Immune Cytolytic Activity for Comprehensive Understanding of Immune Landscape in Hepatocellular Carcinoma

 , ,
, , 
Abstract
1. Introduction
2. Results
2.1. Patient Demographics
2.2. CYT-High Hccs were Associated with Significantly Higher Gene Expressions of Apolipoprotein B mrna Editing Enzyme Catalytic Polypeptide-Like 3 (APOBEC3) Family Members
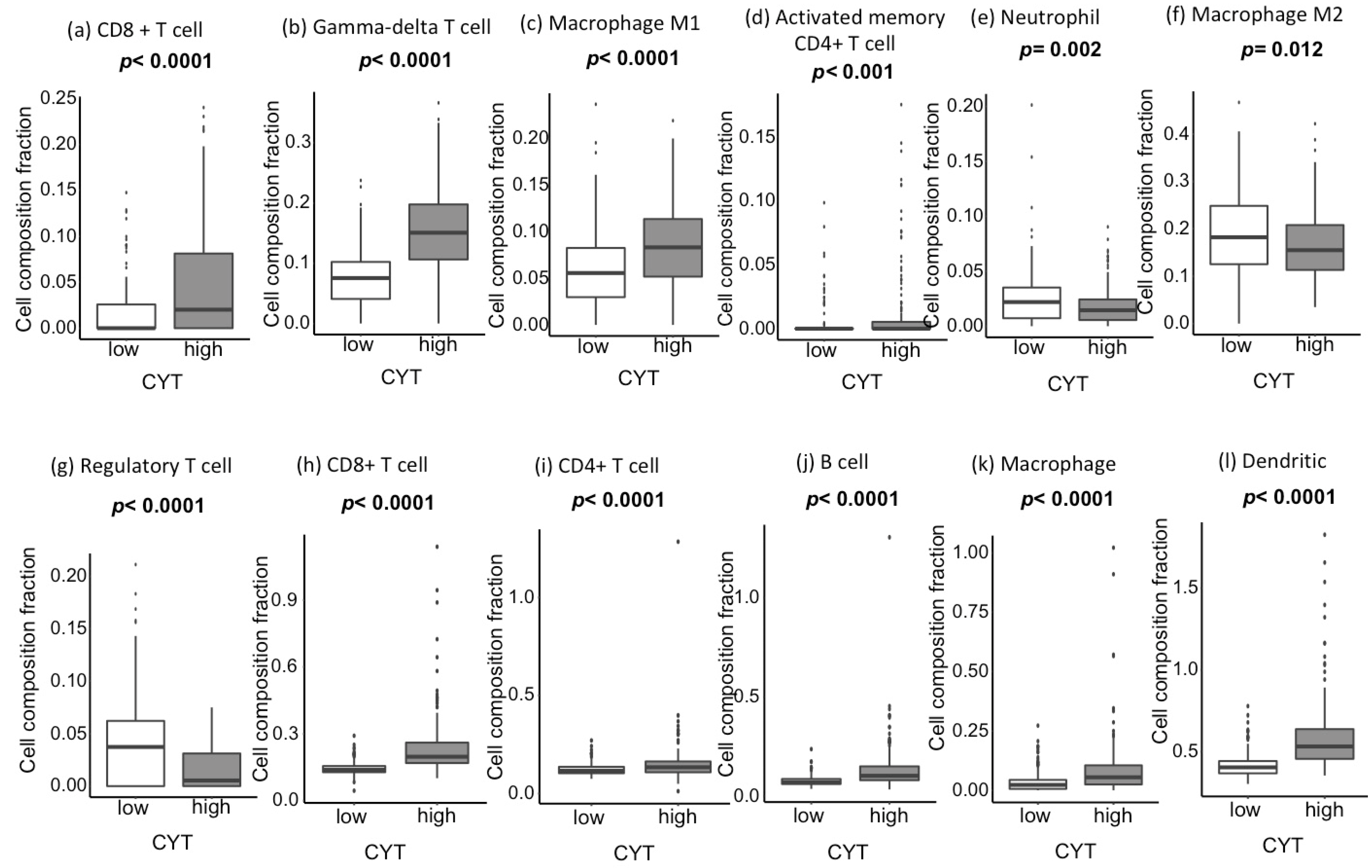
2.3. CYT-High Tumors Have Significantly Higher Infiltration of Anticancer Immune Cells and Lower Infiltration of Procancer Immune Cells
2.4. CYT was Associated with Tumor-Infiltrating Lymphocyte Receptor Genetic Diversity
2.5. CYT was Associated with the Expression of T Cell Exhaustion Markers
2.6. Gene Set Enrichment Analysis (GSEA) Demonstrated That Immune Response-Related Gene Sets were Significantly Enriched in CYT-High Hccs
2.7. Patients with CYT-High HCCs Have Significantly Longer PFI and DFI and Better Prognosis than Patients with CYT-low HCCs
3. Discussion
4. Material and Methods
4.1. Patient Cohort and Genomic Data Processing
4.2. Cytolytic Activity Score (CYT)
4.3. CIBERSORT and Tumor Immune Estimation Resource (TIMER)
4.4. Gene Set Enrichment Analysis (GSEA)
4.5. T cell receptor (TCR) Richness, B cell Receptor (BCR) Richness, and TCR Diversity
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet (Lond. Engl.) 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef]
- Swanton, C.; McGranahan, N.; Starrett, G.J.; Harris, R.S. APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov. 2015, 5, 704–712. [Google Scholar] [CrossRef]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; de Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017, 171, 1042–1056.e10. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef]
- Ferlay, J.E.M.; Lam, F.; Colombet, M.; Pary, L.; Pineros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today; International Agency for Research on Cancer: Lyon, France, 2018; Available online: https://gco.iarc.fr/today/data/factsheets/cancers/11-Liver-fact-sheet.pdf (accessed on 25 November 2019).
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet (Lond. Engl.) 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M. Systemic Therapy for Hepatocellular Carcinoma: Latest Advances. Cancers 2018, 10, 412. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef]
- Balli, D.; Rech, A.J.; Stanger, B.Z.; Vonderheide, R.H. Immune Cytolytic Activity Stratifies Molecular Subsets of Human Pancreatic Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3129–3138. [Google Scholar] [CrossRef]
