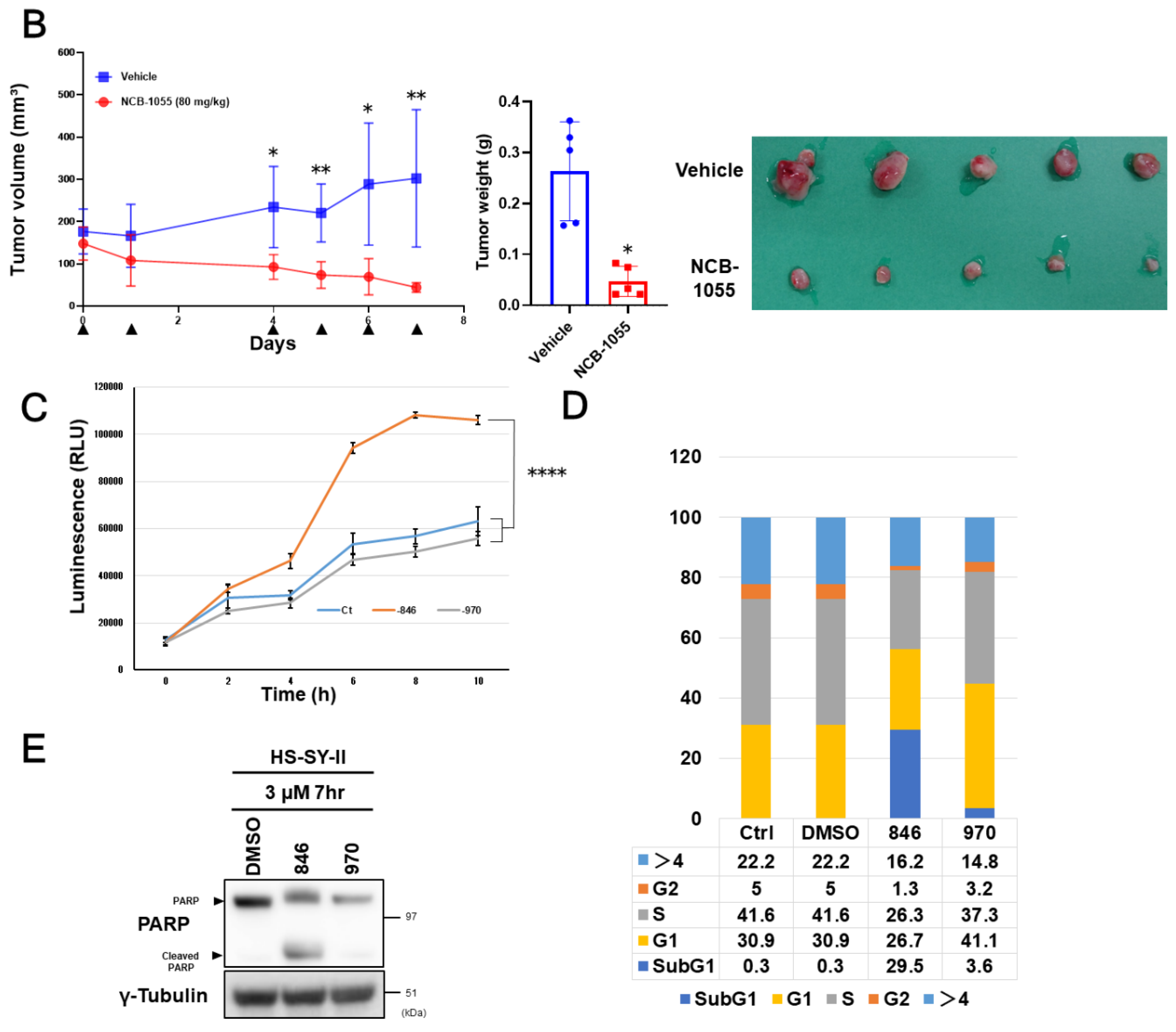
2.3. Sensitivity of Synovial Sarcoma to NCB-0846
2.4. Gene Expression Profiling
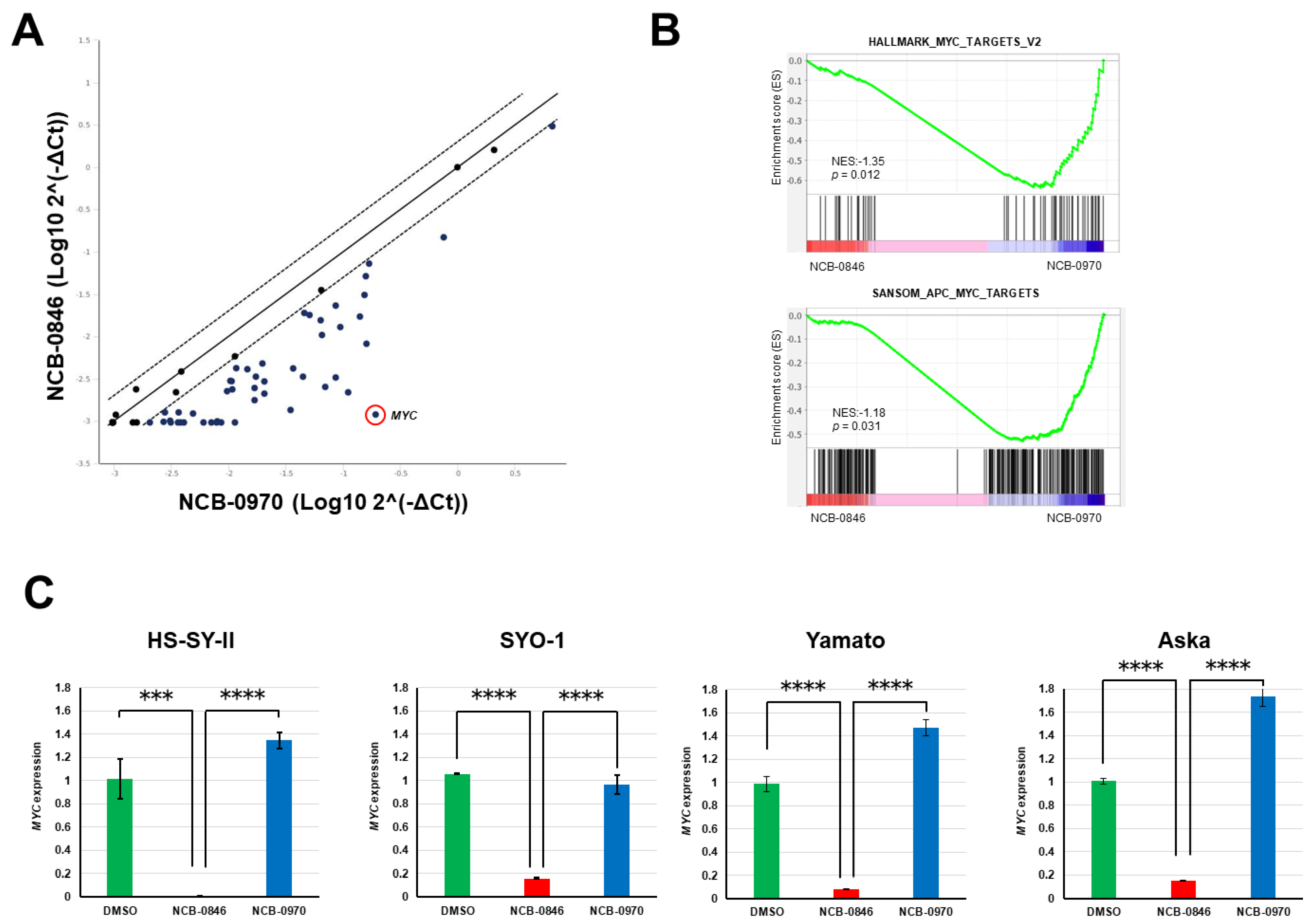
2.5. NCB-0846 Suppresses MYC Gene Expression
2.6. Dependency of Synovial Sarcoma Cells on MYC
3. Discussion
4. Materials and Methods
4.1. Ethical Issues
4.2. Cell Lines
4.3. Luciferase Reporter Assay
4.4. Antibodies
4.5. Immunoblot Analysis
4.6. Immunofluorescence Microscopy
4.7. Patients and Tumor Samples
4.8. Immunohistochemistry
4.9. Gene Silencing by RNA Interference
4.10. Real-Time RT-PCR
4.11. Digital PCR
4.12. Real-Time Cell Analysis (RTCA)
4.13. Real-Time Monitoring of Transcriptional Activity
4.14. Drug Sensitivity
4.15. Xenografts
4.16. Real-Time Monitoring of Apoptosis Induction
4.17. Cell Cycle Analysis
4.18. RNA Sequencing
4.19. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Herzog, C.E. Overview of sarcomas in the adolescent and young adult population. J. Pediatr. Hematol. Oncol. 2005, 27, 215–218. [Google Scholar] [CrossRef]
- Ferrari, A.; Dirksen, U.; Bielack, S. Sarcomas of Soft Tissue and Bone. Prog. Tumor Res. 2016, 43, 128–141. [Google Scholar]
- Ladanyi, M.; Antonescu, C.R.; Leung, D.H.; Woodruff, J.M.; Kawai, A.; Healey, J.H.; Brennan, M.F.; Bridge, J.A.; Neff, J.R.; Barr, F.G.; et al. Impact of SYT-SSX fusion type on the clinical behavior of synovial sarcoma: A multi-institutional retrospective study of 243 patients. Cancer Res. 2002, 62, 135–140. [Google Scholar]
- Al-Hussaini, H.; Hogg, D.; Blackstein, M.E.; O’Sullivan, B.; Catton, C.N.; Chung, P.W.; Griffin, A.M.; Hodgson, D.; Hopyan, S.; Kandel, R.; et al. Clinical features, treatment, and outcome in 102 adult and pediatric patients with localized high-grade synovial sarcoma. Sarcoma 2011, 2011, 231789. [Google Scholar] [CrossRef] [PubMed]
- Outani, H.; Kakunaga, S.; Hamada, K.; Takenaka, S.; Imura, Y.; Nagata, S.; Tanaka, T.; Tamiya, H.; Oshima, K.; Naka, N.; et al. Favorable outcomes of localized synovial sarcoma patients with a high utilization rate of neoadjuvant and/or adjuvant chemotherapy. Mol. Clin. Oncol. 