Conditionally Replicative Adenovirus Controlled by the Stabilization System of AU-Rich Elements Containing mRNA

 , and
, and 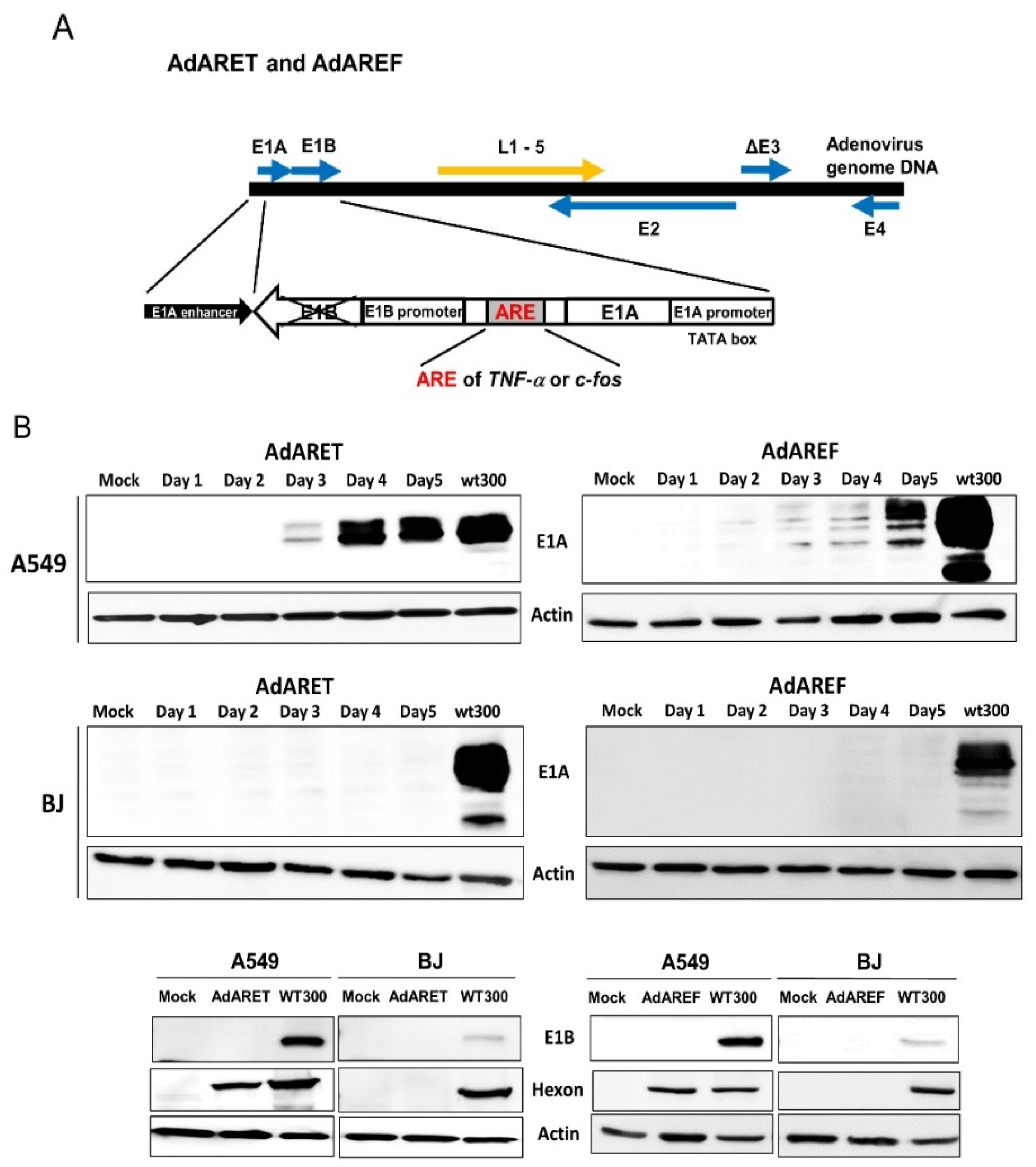{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Construction of an Adenovirus Including an ARE in the 3′-UTR of the E1A Gene and the Resulting Features of AdARET and AdAREF
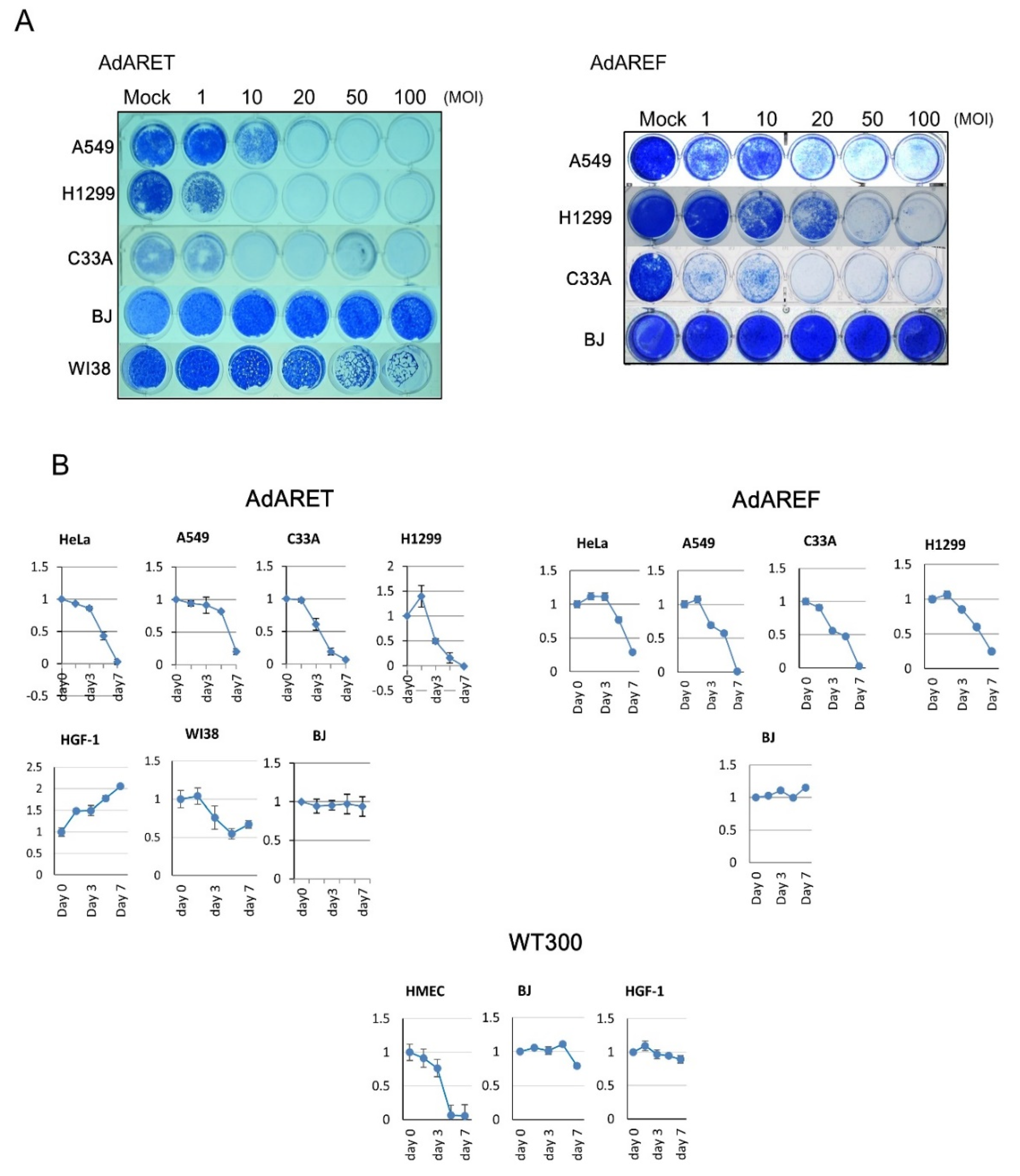
2.2. Selective Replication of AdARET and AdAREF in Cancer Cells
2.3. Requirement of HuR for AdARET and AdAREF Replication
2.4. Cytolytic Potential of AdARET and AdAREF
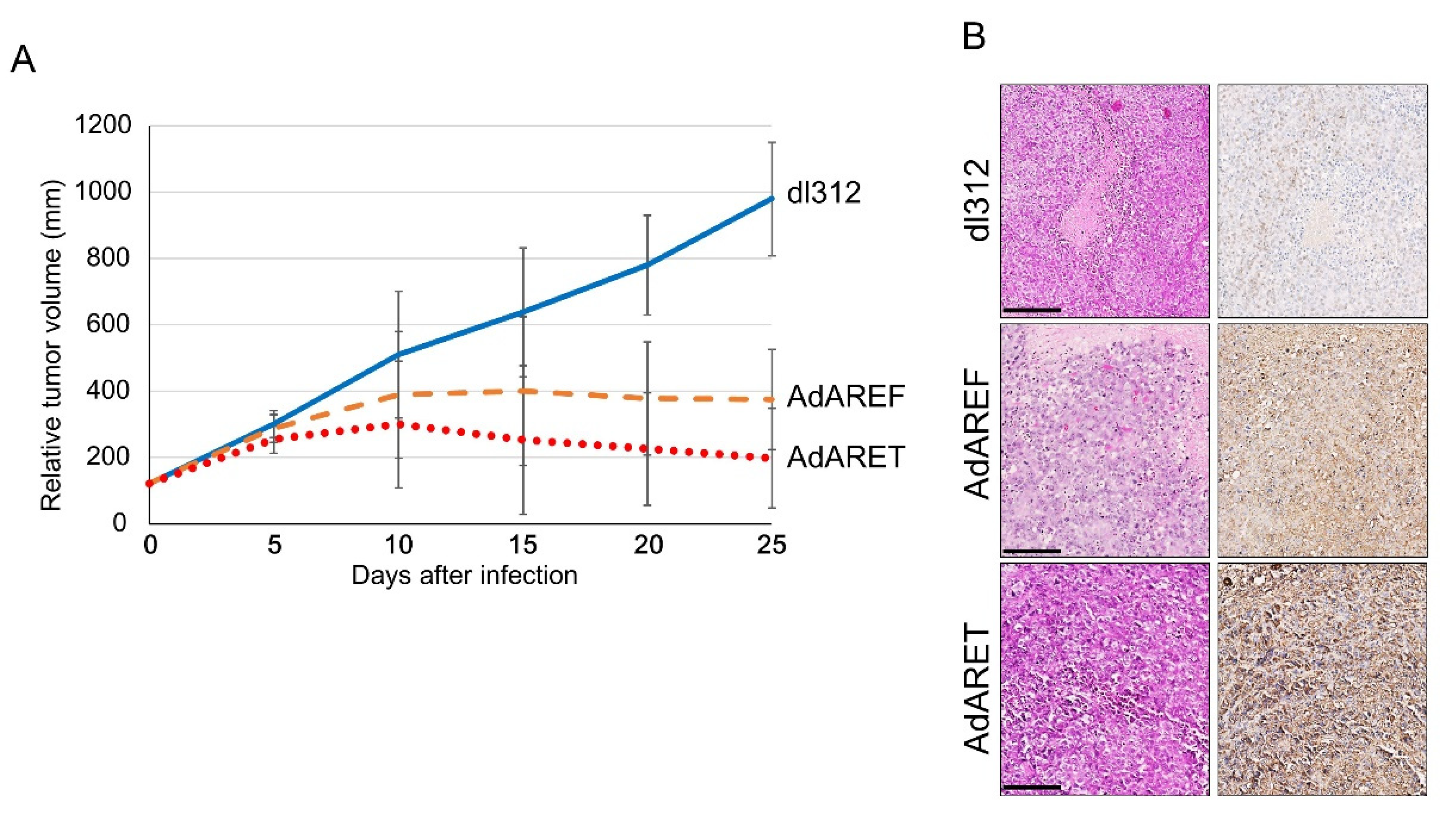
2.5. In Vivo Effects of AdARET and AdAREF
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Antibodies
4.2. Construction of AdARET and AdAREF
4.3. Western Blot Analysis
4.4. In Vitro Virus Proliferation Assay
4.5. HuR Manipulation
4.6. Immunocytochemistry
4.7. Cytopathic Effect Assay and Cell Viability Assay
4.8. In Vivo Human Tumor Model
4.9. Immunohistochemistry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Larson, C.; Oronsky, B.; Scicinski, J.; Fanger, G.R.; Stirn, M.; Oronsky, A. Going viral: A review of replication-selective oncolytic adenoviruses. Oncotarget 2015, 6, 19976–19989. [Google Scholar] [CrossRef] [PubMed]
- Bressy, C.; Benihoud, K. Association of oncolytic adenoviruses with chemotherapies: An overview and future directions. Biochem. Pharmacol. 2014, 90, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, J.R.; Kirn, D.H.; Williams, A.; Heise, C.; Horn, S.; Muna, M. