Establishment and Validation of an Individualized Cell Cycle Process-Related Gene Signature to Predict Cancer-Specific Survival in Patients with Bladder Cancer

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset Preparation and Data Processing
2.2. Candidate Selection and Signature Establishment
2.3. Additional Bioinformatic and Statistical Analyses
3. Results
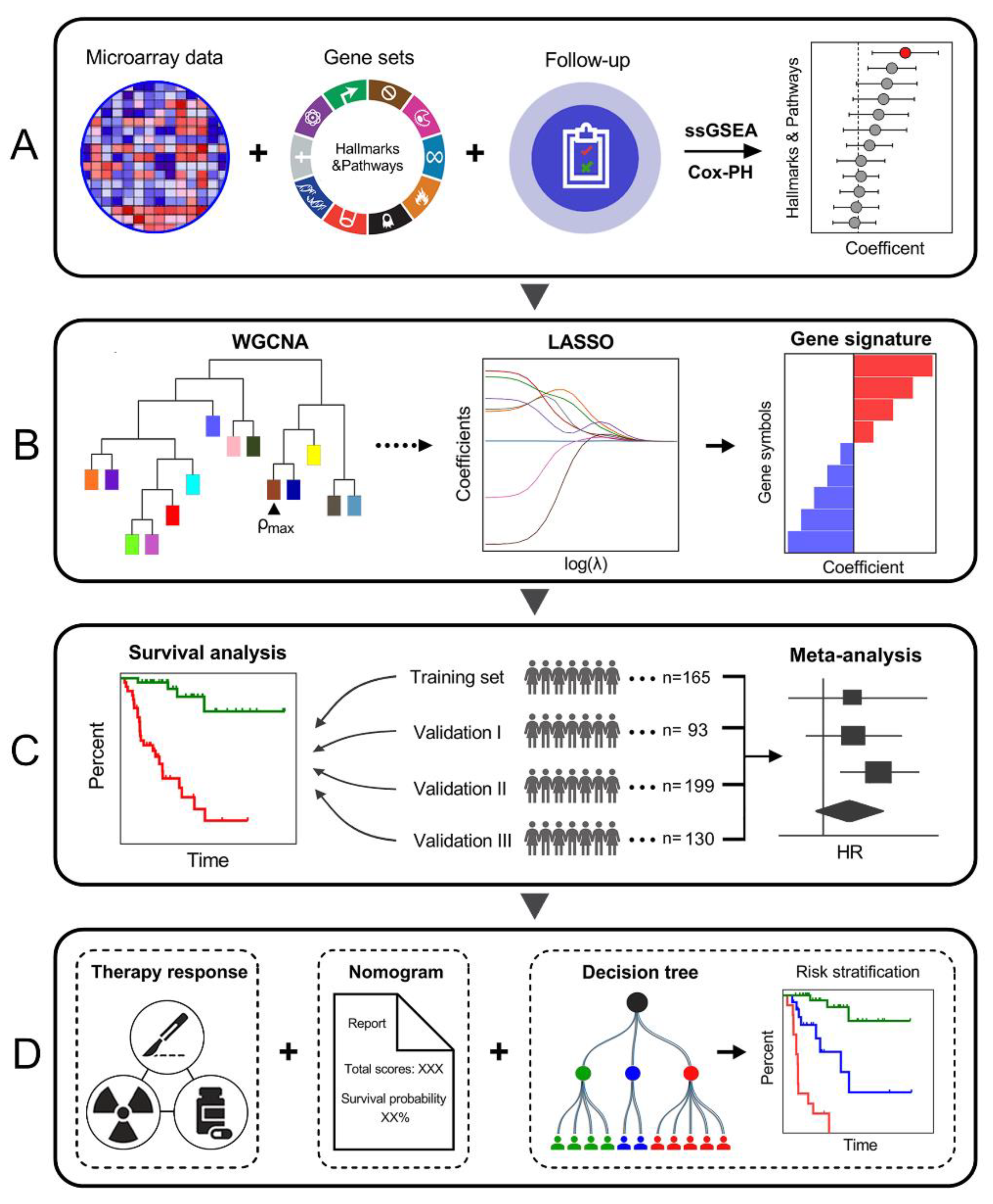
3.1. Schematic Diagram of the Study Design
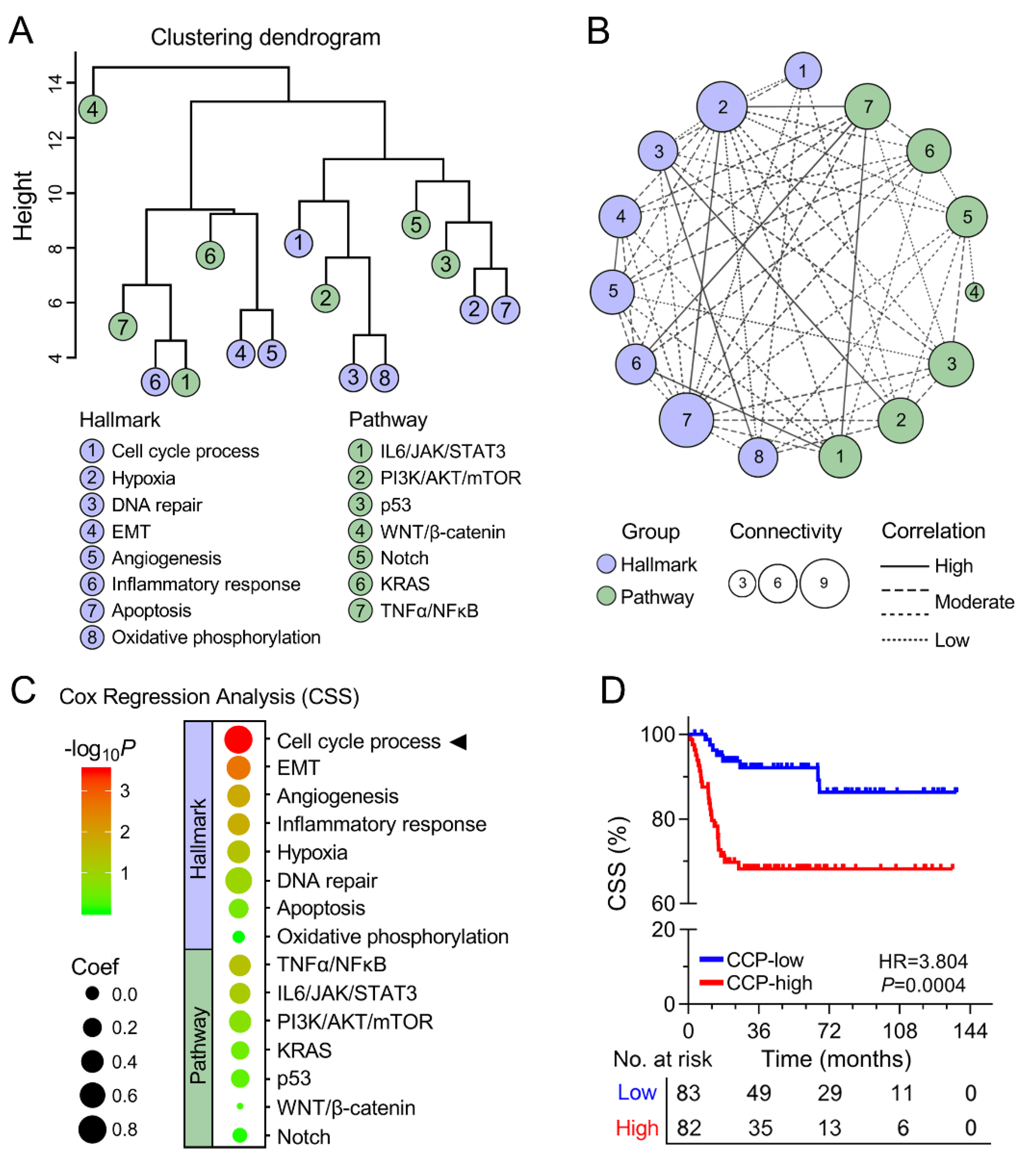
3.2. Cell Cycle Process was Identified as the Primary Risk Factor for CSS
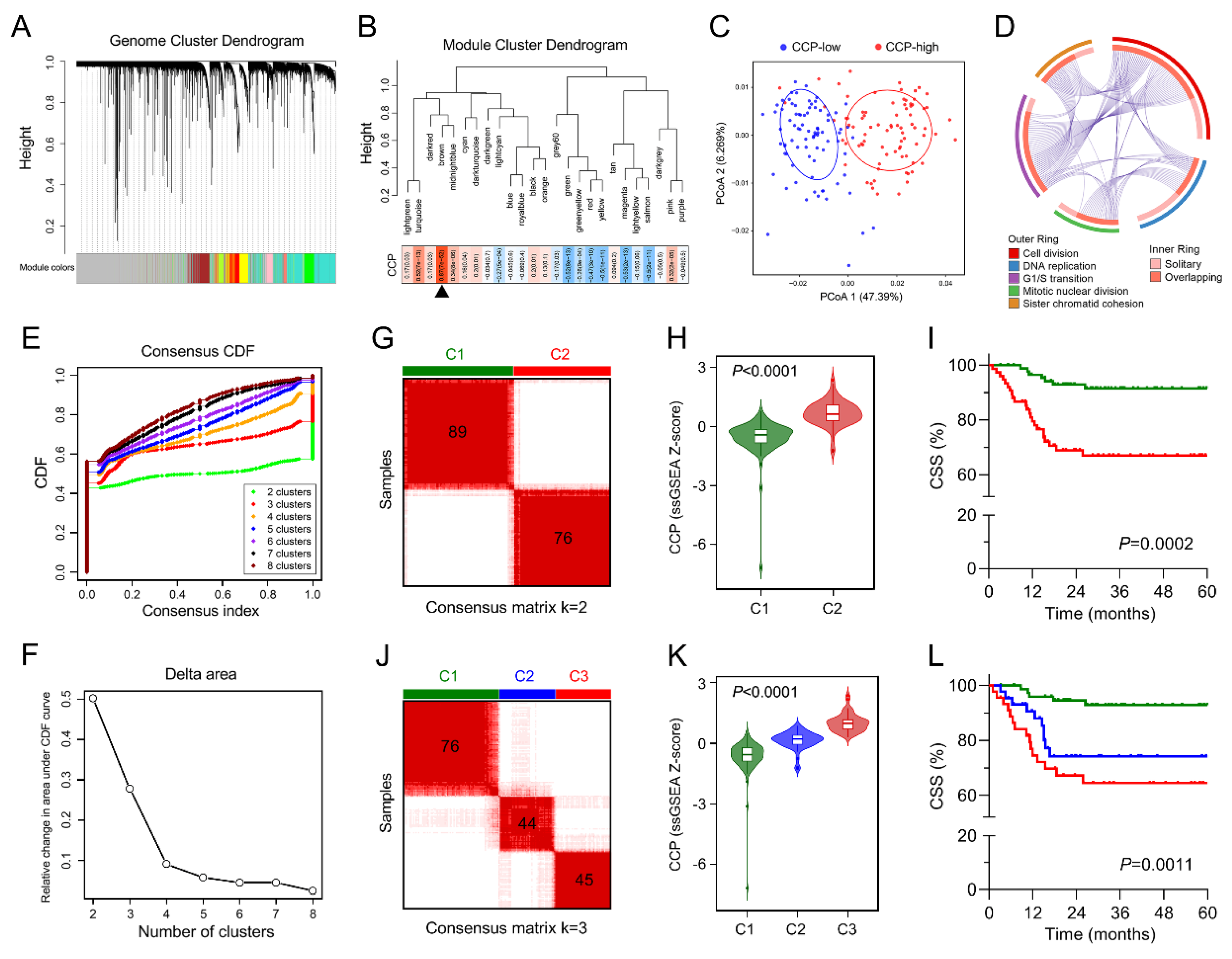
3.3. Identification of a Cell Cycle Process-Related Gene Module
3.4. Establishment of a CCP-Related Gene Signature for CSS
3.5. Copy Number Alteration is Closely Correlated with Dysregulated Expression of the Gene Signature
3.6. Higher CCPRS Predicts Worse CSS in BCa
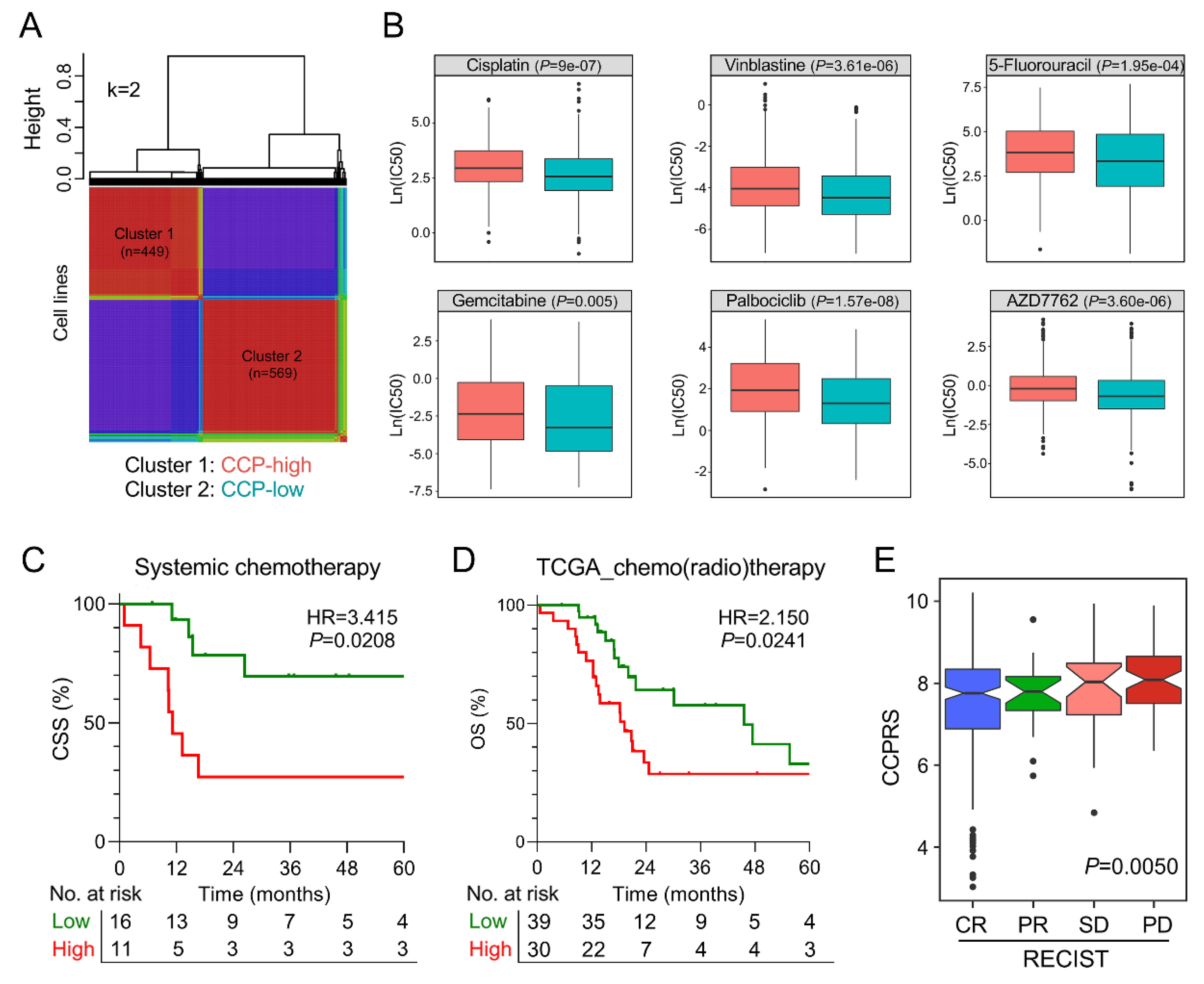
3.7. The Gene Signature Serves as A Promising Marker for Therapeutic Resistance
3.8. Combination of CCPRS and Clinical Variables Improves Risk Assessment and Stratification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BCa | Bladder cancer |
| CCP | Cell cycle process |
| CSS | Cancer-specific survival |
| MI | Muscle invasive |
| TNM | Tumor-Node-Metastasis |
| GEO | Gene Expression Omnibus |
| TCGA | The Cancer Genome Atlas |
| CCLE | Cancer Cell Line Encyclopedia |
| GDSC | Genomics of Drug Sensitivity in Cancer |
| ssGSEA | single-sample gene set enrichment analysis |
