SNAI1-Driven Sequential EMT Changes Attributed by Selective Chromatin Enrichment of RAD21 and GRHL2

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
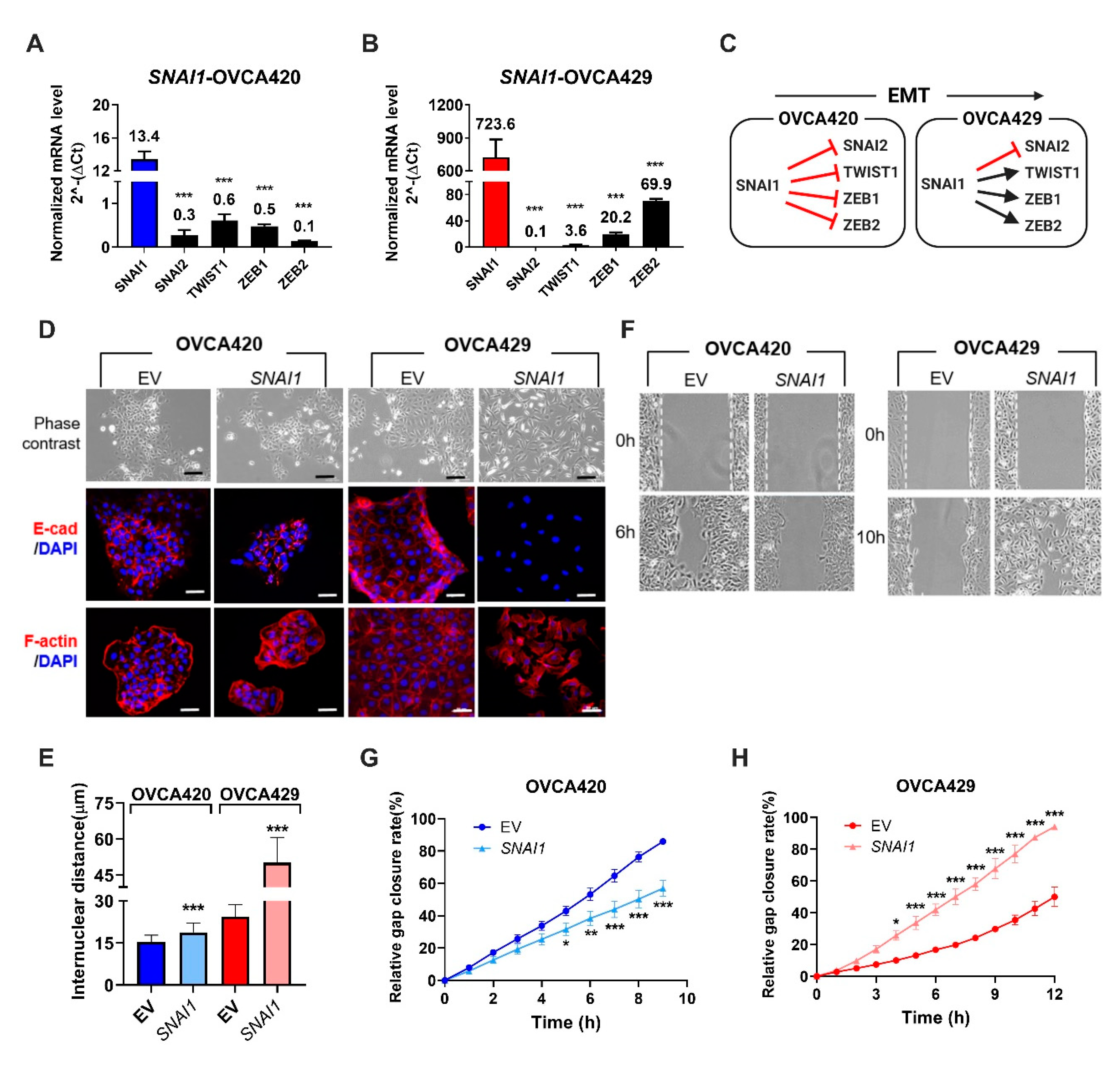
2.1. Expression of SNAI1 Uncovers the Presence of Sequential EMT Changes Along the EMT Spectrum
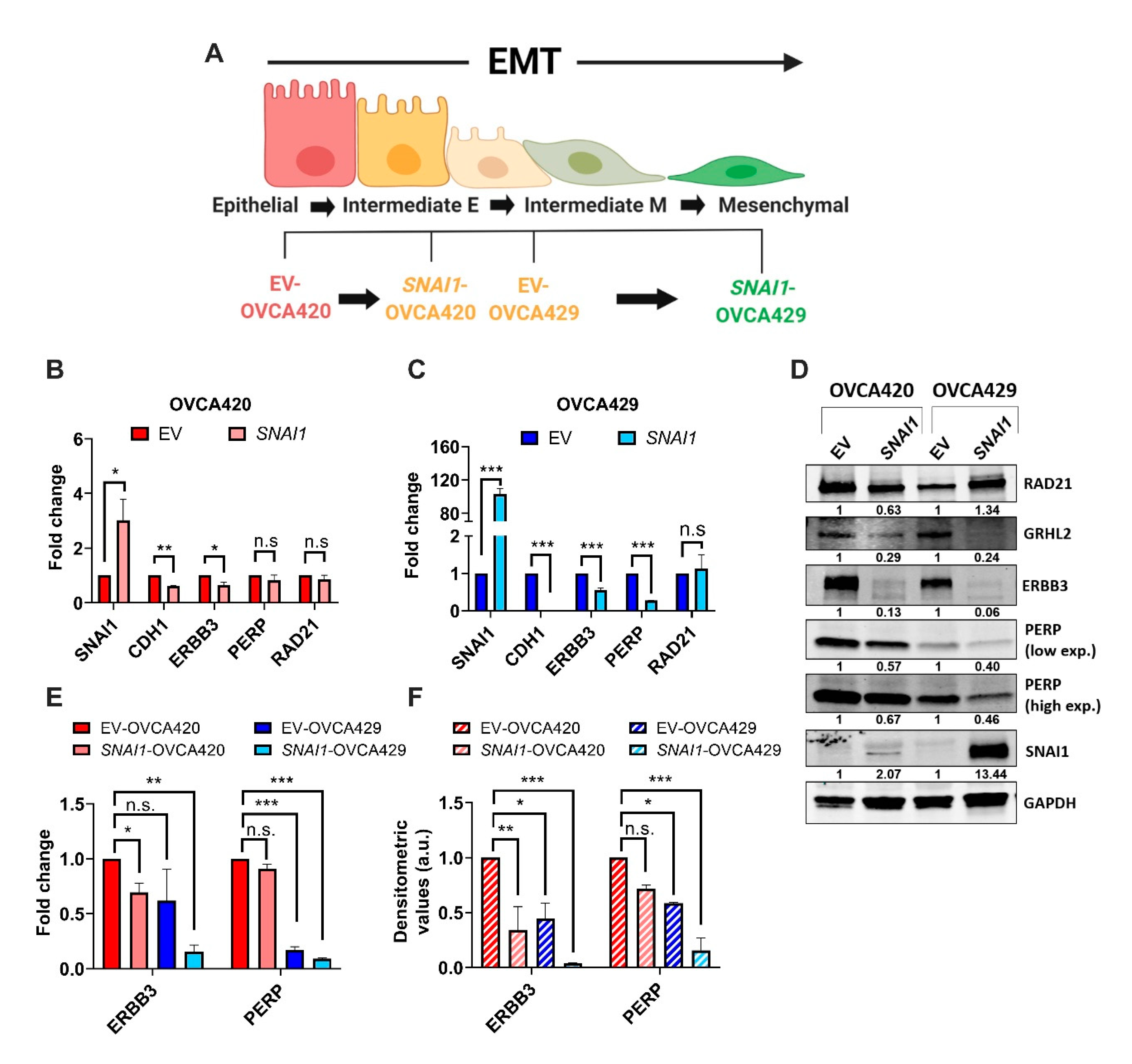
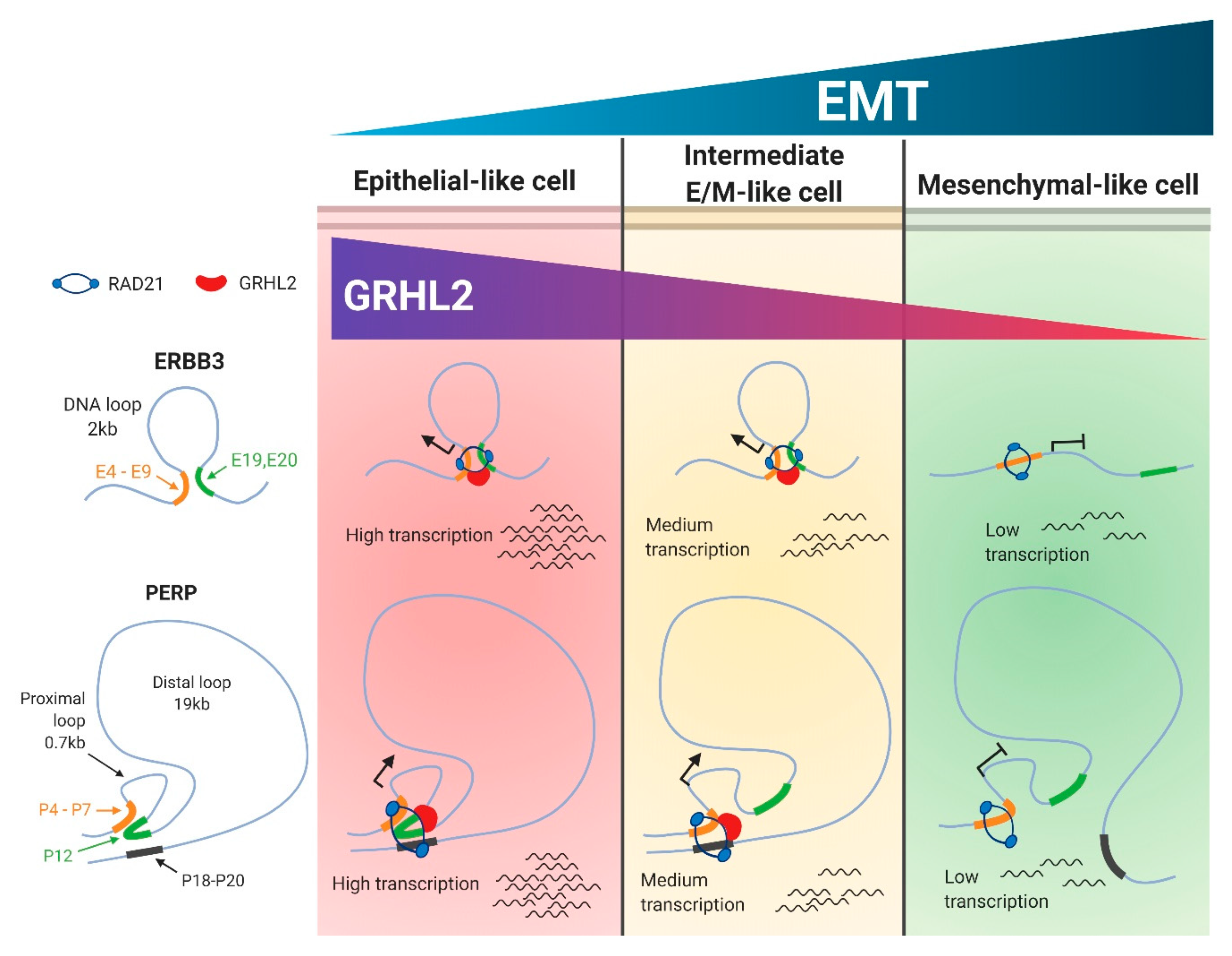
2.2. ERBB3 and PERP as Targets of SNAI1 Induced Sequential EMT Changes
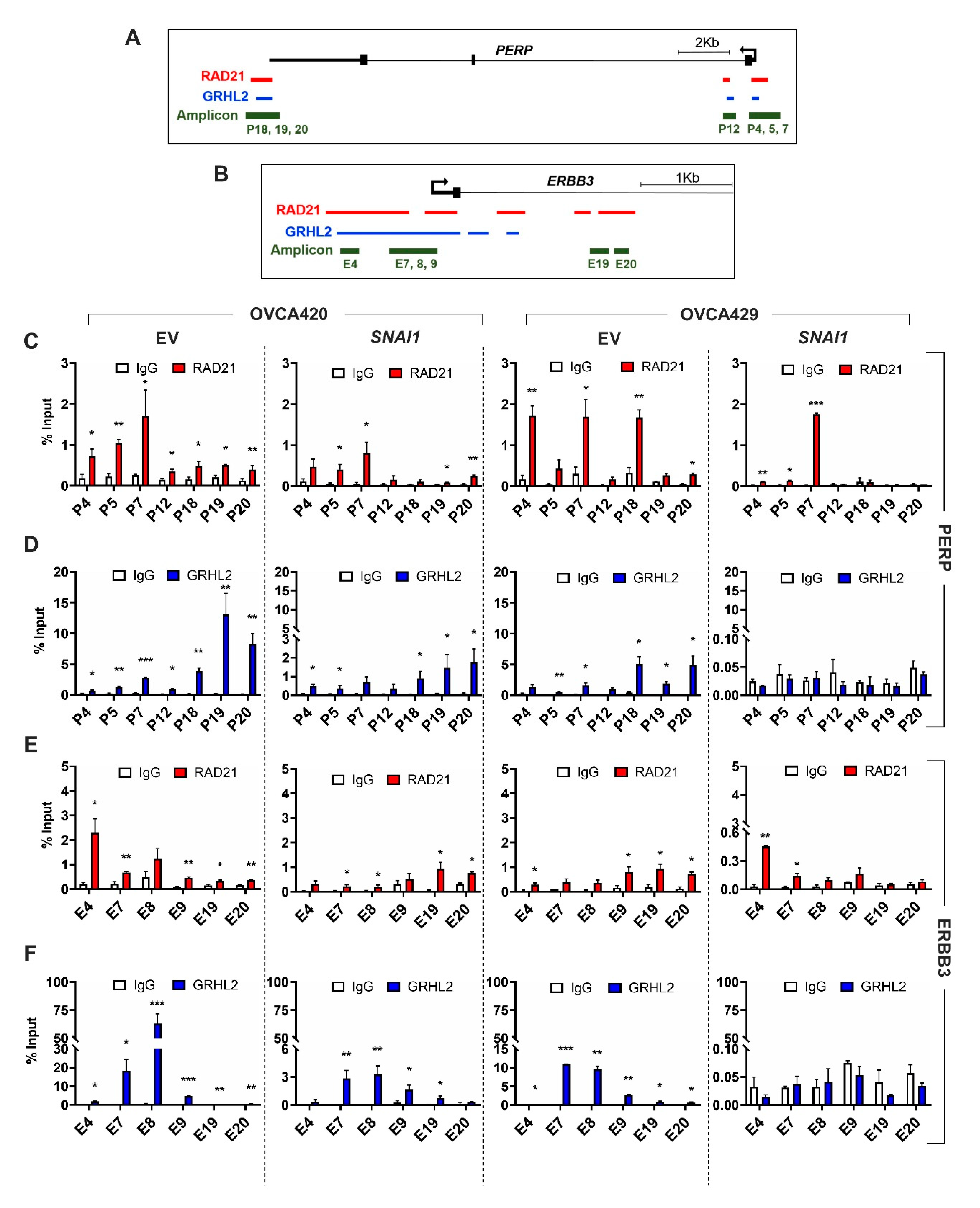
2.3. Selective Enrichment of Cohesin-Complex Component RAD21 and Transcription Factor GRHL2 on PERP and ERBB3 Loci Modulate Target Gene Transcription along the EMT Spectrum
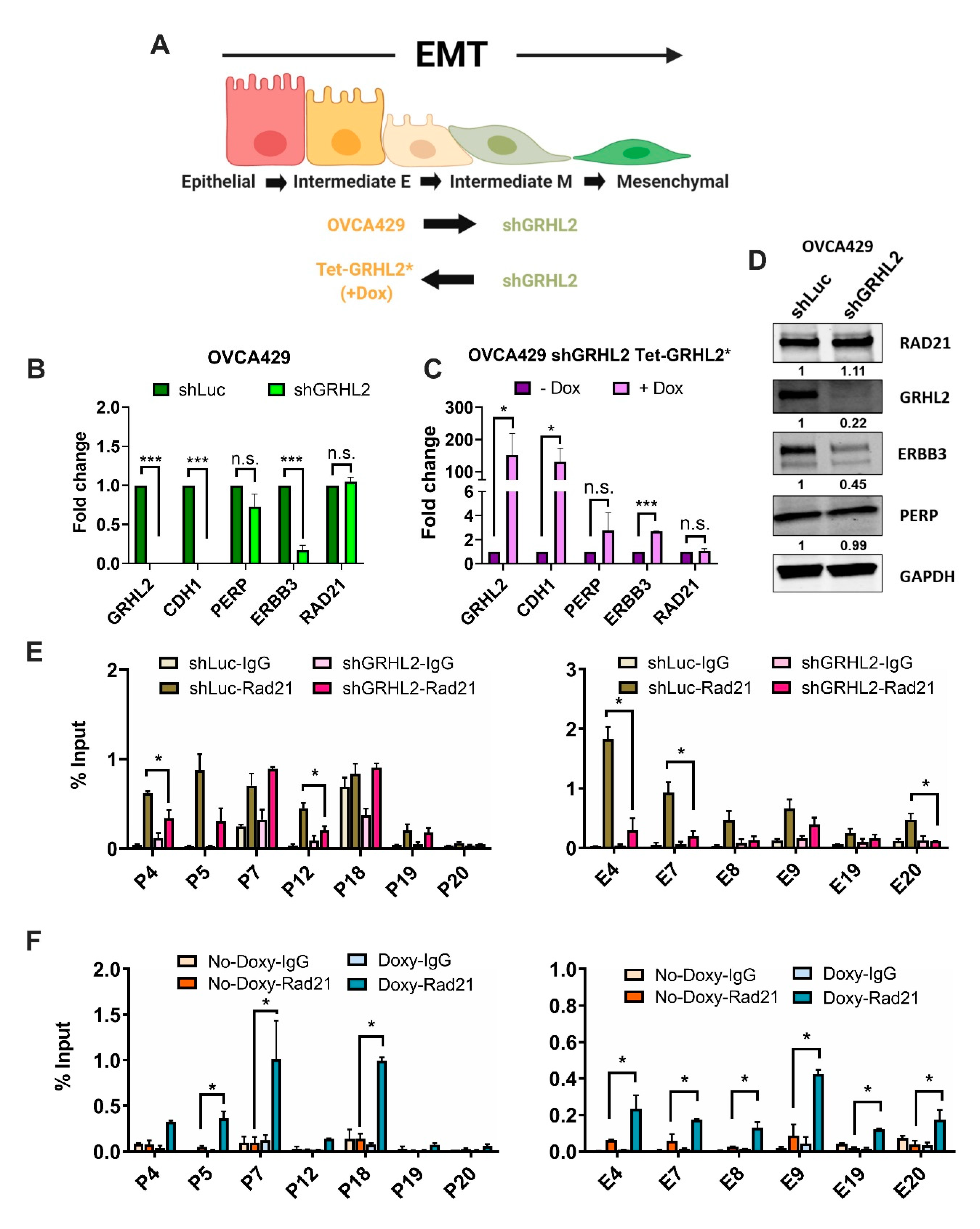
2.4. Enrichment of RAD21 on PERP and ERBB3 Loci Is Partially Dependent on the Abundance of GRHL2 Expression
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Generation of Stable Cell Lines
4.2. Real Time-Quantitative PCR (RT-qPCR) and Chromatin Immunoprecipitation-qPCR (ChIP-qPCR)