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 1996, 274, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Heise, C.; Hermiston, T.; Johnson, L.; Brooks, G.; Sampson-Johannes, A.; Williams, A. An adenovirus E1A mutant that demonstrates potent and selective systemic anti-tumor efficacy. Nat. Med. 2000, 6, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, T.; Kagawa, S.; Kobayashi, N.; Shirakiya, Y.; Umeoka, T.; Teraishi, F. Telomerase-specific replication-selective virotherapy for human cancer. Clin. Cancer Res. 2004, 10, 285–292. [Google Scholar] [CrossRef]
- Rodriguez, R.; Schuur, E.R.; Lim, H.Y.; Henderson, G.A.; Simons, J.W.; Henderson, D.R. Prostate attenuated replication competent adenovirus (ARCA) CN706: A selective cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Res. 1997, 57, 2559–2563. [Google Scholar]
- Chen, C.Y.; Shyu, A.B. AU-rich elements: Characterization and importance in mRNA degradation. Trends Biochem. Sci. 1995, 20, 465–470. [Google Scholar] [CrossRef]
- Jacobson, A.; Peltz, S.W. Interrelationships of the pathways of mRNA decay and translation in eukaryotic cells. Annu. Rev. Biochem. 1996, 65, 693–739. [Google Scholar] [CrossRef]
- Brennan, C.M.; Steitz, J.A. HuR and mRNA stability. Cell Mol. Life Sci. 2001, 58, 266–277. [Google Scholar] [CrossRef]
- Hinman, M.N.; Lou, H. Diverse molecular functions of Hu proteins. Cell Mol. Life Sci. 2008, 65, 3168–3181. [Google Scholar] [CrossRef]
- Lopez de Silanes, I.; Lal, A.; Gorospe, M. HuR: Post-transcriptional paths to malignancy. RNA Biol. 2005, 2, 11–13. [Google Scholar] [CrossRef] [PubMed]
- Higashino, F.; Aoyagi, M.; Takahashi, A.; Ishino, M.; Taoka, M.; Isobe, T. Adenovirus E4orf6 targets pp32/LANP to control the fate of ARE-containing mRNA by perturbing the CRM1-dependent mechanism. J. Cell Biol. 2005, 170, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Kuroshima, T.; Aoyagi, M.; Yasuda, M.; Kitamura, T.; Jehung, J.P.; Ishikawa, M. Viral-mediated stabilization of AU-rich element containing mRNA contributes to cell transformation. Oncogene 2011, 30, 2912–2920. [Google Scholar] [CrossRef] [PubMed]
- Yanagawa-Matsuda, A.; Mikawa, Y.; Habiba, U.; Kitamura, T.; Yasuda, M.; Towfik-Alam, M.; Kitagawa, Y.; Minowa, K.; Shindoh, M.; Higashino, F. Oncolytic potential of an E4-deficient adenovirus that can recognize the stabilization of AU-rich element containing mRNA in cancer cells. Oncol. Rep. 2019, 41, 954–960. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, Y.; Chu, H.; Guan, Y.; Bi, J.; Wang, B. Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis. Int. J. Mol. Sci. 2013, 14, 10015–10041. [Google Scholar] [CrossRef]
- Habiba, U.; Kitamura, T.; Yanagawa-Matsuda, A.; Hida, K.; Higashino, F.; Ohiro, Y.; Totsuka, Y.; Shindoh, M. Cytoplasmic expression of HuR may be a valuable diagnostic tool for determining the potential for malignant transformation of oral verrucous borderline lesions. Oncol. Rep. 2014, 31, 1547–1554. [Google Scholar] [CrossRef]
- Ahmed, A.; Thompson, J.; Emiliusen, L.; Murphy, S.; Beauchamp, R.D.; Suzuki, K. A conditionally replicating adenovirus targeted to tumor cells through activated RAS/P-MAPK-selective mRNA stabilization. Nat. Biotechnol. 2003, 21, 771–777. [Google Scholar] [CrossRef]
- Lang, S.; Hearing, P. The adenovirus E1A oncoprotein recruits the cellular TRRAP/GCN5 histone acetyltransferase complex. Oncogene 2003, 22, 2836–2841. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Srikantan, S.; Yang, X.; Lal, A.; Kim, H.H.; Kuwano, Y. Ubiquitin-mediated proteolysis of HuR by heat shock. EMBO J. 2009, 28, 1271–1282. [Google Scholar] [CrossRef]