| LASSO | least absolute shrinkage and selection operator |
| WGCNA | Weighted correlation network analysis |
| GS | Gene significance |
| MM | Module membership |
| CCPRS | Cell cycle process-related risk score |
| LNM | Lymph node metastasis |
| RECIST | Response Evaluation Criteria in Solid Tumors |
| CR | Complete remission |
| PR | Partial remission |
| SD | Stable disease |
| PD | Progressive disease |
| NMF | Non-negative matrix factorization |
| tROC | Time-dependent receiver operating characteristic |
References
- Kamat, A.M.; Hahn, N.M.; Efstathiou, J.A.; Lerner, S.P.; Malmstrom, P.U.; Choi, W.; Guo, C.C.; Lotan, Y.; Kassouf, W. Bladder cancer. Lancet 2016, 388, 2796–2810. [Google Scholar] [CrossRef]
- Mak, R.H.; Hunt, D.; Shipley, W.U.; Efstathiou, J.A.; Tester, W.J.; Hagan, M.P.; Kaufman, D.S.; Heney, N.M.; Zietman, A.L. Long-term outcomes in patients with muscle-invasive bladder cancer after selective bladder-preserving combined-modality therapy: A pooled analysis of Radiation Therapy Oncology Group protocols 8802, 8903, 9506, 9706, 9906, and 0233. J. Clin. Oncol. 2014, 32, 3801–3809. [Google Scholar] [CrossRef]
- Fonteyne, V.; Ost, P.; Bellmunt, J.; Droz, J.P.; Mongiat-Artus, P.; Inman, B.; Paillaud, E.; Saad, F.; Ploussard, G. Curative Treatment for Muscle Invasive Bladder Cancer in Elderly Patients: A Systematic Review. Eur. Urol. 2018, 73, 40–50. [Google Scholar] [CrossRef]