4.3. Western Blotting
4.4. Immunofluorescence Staining and Analysis of Internuclear Distance
4.5. Gap Closure Migration Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial–mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Invest. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Jordan, N.V.; Johnson, G.L.; Abell, A.N. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle 2011, 10, 2865–2873. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Huang, R.Y.-J.; Wong, M.K.; Tan, T.Z.; Kuay, K.T.; Ng, A.H.C.; Chung, V.Y.; Chu, Y.-S.; Matsumura, N.; Lai, H.-C.; Lee, Y.F.; et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death Dis. 2013, 4, e915. [Google Scholar] [CrossRef]
- Fustaino, V.; Presutti, D.; Colombo, T.; Cardinali, B.; Papoff, G.; Brandi, R.; Bertolazzi, P.; Felici, G.; Ruberti, G. Characterization of epithelial-mesenchymal transition intermediate/hybrid phenotypes associated to resistance to EGFR inhibitors in non-small cell lung cancer cell lines. Oncotarget 2017, 8, 103340–103363. [Google Scholar] [CrossRef]
- Grosse-Wilde, A.; Fouquier d’Hérouël, A.; McIntosh, E.; Ertaylan, G.; Skupin, A.; Kuestner, R.E.; del Sol, A.; Walters, K.-A.; Huang, S. Stemness of the hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival. PLoS ONE 2015, 10, e0126522. [Google Scholar] [CrossRef]
- Ruscetti, M.; Quach, B.; Dadashian, E.L.; Mulholland, D.J.; Wu, H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015, 75, 2749–2759. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, N.; Tokunaga, E.; Iimori, M.; Inoue, Y.; Tanaka, K.; Kitao, H.; Saeki, H.; Oki, E.; Maehara, Y. Epithelial Paradox: Clinical Significance of Coexpression of E-cadherin and Vimentin With Regard to Invasion and Metastasis of Breast Cancer. Clin. Breast Cancer 2018, 18, e1003–e1009. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M.; et al. Circulating Breast Tumor Cells Exhibit Dynamic Changes in Epithelial and Mesenchymal Composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Puisieux, A.; Brabletz, T.; Caramel, J. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014, 16, 488–494. [Google Scholar] [CrossRef]
- Stemmler, M.P.; Eccles, R.L.; Brabletz, S.; Brabletz, T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019, 21, 102–112. [Google Scholar] [CrossRef]
- Nieto, M.A. The snail superfamily of zinc-finger transcription factors. Nat. Rev. Mol. Cell Biol. 2002, 3, 155–166. [Google Scholar] [CrossRef]
- Carver, E.A.; Jiang, R.; Lan, Y.; Oram, K.F.; Gridley, T. The Mouse Snail Gene Encodes a Key Regulator of the Epithelial-Mesenchymal Transition. Mol. Cell. Biol. 2001, 21, 8184–8188. [Google Scholar] [CrossRef]
- Pascual-Reguant, L.; Blanco, E.; Galan, S.; Le Dily, F.; Cuartero, Y.; Serra-Bardenys, G.; Di Carlo, V.; Iturbide, A.; Cebrià-Costa, J.P.; Nonell, L.; et al. Lamin B1 mapping reveals the existence of dynamic and functional euchromatin lamin B1 domains. Nat. Commun. 2018, 9, 3420. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Song, S.-H.; Kim, H.-P.; Han, S.-W.; Yi, E.C.; Kim, T.-Y. Dynamic cohesin-mediated chromatin architecture controls epithelial–mesenchymal plasticity in cancer. EMBO Rep. 2016, 17, 1343–1359. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, H.; Basilicata, M.F.; Shehwana, H.; Kosowan, T.; Schreck, I.; Braeutigam, C.; Konu, O.; Brabletz, T.; Stemmler, M.P. Enhancer cooperativity as a novel mechanism underlying the transcriptional regulation of E-cadherin during mesenchymal to epithelial transition. Biochim. Biophys. Acta BBA Gene Regul. Mech. 2015, 1849, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Sundararajan, V.; Tan, M.; Tan, T.Z.; Ye, J.; Thiery, J.P.; Huang, R.Y.-J. SNAI1 recruits HDAC1 to suppress SNAI2 transcription during epithelial to mesenchymal transition. Sci. Rep. 2019, 9, 8295. [Google Scholar] [CrossRef] [PubMed]
