T Cells Expressing a TCR-Like Antibody Selected Against the Heteroclitic Variant of a Shared MAGE-A Epitope Do Not Recognise the Cognate Epitope


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Peptides and pMHC Complexes
2.3. T2 Assay
2.4. Construction of Phage-Fab Display Library
2.5. Selection of TCR-Like Antibody-Expressing Phages
2.6. Testing Phages for pMHC Binding, Clonality and Binding to Target Cells
2.7. Generation of CAR T Cells
2.8. Testing CAR T Cells for Specificity and Responsiveness
2.9. Statistical Analyses
3. Results
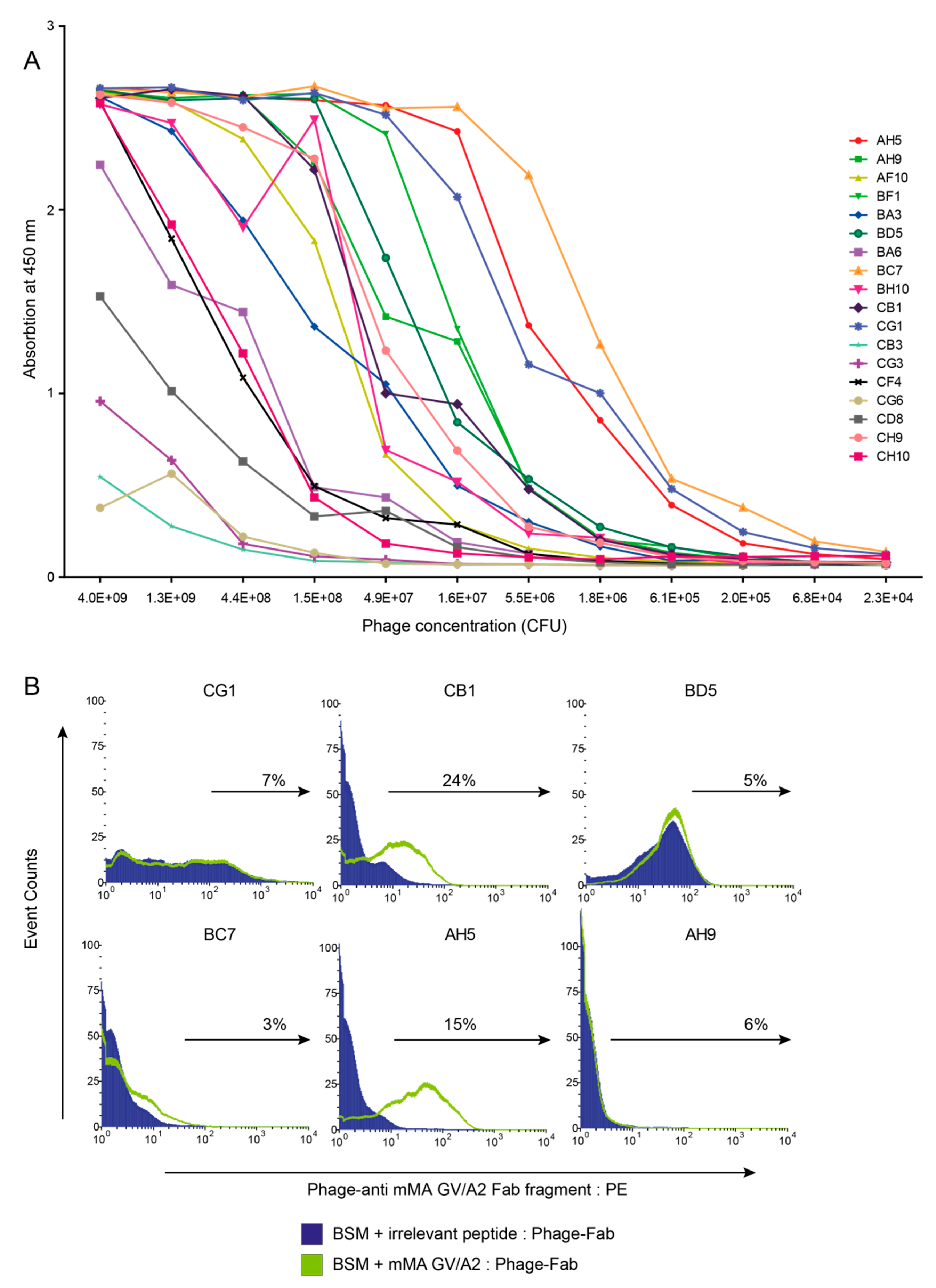
3.1. Combinatorial Phage-Fab Library with High Level of Diversity Enables Selection of TCR-Like Antibody Clones
3.2. Phage-Fab Selections Against Heteroclitic mMA Peptide Revealed Unique AH5 Clone
3.3. mMA-GV/A2-Specific AH5 Antibody Does Not Recognize mMA wt Peptide nor mMA-Positive Tumor Cells
3.4. AH5 CAR Expressing T Cells Do Not Respond to Tumor Cells Endogenously Presenting mMA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bolli, M.; Schultz-Thater, E.; Zajac, P.; Guller, U.; Feder, C.; Sanguedolce, F.; Carafa, V.; Terracciano, L.; Hudolin, T.; Spagnoli, G.C.; et al. NY-ESO-1/LAGE-1 coexpression with MAGE-A cancer/testis antigens: A tissue microarray study. Int. J. Cancer. 2005, 115, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Boon, T.; Van Der Bruggen, P. Human tumor antigens recognized by T lymphocytes. J. Exp. Med. 1996, 183, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Borbulevych, O.Y.; Baxter, T.K.; Yu, Z.; Restifo, N.P.; Baker, B.M. Increased Immunogenicity of an Anchor-Modified Tumor-Associated Antigen Is Due to the Enhanced Stability of the Peptide/MHC Complex: Implications for Vaccine Design1. J. Immunol. 2005, 174, 4812–4820. [Google Scholar] [CrossRef] [PubMed]
- Buhrman, J.D.; Slansky, J.E. Improving T cell responses to modified peptides in tumor vaccines. Immunol. Res. 2013, 55, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Chames, P.; Hufton, S.E.; Coulie, P.G.; Uchanska-Ziegler, B.; Hoogenboom, H.R. Direct selection of a human antibody fragment directed against the tumor T-cell epitope HLA-A1–MAGE-A1 from a nonimmunized phage-Fab library. Proc. Natl. Acad. Sci. USA 2000, 97, 7969–7974. [Google Scholar] [CrossRef]
- Chen, J.-L.; Stewart-Jones, G.; Bossi, G.; Lissin, N.M.; Wooldridge, L.; Choi, E.M.L.; Held, G.; Dunbar, P.R.; Esnouf, R.M.; Sami, M.; et al. Structural and kinetic basis for heightened immunogenicity of T cell vaccines. J. Exp. Med. 2005, 201, 1243–1255. [Google Scholar] [CrossRef]