- Kogenaru, S.; Qing, Y.; Guo, Y.; Wang, N. RNA-seq and microarray complement each other in transcriptome profiling. BMC Genom. 2012, 13, 629. [Google Scholar] [CrossRef]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA sequencing into clinical diagnostics: Opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar] [CrossRef]
- Lee, J.S.; Leem, S.H.; Lee, S.Y.; Kim, S.C.; Park, E.S.; Kim, S.B.; Kim, S.K.; Kim, Y.J.; Kim, W.J.; Chu, I.S. Expression signature of E2F1 and its associated genes predict superficial to invasive progression of bladder tumors. J. Clin. Oncol. 2010, 28, 2660–2667. [Google Scholar] [CrossRef]
- Van der Heijden, A.G.; Mengual, L.; Lozano, J.J.; Ingelmo-Torres, M.; Ribal, M.J.; Fernandez, P.L.; Oosterwijk, E.; Schalken, J.A.; Alcaraz, A.; Witjes, J.A. A five-gene expression signature to predict progression in T1G3 bladder cancer. Eur. J. Cancer 2016, 64, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Le Goux, C.; Vacher, S.; Pignot, G.; Sibony, M.; Barry Delongchamps, N.; Terris, B.; Piaggio, E.; Zerbib, M.; Damotte, D.; Bieche, I. mRNA Expression levels of genes involved in antitumor immunity: Identification of a 3-gene signature associated with prognosis of muscle-invasive bladder cancer. Oncoimmunology 2017, 6, e1358330. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Lee, D.D.; Seung, H.S. Learning the parts of objects by non-negative matrix factorization. Nature 1999, 401, 788–791. [Google Scholar] [CrossRef]