2019, 11, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Sultan, I.; Rodriguez-Galindo, C.; Saab, R.; Yasir, S.; Casanova, M.; Ferrari, A. Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005: An analysis of 1268 patients. Cancer 2009, 115, 3537–3547. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.; Rocques, P.J.; Crew, A.J.; Gill, S.; Shipley, J.; Chan, A.M.; Gusterson, B.A.; Cooper, C.S. Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat. Genet. 1994, 7, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Mertens, F.; Isaksson, M.; Limon, J.; Gustafson, P.; Skytting, B.; Akerman, M.; Sciot, R.; Dal Cin, P.; Samson, I.; et al. Clinical impact of molecular and cytogenetic findings in synovial sarcoma. Genes Chromosomes Cancer 2001, 31, 362–372. [Google Scholar] [CrossRef]
- Carmody Soni, E.E.; Schlottman, S.; Erkizan, H.V.; Uren, A.; Toretsky, J.A. Loss of SS18-SSX1 inhibits viability and induces apoptosis in synovial sarcoma. Clin. Orthop. Relat. Res. 2014, 472, 874–882. [Google Scholar] [CrossRef]
- McBride, M.J.; Pulice, J.L.; Beird, H.C.; Ingram, D.R.; D’Avino, A.R.; Shern, J.F.; Charville, G.W.; Hornick, J.L.; Nakayama, R.T.; Garcia-Rivera, E.M.; et al. The SS18-SSX Fusion Oncoprotein Hijacks BAF Complex Targeting and Function to Drive Synovial Sarcoma. Cancer Cell 2018, 33, 1128–1141. [Google Scholar] [CrossRef]
- Banito, A.; Li, X.; Laporte, A.N.; Roe, J.S.; Sanchez-Vega, F.; Huang, C.H.; Dancsok, A.R.; Hatzi, K.; Chen, C.C.; Tschaharganeh, D.F.; et al. The SS18-SSX Oncoprotein Hijacks KDM2B-PRC1.1 to Drive Synovial Sarcoma. Cancer Cell 2018, 34, 346–348. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 2013, 13, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Hasegawa, T.; Kanai, Y.; Tsutsumi, Y.; Osamura, Y.; Abe, Y.; Sakai, H.; Hirohashi, S. Expression of cadherins and their undercoat proteins (α-, β-, and γ-catenins and p120) and accumulation of β-catenin with no gene mutations in synovial sarcoma. Virchows Arch. 2001, 438, 23–30. [Google Scholar] [PubMed]