- Cole, D.K.; Edwards, E.S.; Wynn, K.K.; Clement, M.; Miles, J.J.; Ladell, K.; Ekeruche, J.; Gostick, E.; Adams, K.J.; Skowera, A.; et al. Modification of MHC anchor residues generates heteroclitic peptides that alter TCR binding and T cell recognition. J. Immunol. 2010, 185, 2600–2610. [Google Scholar] [CrossRef]
- Dahan, R.; Reiter, Y. T-cell-receptor-like antibodies—Generation, function and applications. Expert Rev. Mol. Med. 2012, 14, 6. [Google Scholar] [CrossRef]
- De Haard, H.J.W.; Van Neer, N.; Reurs, A.; Hufton, S.E.; Roovers, R.C.; Henderikx, P.; de Bruïne, A.P.; Arends, J.-W.; Hoogenboom, H.R.; De Bruïne, A.P. A Large Non-immunized Human Fab Fragment Phage Library That Permits Rapid Isolation and Kinetic Analysis of High Affinity Antibodies. J. Boil. Chem. 1999, 274, 18218–18230. [Google Scholar] [CrossRef]
- Debets, R.; Donnadieu, E.; Chouaib, S.; Coukos, G. TCR-engineered T cells to treat tumors: Seeing but not touching? Semin. Immunol. 2016, 28, 10–21. [Google Scholar] [CrossRef]
- Denkberg, G.; Cohen, C.J.; Lev, A.; Chames, P.; Hoogenboom, H.R.; Reiter, Y. Direct visualization of distinct T cell epitopes derived from a melanoma tumor-associated antigen by using human recombinant antibodies with MHC- restricted T cell receptor-like specificity. Proc. Natl. Acad. Sci. USA 2002, 99, 9421–9426. [Google Scholar] [CrossRef] [PubMed]
- Denkberg, G.; Klechevsky, E.; Reiter, Y. Modification of a tumor-derived peptide at an HLA-A2 anchor residue can alter the conformation of the MHC-peptide complex: probing with TCR-like recombinant antibodies. J. Immunol. 2002, 169, 4399–4407. [Google Scholar] [CrossRef] [PubMed]
- Epel, M.; Carmi, I.; Soueid-Baumgarten, S.; Oh, S.; Bera, T.; Pastan, I.; Berzofsky, J.; Reiter, Y. Targeting TARP, a novel breast and prostate tumor-associated antigen, with T cell receptor-like human recombinant antibodies. Eur. J. Immunol. 2008, 38, 1706–1720. [Google Scholar] [CrossRef] [PubMed]
- Even-Desrumeaux, K.; Chames, P. Phage Display and Selections on Cells. Advanced Structural Safety Studies 2012, 907, 225–235. [Google Scholar] [CrossRef]
- Gilham, D.E.; Debets, R.; Pule, M.; Hawkins, R.E.; Abken, H. CAR–T cells and solid tumors: tuning T cells to challenge an inveterate foe. Trends Mol. Med. 2012, 18, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Gjertsen, M.K.; Bakka, A.; Breivik, J.; Saeterdal, I.; Gedde-Dahl, T.; Stokke, K.T.; Solheim, B.G.; Egge, T.S.; Søreide, O.; Thorsby, E.; et al. Ex vivo ras peptide vaccination in patients with advanced pancreatic cancer: Results of a phase I/II study. Int. J. Cancer 1996, 65, 450–453. [Google Scholar] [CrossRef]