- Strobl, C.; Malley, J.; Tutz, G. An introduction to recursive partitioning: Rationale, application, and characteristics of classification and regression trees, bagging, and random forests. Psychol. Methods 2009, 14, 323–348. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.H.; Stoeber, K. The cell cycle and cancer. J. Pathol. 2012, 226, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Ooi, A.; Oyama, T.; Nakamura, R.; Tajiri, R.; Ikeda, H.; Fushida, S.; Dobashi, Y. Gene amplification of CCNE1, CCND1, and CDK6 in gastric cancers detected by multiplex ligation-dependent probe amplification and fluorescence in situ hybridization. Hum. Pathol. 2017, 61, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Roh, Y.G.; Kim, S.K.; Lee, J.S.; Seol, S.Y.; Lee, H.H.; Kim, W.T.; Kim, W.J.; Heo, J.; Cha, H.J.; et al. Activation of EZH2 and SUZ12 Regulated by E2F1 Predicts the Disease Progression and Aggressive Characteristics of Bladder Cancer. Clin. Cancer Res. 2015, 21, 5391–5403. [Google Scholar] [CrossRef]
- Watters, A.D.; Latif, Z.; Forsyth, A.; Dunn, I.; Underwood, M.A.; Grigor, K.M.; Bartlett, J.M. Genetic aberrations of c-myc and CCND1 in the development of invasive bladder cancer. Br. J. Cancer 2002, 87, 654–658. [Google Scholar] [CrossRef]
- Mitra, A.P.; Hansel, D.E.; Cote, R.J. Prognostic value of cell-cycle regulation biomarkers in bladder cancer. Semin. Oncol. 2012, 39, 524–533. [Google Scholar] [CrossRef][Green Version]