- Trautmann, M.; Sievers, E.; Aretz, S.; Kindler, D.; Michels, S.; Friedrichs, N.; Renner, M.; Kirfel, J.; Steiner, S.; Huss, S.; et al. SS18-SSX fusion protein-induced Wnt/β-catenin signaling is a therapeutic target in synovial sarcoma. Oncogene 2014, 33, 5006–5016. [Google Scholar] [CrossRef] [PubMed]
- Pretto, D.; Barco, R.; Rivera, J.; Neel, N.; Gustavson, M.D.; Eid, J.E. The synovial sarcoma translocation protein SYT-SSX2 recruits β-catenin to the nucleus and associates with it in an active complex. Oncogene 2006, 25, 3661–3669. [Google Scholar] [CrossRef] [PubMed]
- Cironi, L.; Petricevic, T.; Fernandes Vieira, V.; Provero, P.; Fusco, C.; Cornaz, S.; Fregni, G.; Letovanec, I.; Aguet, M.; Stamenkovic, I. The fusion protein SS18-SSX1 employs core Wnt pathway transcription factors to induce a partial Wnt signature in synovial sarcoma. Sci. Rep. 2016, 6, 22113. [Google Scholar] [CrossRef]
- Barham, W.; Frump, A.L.; Sherrill, T.P.; Garcia, C.B.; Saito-Diaz, K.; VanSaun, M.N.; Fingleton, B.; Gleaves, L.; Orton, D.; Capecchi, M.R.; et al. Targeting the Wnt pathway in synovial sarcoma models. Cancer Discov. 2013, 3, 1286–1301. [Google Scholar] [CrossRef]
- Shitashige, M.; Naishiro, Y.; Idogawa, M.; Honda, K.; Ono, M.; Hirohashi, S.; Yamada, T. Involvement of splicing factor-1 in β-catenin/T-cell factor-4-mediated gene transactivation and pre-mRNA splicing. Gastroenterology 2007, 132, 1039–1054. [Google Scholar] [CrossRef]
- Shitashige, M.; Satow, R.; Jigami, T.; Aoki, K.; Honda, K.; Shibata, T.; Ono, M.; Hirohashi, S.; Yamada, T. Traf2- and Nck-interacting kinase is essential for Wnt signaling and colorectal cancer growth. Cancer Res. 2010, 70, 5024–5033. [Google Scholar] [CrossRef]
- Masuda, M.; Sawa, M.; Yamada, T. Therapeutic targets in the Wnt signaling pathway: Feasibility of targeting TNIK in colorectal cancer. Pharmacol. Ther. 2015, 156, 1–9. [Google Scholar] [CrossRef]
- Masuda, M.; Uno, Y.; Ohbayashi, N.; Ohata, H.; Mimata, A.; Kukimoto-Niino, M.; Moriyama, H.; Kashimoto, S.; Inoue, T.; Goto, N.; et al. TNIK inhibition abrogates colorectal cancer stemness. Nat. Commun. 2016, 7, 12586. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Masuda, M. Emergence of TNIK inhibitors in cancer therapeutics. Cancer Sci. 2017, 108, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Knoepfler, P.S. Myc goes global: New tricks for an old oncogene. Cancer Res. 2007, 67, 5061–5063. [Google Scholar] [CrossRef] [PubMed]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef]