- Graff-Dubois, S.; Faure, O.; Gross, D.; Alves, P.; Scardino, A.; Chouaib, S.; Lemonnier, F.A.; Kosmatopoulos, K. Generation of CTL recognizing an HLA-A*0201-restricted epitope shared by MAGE-A1, -A2, -A3, -A4, -A6, -A10, and -A12 tumor antigens: implication in a broad-spectrum tumor immunotherapy. J. Immunol. 2002, 169, 575–580. [Google Scholar] [CrossRef]
- Hege, K.M.; Bergsland, E.K.; Fisher, G.A.; Nemunaitis, J.J.; Warren, R.S.; McArthur, J.G.; Lin, A.A.; Schlom, J.; June, C.H.; Sherwin, S.A. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J. Immunother. Cancer 2017, 5, 22. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. New Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Hofmann, O.; Caballero, O.L.; Stevenson, B.J.; Chen, Y.-T.; Cohen, T.; Chua, R.; Maher, C.A.; Panji, S.; Schaefer, U.; Kruger, A.; et al. Genome-wide analysis of cancer/testis gene expression. Proc. Natl. Acad. Sci. USA 2008, 105, 20422–20427. [Google Scholar] [CrossRef]
- Hudolin, T.; Kastelan, Z.; Ilic, I.; Levarda-Hudolin, K.; Basic-Jukic, N.; Rieken, M.; Spagnoli, G.C.; Juretić, A.; Mengus, C. Immunohistochemical analysis of the expression of MAGE-A and NY-ESO-1 cancer/testis antigens in diffuse large B-cell testicular lymphoma. J. Transl. Med. 2013, 11, 123. [Google Scholar] [CrossRef] [PubMed]
- A Johnson, L.; June, C.H. Driving gene-engineered T cell immunotherapy of cancer. Cell Res. 2016, 27, 38–58. [Google Scholar] [CrossRef] [PubMed]
- Klechevsky, E.; Gallegos, M.; Denkberg, G.; Palucka, K.; Banchereau, J.; Cohen, C.; Reiter, Y. Antitumor activity of immunotoxins with T-cell receptor-like specificity against human melanoma xenografts. Cancer Res. 2008, 68, 6360–6367. [Google Scholar] [CrossRef] [PubMed]
- Kunert, A.; Straetemans, T.; Govers, C.; Lamers, C.; Mathijssen, R.; Sleijfer, S.; Debets, R. TCR-Engineered T Cells Meet New Challenges to Treat Solid Tumors: Choice of Antigen, T Cell Fitness, and Sensitization of Tumor Milieu. Front. Immunol. 2013, 4, 363. [Google Scholar] [CrossRef]
- Lamers, C.H.; Sleijfer, S.; Vulto, A.G.; Kruit, W.H.; Kliffen, M.; Debets, R.; Gratama, J.W.; Stoter, G.; Oosterwijk, E. Treatment of Metastatic Renal Cell Carcinoma With Autologous T-Lymphocytes Genetically Retargeted Against Carbonic Anhydrase IX: First Clinical Experience. J. Clin. Oncol. 2006, 24, e20–e22. [Google Scholar] [CrossRef]
- Lamers, C.H.J.; Willemsen, R.; Van Elzakker, P.; Van Steenbergen-Langeveld, S.; Broertjes, M.; Oosterwijk-Wakka, J.; Oosterwijk, E.; Sleijfer, S.; Debets, R.; Gratama, J.W. Immune responses to transgene and retroviral vector in patients treated with ex vivo–engineered T cells. Blood 2011, 117, 72–82. [Google Scholar] [CrossRef]
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740. [Google Scholar] [CrossRef]