- Cuzick, J.; Swanson, G.P.; Fisher, G.; Brothman, A.R.; Berney, D.M.; Reid, J.E.; Mesher, D.; Speights, V.O.; Stankiewicz, E.; Foster, C.S.; et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: A retrospective study. Lancet Oncol. 2011, 12, 245–255. [Google Scholar] [CrossRef]
- Sommariva, S.; Tarricone, R.; Lazzeri, M.; Ricciardi, W.; Montorsi, F. Prognostic Value of the Cell Cycle Progression Score in Patients with Prostate Cancer: A Systematic Review and Meta-analysis. Eur. Urol. 2016, 69, 107–115. [Google Scholar] [CrossRef]
- Hollander, L.L.; Guo, X.; Salem, R.R.; Cha, C.H. The novel tumor angiogenic factor, adrenomedullin-2, predicts survival in pancreatic adenocarcinoma. J. Surg. Res. 2015, 197, 219–224. [Google Scholar] [CrossRef]
- Wang, L.J.; Xiao, F.; Kong, L.M.; Wang, D.N.; Li, H.Y.; Wei, Y.G.; Tan, C.; Zhao, H.; Zhang, T.; Cao, G.Q.; et al. Intermedin Enlarges the Vascular Lumen by Inducing the Quiescent Endothelial Cell Proliferation. Arter. Thromb. Vasc. Biol. 2018, 38, 398–413. [Google Scholar] [CrossRef]
- Bergo, M.O.; Ambroziak, P.; Gregory, C.; George, A.; Otto, J.C.; Kim, E.; Nagase, H.; Casey, P.J.; Balmain, A.; Young, S.G. Absence of the CAAX endoprotease Rce1: Effects on cell growth and transformation. Mol. Cell Biol. 2002, 22, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Li, M.; Wang, D.; He, J.; Wu, W.; Zeng, Q.; Li, J.; Xiao, M.; Hu, J.; He, Y.; et al. Overexpressed Rce1 is positively correlated with tumor progression and predicts poor prognosis in prostate cancer. Hum. Pathol. 