- Sansom, O.J.; Meniel, V.S.; Muncan, V.; Phesse, T.J.; Wilkins, J.A.; Reed, K.R.; Vass, J.K.; Athineos, D.; Clevers, H.; Clarke, A.R. Myc deletion rescues Apc deficiency in the small intestine. Nature 2007, 446, 676–679. [Google Scholar] [CrossRef]
- Barrios, C.; Castresana, J.S.; Ruiz, J.; Kreicbergs, A. Amplification of the c-myc proto-oncogene in soft tissue sarcomas. Oncology 1994, 51, 13–17. [Google Scholar] [CrossRef]
- Vlenterie, M.; Litière, S.; Rizzo, E.; Marréaud, S.; Judson, I.; Gelderblom, H.; Le Cesne, A.; Wardelmann, E.; Messiou, C.; Gronchi, A.; et al. Outcome of chemotherapy in advanced synovial sarcoma patients: Review of 15 clinical trials from the European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group; setting a new landmark for studies in this entity. Eur. J. Cancer 2016, 58, 62–72. [Google Scholar] [CrossRef]
- van der Graaf, W.T.; Blay, J.Y.; Chawla, S.P.; Kim, D.W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Lee, A.T.J.; Jones, R.L.; Huang, P.H. Pazopanib in advanced soft tissue sarcomas. Signal Transduct. Target. Ther. 2019, 4, 16. [Google Scholar] [CrossRef]
- Nakamura, T.; Matsumine, A.; Kawai, A.; Araki, N.; Goto, T.; Yonemoto, T.; Sugiura, H.; Nishida, Y.; Hiraga, H.; Honoki, K.; et al. The clinical outcome of pazopanib treatment in Japanese patients with relapsed soft tissue sarcoma: A Japanese Musculoskeletal Oncology Group (JMOG) study. Cancer 2016, 122, 1408–1416. [Google Scholar] [CrossRef]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 (c259)T Cells in Synovial Sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Nagayama, S.; Fukukawa, C.; Katagiri, T.; Okamoto, T.; Aoyama, T.; Oyaizu, N.; Imamura, M.; Toguchida, J.; Nakamura, Y. Therapeutic potential of antibodies against FZD 10, a cell-surface protein, for synovial sarcomas. Oncogene 2005, 24, 6201–6212. [Google Scholar] [CrossRef] [PubMed]