- Marincola, F.M.; Hijazi, Y.M.; Fetsch, P.; Salgaller, M.L.; Rivoltini, L.; Cormier, J.; Simonis, T.B.; Duray, P.H.; Herlyn, M.; Kawakami, Y.; et al. Analysis of Expression of the Melanoma-Associated Antigens MART-1 and gp lOO in Metastatic Melanoma Cell Lines and in In Situ Lesions. J. Immunother. 1996, 19, 192–205. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. New Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Miles, K.M.; Miles, J.J.; Madura, F.; Sewell, A.K.; Cole, D.K. Real time detection of peptide–MHC dissociation reveals that improvement of primary MHC-binding residues can have a minimal, or no, effect on stability. Mol. Immunol. 2010, 48, 728–732. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. New Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Oren, R.; Hod-Marco, M.; Haus-Cohen, M.; Thomas, S.; Blat, D.; Duvshani, N.; Denkberg, G.; Elbaz, Y.; Benchetrit, F.; Eshhar, Z.; et al. Functional Comparison of Engineered T Cells Carrying a Native TCR versus TCR-like Antibody–Based Chimeric Antigen Receptors Indicates Affinity/Avidity Thresholds. J. Immunol. 2014, 193, 5733–5743. [Google Scholar] [CrossRef] [PubMed]
- Renkvist, N.; Castelli, C.; Robbins, P.F.; Parmiani, G. A listing of human tumor antigens recognized by T cells. Cancer Immunol. Immunother. 2001, 50, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. Progress in human tumour immunology and immunotherapy. Nature 2001, 411, 380–384. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Schwartzentruber, U.J.; Hwu, P.; Marincola, F.M.; Topalian, S.L.; Restifo, N.P.; Dudley, M.E.; Schwarz, S.L.; Spiess, P.J.; et al. Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat. Med. 1998, 4, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Speiser, D.E.; Baumgaertner, P.; Voelter, V.; Devevre, E.; Barbey, C.; Rufer, N.; Romero, P. Unmodified self antigen triggers human CD8 T cells with stronger tumor reactivity than altered antigen. Proc. Natl. Acad. Sci. USA 2008, 105, 3849–3854. [Google Scholar] [CrossRef]
- Stauss, H.J.; Cesco-Gaspere, M.; Thomas, S.; Hart, D.P.; Xue, S.-A.; Holler, A.; Wright, G.; Perro, M.; Little, A.-M.; Pospori, C.; et al. Monoclonal T-Cell Receptors: New Reagents for Cancer Therapy. Mol. Ther. 2007, 15, 1744–1750. [Google Scholar] [CrossRef]
- Stoiber, S.; Cadilha, B.L.; Benmebarek, M.-R.; Lesch, S.; Endres, S.; Kobold, S. Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells 2019, 8, 472. [Google Scholar] [CrossRef]
- Weidanz, J.A.; Hawkins, O.; Verma, B.; Hildebrand, W.H. TCR-Like Biomolecules Target Peptide/MHC Class I Complexes on the Surface of Infected and Cancerous Cells. Int. Rev. Immunol. 2011, 30, 328–340. [Google Scholar] [CrossRef][Green Version]