2016, 47, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Wang, Y.; He, L.; Huang, G.; Du, Y.; Zhang, G.; Yan, X.; Xia, P.; Ye, B.; Wang, S.; et al. ZIC2-dependent OCT4 activation drives self-renewal of human liver cancer stem cells. J. Clin. Investig. 2015, 125, 3795–3808. [Google Scholar] [CrossRef]
- Bilguvar, K.; Ozturk, A.K.; Louvi, A.; Kwan, K.Y.; Choi, M.; Tatli, B.; Yalnizoglu, D.; Tuysuz, B.; Caglayan, A.O.; Gokben, S.; et al. Whole-exome sequencing identifies recessive WDR62 mutations in severe brain malformations. Nature 2010, 467, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Lim, N.R.; Shohayeb, B.; Zaytseva, O.; Mitchell, N.; Millard, S.S.; Ng, D.C.H.; Quinn, L.M. Glial-Specific Functions of Microcephaly Protein WDR62 and Interaction with the Mitotic Kinase AURKA Are Essential for Drosophila Brain Growth. Stem Cell Rep. 2017, 9, 32–41. [Google Scholar] [CrossRef]
- Zeng, S.; Tao, Y.; Huang, J.; Zhang, S.; Shen, L.; Yang, H.; Pei, H.; Zhong, M.; Zhang, G.; Liu, T.; et al. WD40 repeat-containing 62 overexpression as a novel indicator of poor prognosis for human gastric cancer. Eur. J. Cancer 2013, 49, 3752–3762. [Google Scholar] [CrossRef]
- Shinmura, K.; Kato, H.; Kawanishi, Y.; Igarashi, H.; Inoue, Y.; Yoshimura, K.; Nakamura, S.; Fujita, H.; Funai, K.; Tanahashi, M.; et al. WDR62 overexpression is associated with a poor prognosis in patients with lung adenocarcinoma. Mol. Carcinog. 2017, 56, 1984–1991. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, R.; Bao, X.; Rogowski, P.; Schäfer, C.; Schmidt-Hegemann, N.