- Li, H.K.; Sugyo, A.; Tsuji, A.B.; Morokoshi, Y.; Minegishi, K.; Nagatsu, K.; Kanda, H.; Harada, Y.; Nagayama, S.; Katagiri, T.; et al. α-particle therapy for synovial sarcoma in the mouse using an astatine-211-labeled antibody against frizzled homolog 10. Cancer Sci. 2018, 109, 2302–2309. [Google Scholar] [CrossRef]
- Giraudet, A.L.; Cassier, P.A.; Iwao-Fukukawa, C.; Garin, G.; Badel, J.N.; Kryza, D.; Chabaud, S.; Gilles-Afchain, L.; Clapisson, G.; Desuzinges, C.; et al. A first-in-human study investigating biodistribution, safety and recommended dose of a new radiolabeled MAb targeting FZD10 in metastatic synovial sarcoma patients. BMC Cancer 2018, 18, 646. [Google Scholar] [CrossRef]
- Kadoch, C.; Crabtree, G.R. Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 2013, 153, 71–85. [Google Scholar] [CrossRef]
- Middeljans, E.; Wan, X.; Jansen, P.W.; Sharma, V.; Stunnenberg, H.G.; Logie, C. SS18 together with animal-specific factors defines human BAF-type SWI/SNF complexes. PLoS ONE 2012, 7, e33834. [Google Scholar] [CrossRef]
- Mora-Blanco, E.L.; Mishina, Y.; Tillman, E.J.; Cho, Y.J.; Thom, C.S.; Pomeroy, S.L.; Shao, W.; Roberts, C.W. Activation of β-catenin/TCF targets following loss of the tumor suppressor SNF5. Oncogene 2014, 33, 933–938. [Google Scholar] [CrossRef]
- Barrott, J.J.; Illum, B.E.; Jin, H.; Zhu, J.F.; Mosbruger, T.; Monument, M.J.; Smith-Fry, K.; Cable, M.G.; Wang, Y.; Grossmann, A.H.; et al. β-Catenin stabilization enhances SS18-SSX2-driven synovial sarcomagenesis and blocks the mesenchymal to epithelial transition. Oncotarget 2015, 6, 22758–22766. [Google Scholar] [CrossRef]
- Mahmoudi, T.; Li, V.S.; Ng, S.S.; Taouatas, N.; Vries, R.G.; Mohammed, S.; Heck, A.J.; Clevers, H. The kinase TNIK is an essential activator of Wnt target genes. EMBO J. 2009, 28, 3329–3340. [Google Scholar] [CrossRef]
- Meichle, A.; Philipp, A.; Eilers, M. The functions of Myc proteins. Biochim. Biophys. Acta 1992, 1114, 129–146. [Google Scholar] [CrossRef]
- Levens, D. Cellular MYCro economics: Balancing MYC function with MYC expression. Cold Spring Harb. Perspect. Med. 2013, 3, a014233. [Google Scholar] [CrossRef]