- Willemsen, R.A.; Ronteltap, C.; Chames, P.; Debets, R.; Bolhuis, R.L.H. T cell retargeting with MHC class I-restricted antibodies: the CD28 costimulatory domain enhances antigen-specific cytotoxicity and cytokine production. J. Immunol. 2005, 174, 7853–7858. [Google Scholar] [CrossRef]
- Willemsen, R.A.; Weijtens, M.E.M.; Ronteltap, C.; Eshhar, Z.; Gratama, J.W.; Chames, P.; Bolhuis, R.L.H. Grafting primary human T lymphocytes with cancer-specific chimeric single chain and two chain TCR. Gene Ther. 2000, 7, 1369–1377. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigena | Selection Round | Phage Inputb | Phage Outputb | Enrichment | (Pan-)MHC Bindersc | TCR-like Bindersd | Unique Patternse |
|---|---|---|---|---|---|---|---|
| mM-A(GV)/A2 (combined w/ melanoma cells in round 2) | 1 | 1 × 1012 | 9 × 106 | - | - | - | - |
| 2f | 1× 1012 | 9 × 106 | 1 | - | - | - | |
| 3 | 2 × 1012 | 5 × 108 | 56 | - | - | - | |
| 4 | 6 × 1012 | 2 × 1010 | 2222 | 2/94 (2%) | 80/94 (85%) | 5 | |
| mM-A(GV)/A2 (combined w/ melanoma cells in round 4) | 1 | 1 × 1012 | 9 × 106 | - | - | - | - |
| 2 | 1 × 1013 | 7 × 107 | 7,7 | - | - | - | |
| 3 | 1 × 1013 | 2 × 1010 | 2222 | 57/282 (20%) | 46/282 (16%) | 18 | |
| 4f | 2 × 1013 | 8 × 108 | 89 | 6/282 (2%) | 14/282 (5%) | 9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeed, M.; Schooten, E.; van Brakel, M.; K. Cole, D.; ten Hagen, T.L.M.; Debets, R. T Cells Expressing a TCR-Like Antibody Selected Against the Heteroclitic Variant of a Shared MAGE-A Epitope Do Not Recognise the Cognate Epitope. Cancers 2020, 12, 1255. https://doi.org/10.3390/cancers12051255
Saeed M, Schooten E, van Brakel M, K. Cole D, ten Hagen TLM, Debets R. T Cells Expressing a TCR-Like Antibody Selected Against the Heteroclitic Variant of a Shared MAGE-A Epitope Do Not Recognise the Cognate Epitope. Cancers. 2020; 12(5):1255. https://doi.org/10.3390/cancers12051255
Chicago/Turabian StyleSaeed, Mesha, Erik Schooten, Mandy van Brakel, David K. Cole, Timo L. M. ten Hagen, and Reno Debets. 2020. "T Cells Expressing a TCR-Like Antibody Selected Against the Heteroclitic Variant of a Shared MAGE-A Epitope Do Not Recognise the Cognate Epitope" Cancers 12, no. 5: 1255. https://doi.org/10.3390/cancers12051255
APA StyleSaeed, M., Schooten, E., van Brakel, M., K. Cole, D., ten Hagen, T. L. M., & Debets, R. (2020). T Cells Expressing a TCR-Like Antibody Selected Against the Heteroclitic Variant of a Shared MAGE-A Epitope Do Not Recognise the Cognate Epitope. Cancers, 12(5), 1255. https://doi.org/10.3390/cancers12051255