-S.; Unger, K.; Lu, S.; Sun, J.; Buchner, A.; Stief, C.; et al. Establishment and Validation of an Individualized Cell Cycle Process-Related Gene Signature to Predict Cancer-Specific Survival in Patients with Bladder Cancer. Cancers 2020, 12, 1146. https://doi.org/10.3390/cancers12051146
Shi R, Bao X, Rogowski P, Schäfer C, Schmidt-Hegemann N-S, Unger K, Lu S, Sun J, Buchner A, Stief C, et al. Establishment and Validation of an Individualized Cell Cycle Process-Related Gene Signature to Predict Cancer-Specific Survival in Patients with Bladder Cancer. Cancers. 2020; 12(5):1146. https://doi.org/10.3390/cancers12051146
Chicago/Turabian StyleShi, Run, Xuanwen Bao, Paul Rogowski, Christian Schäfer, Nina-Sophie Schmidt-Hegemann, Kristian Unger, Shun Lu, Jing Sun, Alexander Buchner, Christian Stief, and et al. 2020. "Establishment and Validation of an Individualized Cell Cycle Process-Related Gene Signature to Predict Cancer-Specific Survival in Patients with Bladder Cancer" Cancers 12, no. 5: 1146. https://doi.org/10.3390/cancers12051146
APA StyleShi, R., Bao, X., Rogowski, P., Schäfer, C., Schmidt-Hegemann, N.-S., Unger, K., Lu, S., Sun, J., Buchner, A., Stief, C., Belka, C., & Li, M. (2020). Establishment and Validation of an Individualized Cell Cycle Process-Related Gene Signature to Predict Cancer-Specific Survival in Patients with Bladder Cancer. Cancers, 12(5), 1146. https://doi.org/10.3390/cancers12051146