- Riou, G.; Barrois, M.; Le, M.G.; George, M.; Le Doussal, V.; Haie, C. C-myc proto-oncogene expression and prognosis in early carcinoma of the uterine cervix. Lancet 1987, 1, 761–763. [Google Scholar] [CrossRef]
- Garte, S.J. The c-myc oncogene in tumor progression. Crit. Rev. Oncog. 1993, 4, 435–449. [Google Scholar]
- Shen, J.; Scotlandi, K.; Baldini, N.; Manara, M.C.; Benini, S.; Cerisano, V.; Picci, P.; Serra, M. Prognostic significance of nuclear accumulation of c-myc and mdm2 proteins in synovial sarcoma of the extremities. Oncology 2000, 58, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, R.; Mitani, S.; Nakagawa, T.; Hasegawa, T.; Kawai, A.; Morioka, H.; Yabe, H.; Toyama, Y.; Ogose, A.; Toguchida, J.; et al. Gene expression profiling of synovial sarcoma: Distinct signature of poorly differentiated type. Am. J. Surg. Pathol. 2010, 34, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Machen, S.K.; Easley, K.A.; Goldblum, J.R. Synovial sarcoma of the extremities: A clinicopathologic study of 34 cases, including semi-quantitative analysis of spindled, epithelial, and poorly differentiated areas. Am. J. Surg. Pathol. 1999, 23, 268–275. [Google Scholar] [CrossRef]
- Naka, N.; Takenaka, S.; Araki, N.; Miwa, T.; Hashimoto, N.; Yoshioka, K.; Joyama, S.; Hamada, K.; Tsukamoto, Y.; Tomita, Y.; et al. Synovial sarcoma is a stem cell malignancy. Stem Cells 2010, 28, 1119–1131. [Google Scholar] [CrossRef]
- Kawai, A.; Naito, N.; Yoshida, A.; Morimoto, Y.; Ouchida, M.; Shimizu, K.; Beppu, Y. Establishment and characterization of a biphasic synovial sarcoma cell line, SYO-1. Cancer Lett. 2004, 204, 105–113. [Google Scholar] [CrossRef]
- Masuda, M.; Chen, W.Y.; Miyanaga, A.; Nakamura, Y.; Kawasaki, K.; Sakuma, T.; Ono, M.; Chen, C.L.; Honda, K.; Yamada, T. Alternative mammalian target of rapamycin (mTOR) signal activation in sorafenib-resistant hepatocellular carcinoma cells revealed by array-based pathway profiling. Mol. Cell. Proteom. 2014, 13, 1429–1438. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Ito, J.; Asano, N.; Kawai, A.; Yoshida, A. The diagnostic utility of reduced immunohistochemical expression of SMARCB1 in synovial sarcomas: A validation study. Hum. Pathol. 