- Lin, W.N.; Lin, C.C.; Cheng, H.Y.; Yang, C.M. Regulation of cyclooxygenase-2 and cytosolic phospholipase A2 gene expression by lipopolysaccharide through the RNA-binding protein HuR: Involvement of NADPH oxidase reactive oxygen species and mitogen-activated protein kinases. Br. J. Pharmacol. 2011, 163, 1691–1706. [Google Scholar] [CrossRef]
- Lin, F.Y.; Chen, Y.H.; Lin, Y.W.; Tsai, J.S.; Chen, J.W.; Wang, H.J.; Chen, Y.L.; Li, C.Y.; Lin, S.J. The role of human antigen R: An RNA-binding protein; in mediating the stabilization of toll-like receptor 4 mRNA induced by endotoxin. a novel mechanism involved in vascular inflammation. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2622–2629. [Google Scholar] [CrossRef] [PubMed]
- Jehung, J.P.; Kitamura, T.; Matsuda, A.Y.; Kuroshima, T.; Alam, M.T.; Yasuda, M.; Sano, H.; Kitagawa, Y.; Minowa, K.; Shindho, M.; et al. Adenovirus infection induces HuR relocalization to facilitate virus replication. BBRC 2018, 495, 1795–1800. [Google Scholar] [CrossRef] [PubMed]
- Jones, N.; Shenk, T. Isolation of adenovirus type 5 host range deletion mutants defective for transformation in rat embryo cells. Cell 1979, 17, 683–689. [Google Scholar] [CrossRef]
- Yu, W.; Fang, H. Clinical trials with oncolytic adenovirus in China. Curr. Cancer Drug Targets 2007, 7, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Higashino, F.; Pipas, J.M.; Shenk, T. Adenovirus E4orf6 oncoprotein modulates the function of the p53-related protein, p73. Proc. Natl. Acad. Sci. USA 1998, 95, 15683–15687. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, M.; Higashino, F.; Yasuda, M.; Takahashi, A.; Sawada, Y.; Totsuka, Y. Nuclear export of the adenovirus E4orf6 protein is necessary for its ability to antagonize the apoptotic activity of the BH3-only proteins. Oncogene 2003, 22, 6919–6927. [Google Scholar] [CrossRef][Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikawa, Y.; Towfik Alam, M.; Hossain, E.; Yanagawa-Matsuda, A.; Kitamura, T.; Yasuda, M.; Habiba, U.; Ahmed, I.; Kitagawa, Y.; Shindoh, M.; et al. Conditionally Replicative Adenovirus Controlled by the Stabilization System of AU-Rich Elements Containing mRNA. Cancers 2020, 12, 1205. https://doi.org/10.3390/cancers12051205
Mikawa Y, Towfik Alam M, Hossain E, Yanagawa-Matsuda A, Kitamura T, Yasuda M, Habiba U, Ahmed I, Kitagawa Y, Shindoh M, et al. Conditionally Replicative Adenovirus Controlled by the Stabilization System of AU-Rich Elements Containing mRNA. Cancers. 2020; 12(5):1205. https://doi.org/10.3390/cancers12051205
Chicago/Turabian StyleMikawa, Yohei, Mohammad Towfik Alam, Elora Hossain, Aya Yanagawa-Matsuda, Tetsuya Kitamura, Motoaki Yasuda, Umma Habiba, Ishraque Ahmed, Yoshimasa Kitagawa, Masanobu Shindoh, and et al. 2020. "Conditionally Replicative Adenovirus Controlled by the Stabilization System of AU-Rich Elements Containing mRNA" Cancers 12, no. 5: 1205. https://doi.org/10.3390/cancers12051205
APA StyleMikawa, Y., Towfik Alam, M., Hossain, E., Yanagawa-Matsuda, A., Kitamura, T., Yasuda, M., Habiba, U., Ahmed, I., Kitagawa, Y., Shindoh, M., & Higashino, F. (2020). Conditionally Replicative Adenovirus Controlled by the Stabilization System of AU-Rich Elements Containing mRNA. Cancers, 12(5), 1205. https://doi.org/10.3390/cancers12051205