- Shrestha, R.; Prithviraj, P.; Anaka, M.; Bridle, K.R.; Crawford, D.H.G.; Dhungel, B.; Steel, J.C.; Jayachandran, A. Monitoring Immune Checkpoint Regulators as Predictive Biomarkers in Hepatocellular Carcinoma. Front. Oncol. 2018, 8, 269. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Galon, J.; Pages, F.; Tartour, E.; Sautes-Fridman, C.; Kroemer, G. Prognostic and predictive impact of intra- and peritumoral immune infiltrates. Cancer Res. 2011, 71, 5601–5605. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Narayanan, S.; Kawaguchi, T.; Yan, L.; Peng, X.; Qi, Q.; Takabe, K. Cytolytic Activity Score to Assess Anticancer Immunity in Colorectal Cancer. Ann. Surg. Oncol. 2018, 25, 2323–2331. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Narayanan, S.; Takabe, K. ASO Author Reflections: “From Computer to Bedside”: A New Translational Approach to Immunogenomics. Ann. Surg. Oncol. 2018, 25 (Suppl. S3), 846–847. [Google Scholar] [CrossRef]
- Irvine, K.M.; Ratnasekera, I.; Powell, E.E.; Hume, D.A. Causes and Consequences of Innate Immune Dysfunction in Cirrhosis. Front. Immunol. 2019, 10, 293. [Google Scholar] [CrossRef]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef]
- Li, B.; Li, T.; Pignon, J.C.; Wang, B.; Wang, J.; Shukla, S.A.; Dou, R.; Chen, Q.; Hodi, F.S.; Choueiri, T.K.; et al. Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat. Genet. 2016, 48, 725–732. [Google Scholar] [CrossRef]
- Lin, K.R.; Deng, F.W.; Jin, Y.B.; Chen, X.P.; Pan, Y.M.; Cui, J.H.; You, Z.X.; Chen, H.W.; Luo, W. T cell receptor repertoire profiling predicts the prognosis of HBV-associated hepatocellular carcinoma. Cancer Med. 2018, 7, 3755–3762. [Google Scholar] [CrossRef]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 15, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Salazar, R.; Tabernero, J. Personalizing colon cancer adjuvant therapy: Selecting optimal treatments for individual patients. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1787–1796. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Sprengers, D.; Boor, P.P.C.; Doukas, M.; Schutz, H.; Mancham, S.; Pedroza-Gonzalez, A.; Polak, W.G.; de Jonge, J.; Gaspersz, M.; et al. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology 2017, 153, 1107–1119.e10. [Google Scholar] [CrossRef] [PubMed]
- Bui, J.D.; Uppaluri, R.; Hsieh, C.S.; Schreiber, R.D. Comparative analysis of regulatory and effector T cells in progressively growing versus rejecting tumors of similar origins. Cancer Res. 2006, 66, 7301–7309. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Qiu, S.J.; Fan, J.; Zhou, J.; Wang, X.Y.; Xiao, Y.S.; Xu, Y.; Li, Y.W.; Tang, Z.Y. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J. Clin. Oncol. 2007, 25, 2586–2593. [Google Scholar] [CrossRef]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef]
- Bruix, J.; Llovet, J.M. Prognostic prediction and treatment strategy in hepatocellular carcinoma. Hepatology (Baltim. Md) 2002, 35, 519–524. [Google Scholar] [CrossRef]