2016, 47, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Amary, M.F.; Berisha, F.; Bernardi Fdel, C.; Herbert, A.; James, M.; Reis-Filho, J.S.; Fisher, C.; Nicholson, A.G.; Tirabosco, R.; Diss, T.C.; et al. Detection of SS18-SSX fusion transcripts in formalin-fixed paraffin-embedded neoplasms: Analysis of conventional RT-PCR, qRT-PCR and dual color FISH as diagnostic tools for synovial sarcoma. Mod. Pathol. 2007, 20, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Miyanaga, A.; Honda, K.; Tsuta, K.; Masuda, M.; Yamaguchi, U.; Fujii, G.; Miyamoto, A.; Shinagawa, S.; Miura, N.; Tsuda, H.; et al. Diagnostic and prognostic significance of the alternatively spliced ACTN4 variant in high-grade neuroendocrine pulmonary tumours. Ann. Oncol. 2013, 24, 84–90. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sekita, T.; Yamada, T.; Kobayashi, E.; Yoshida, A.; Hirozane, T.; Kawai, A.; Uno, Y.; Moriyama, H.; Sawa, M.; Nagakawa, Y.; et al. Feasibility of Targeting Traf2-and-Nck-Interacting Kinase in Synovial Sarcoma. Cancers 2020, 12, 1258. https://doi.org/10.3390/cancers12051258
Sekita T, Yamada T, Kobayashi E, Yoshida A, Hirozane T, Kawai A, Uno Y, Moriyama H, Sawa M, Nagakawa Y, et al. Feasibility of Targeting Traf2-and-Nck-Interacting Kinase in Synovial Sarcoma. Cancers. 2020; 12(5):1258. https://doi.org/10.3390/cancers12051258
Chicago/Turabian StyleSekita, Tetsuya, Tesshi Yamada, Eisuke Kobayashi, Akihiko Yoshida, Toru Hirozane, Akira Kawai, Yuko Uno, Hideki Moriyama, Masaaki Sawa, Yuichi Nagakawa, and et al. 2020. "Feasibility of Targeting Traf2-and-Nck-Interacting Kinase in Synovial Sarcoma" Cancers 12, no. 5: 1258. https://doi.org/10.3390/cancers12051258
APA StyleSekita, T., Yamada, T., Kobayashi, E., Yoshida, A., Hirozane, T., Kawai, A., Uno, Y., Moriyama, H., Sawa, M., Nagakawa, Y., Tsuchida, A., Matsumoto, M., Nakamura, M., Nakayama, R., & Masuda, M. (2020). Feasibility of Targeting Traf2-and-Nck-Interacting Kinase in Synovial Sarcoma. Cancers, 12(5), 1258. https://doi.org/10.3390/cancers12051258