- Yau, T.; Tang, V.Y.; Yao, T.J.; Fan, S.T.; Lo, C.M.; Poon, R.T. Development of Hong Kong Liver Cancer staging system with treatment stratification for patients with hepatocellular carcinoma. Gastroenterology 2014, 146, 1691–1700.e3. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Severson, E.; Pignon, J.C.; Zhao, H.; Li, T.; Novak, J.; Jiang, P.; Shen, H.; Aster, J.C.; Rodig, S.; et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 2016, 17, 174. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kawaguchi, T.; Yan, L.; Young, J.; Qi, Q.; Takabe, K. Clinical Relevance of microRNA Expressions in Breast Cancer Validated Using the Cancer Genome Atlas (TCGA). Ann. Surg. Oncol. 2017, 24, 2943–2949. [Google Scholar] [CrossRef] [PubMed]
- Oshi, M.; Takahashi, H.; Tokumaru, Y.; Yan, L.; Rashid, O.M.; Matsuyama, R.; Endo, I.; Takabe, K. G2M Cell Cycle Pathway Score as a Prognostic Biomarker of Metastasis in Estrogen Receptor (ER)-Positive Breast Cancer. Int. J. Mol. Sci. 2020, 21, 2921. [Google Scholar] [CrossRef] [PubMed]
- Sporn, J.C.; Katsuta, E.; Yan, L.; Takabe, K. Expression of MicroRNA-9 is Associated With Overall Survival in Breast Cancer Patients. J. Surg. Res. 2019, 233, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Tokumaru, Y.; Oshi, M.; Katsuta, E.; Yan, L.; Satyananda, V.; Matsuhashi, N.; Futamura, M.; Akao, Y.; Yoshida, K.; Takabe, K. KRAS signaling enriched triple negative breast cancer is associated with favorable tumor immune microenvironment and better survival. Am. J. Cancer Res. 2020, 10, 897–907. [Google Scholar]
- Young, J.; Kawaguchi, T.; Yan, L.; Qi, Q.; Liu, S.; Takabe, K. Tamoxifen sensitivity-related microRNA-342 is a useful biomarker for breast cancer survival. Oncotarget 2017, 8, 99978–99989. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | CYT Low (186) | CYT High (185) | p-Value |
|---|---|---|---|
| Age | 59.5 ± 13.9 | 59.4 ± 13.1 | 0.68 |
| Gender (Male/Female) | 123/63 | 127/58 | 0.68 |
| AJCC T category (T1/T2/T3/T4) * | 83/54/44/5 | 98/40/36/8 | 0.19 |
| AJCC N category (N0/N1) * | 127/3 | 125/1 | 0.64 |
| Pathological Stage (I/II/III/IV) * | 77/48/45/3 | 94/38/40/2 | 0.34 |
| Adjacent hepatic tissue inflammation extent (None/Mild/Severe) * | 64/50/8 | 53/49/10 | 0.66 |
| Underlying liver disease (No history/Alcohol/Hep B/Hep C/Others) * | 56/52/36/16/16 | 35/65/44/18/14 | 0.12 |
| TIL (Positive/Negative) | 71/115 | 114/71 | <0.001 |
| Gene set Name | ES | NES | p-Value |
|---|---|---|---|
| Defense Response | 0.705 | 2.607 | <0.001 |
| Response To Wounding | 0.636 | 2.565 | <0.001 |
| Immune Response | 0.735 | 2.565 | <0.001 |
| Inflammatory Response | 0.695 | 2.562 | <0.001 |
| Immune System Process | 0.710 | 2.553 | <0.001 |
| Regulation Of Immune System Process | 0.724 | 2.325 | <0.001 |
| Cellular Defense Response | 0.789 | 2.302 | <0.001 |
| Leukocyte Activation | 0.737 | 2.256 | <0.001 |
| Positive Regulation Of Immune System Process | 0.724 | 2.229 | <0.001 |
| Humoral Immune Response | 0.742 | 2.225 | <0.001 |
| Positive Regulation Of Cytokine Biosynthetic Process | 0.739 | 2.139 | 0.001 |
| T-Cell Activation | 0.778 | 2.139 | 0.001 |
| Cytokine Biosynthetic Process | 0.631 | 2.100 | 0.001 |
| Innate Immune Response | 0.607 | 1.892 | 0.017 |
| Variables | Univariate Analysis | Multivariate Analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p-Value | HR | 95% CI | p-Value | |
| Age | 1.03 | 1.005–1.049 | 0.016 | 1.03 | 1.006–1.051 | 0.013 |
| Gender | 0.81 | 0.483–1.364 | 0.432 | |||
| Stage (II vs. I) | 1.83 | 0.948–3.538 | 0.072 | 2.03 | 1.046–3.948 | 0.036 |
| Stage (III vs. I) | 2.46 | 1.340–4.527 | 0.004 | 2.74 | 1.488–5.049 | 0.001 |
| Stage (IV vs. I) | 8.42 | 2.484–28.543 | <0.001 | 12.58 | 3.531–44.853 | <0.001 |
| Adjacent hepatic tissue inflammation extent type (Mild vs. None) | 1.24 | 0.717–2.129 | 0.446 | |||
| Adjacent hepatic tissue inflammation extent type (Severe vs. None) | 0.98 | 0.343–2.778 | 0.964 | |||
| Virus status (Alcohol consumption vs. None) | 0.58 | 0.291–1.175 | 0.132 | |||
| Virus status (Hepatitis B vs. None) | 0.43 | 0.209–0.891 | 0.023 | |||
| Virus status (Hepatitis C vs. None) | 1.05 | 0.424–2.602 | 0.916 | |||
| Virus status (Others vs. None) | 0.94 | 0.400–2.210 | 0.887 | |||
| TIL (positive vs. negative) | 1.11 | 0.662–1.864 | 0.692 | |||
| CYT (high vs. low) | 0.55 | 0.326–0.921 | 0.023 | 0.47 | 0.275–0.819 | 0.007 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahashi, H.; Kawaguchi, T.; Yan, L.; Peng, X.; Qi, Q.; Morris, L.G.T.; Chan, T.A.; Tsung, A.; Otsuji, E.; Takabe, K. Immune Cytolytic Activity for Comprehensive Understanding of Immune Landscape in Hepatocellular Carcinoma. Cancers 2020, 12, 1221. https://doi.org/10.3390/cancers12051221
Takahashi H, Kawaguchi T, Yan L, Peng X, Qi Q, Morris LGT, Chan TA, Tsung A, Otsuji E, Takabe K. Immune Cytolytic Activity for Comprehensive Understanding of Immune Landscape in Hepatocellular Carcinoma. Cancers. 2020; 12(5):1221. https://doi.org/10.3390/cancers12051221
Chicago/Turabian StyleTakahashi, Hideo, Tsutomu Kawaguchi, Li Yan, Xuan Peng, Qianya Qi, Luc G.T. Morris, Timothy A. Chan, Allan Tsung, Eigo Otsuji, and Kazuaki Takabe. 2020. "Immune Cytolytic Activity for Comprehensive Understanding of Immune Landscape in Hepatocellular Carcinoma" Cancers 12, no. 5: 1221. https://doi.org/10.3390/cancers12051221
APA StyleTakahashi, H., Kawaguchi, T., Yan, L., Peng, X., Qi, Q., Morris, L. G. T., Chan, T. A., Tsung, A., Otsuji, E., & Takabe, K. (2020). Immune Cytolytic Activity for Comprehensive Understanding of Immune Landscape in Hepatocellular Carcinoma. Cancers, 12(5), 1221. https://doi.org/10.3390/cancers12051221