- Asad, M.; Wong, M.K.; Tan, T.Z.; Choolani, M.; Low, J.; Mori, S.; Virshup, D.; Thiery, J.P.; Huang, R.Y.-J. FZD7 drives in vitro aggressiveness in Stem-A subtype of ovarian cancer via regulation of non-canonical Wnt/PCP pathway. Cell Death Dis. 2014, 5, e1346. [Google Scholar] [CrossRef] [PubMed]
- Ihrie, R.A.; Marques, M.R.; Nguyen, B.T.; Horner, J.S.; Papazoglu, C.; Bronson, R.T.; Mills, A.A.; Attardi, L.D. Perp is a p63-regulated gene essential for epithelial integrity. Cell 2005, 120, 843–856. [Google Scholar] [CrossRef]
- Chung, V.Y.; Tan, T.Z.; Tan, M.; Wong, M.K.; Kuay, K.T.; Yang, Z.; Ye, J.; Muller, J.; Koh, C.M.; Guccione, E.; et al. GRHL2-miR-200-ZEB1 maintains the epithelial status of ovarian cancer through transcriptional regulation and histone modification. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Werner, S.; Frey, S.; Riethdorf, S.; Schulze, C.; Alawi, M.; Kling, L.; Vafaizadeh, V.; Sauter, G.; Terracciano, L.; Schumacher, U.; et al. Dual Roles of the Transcription Factor Grainyhead-like 2 (GRHL2) in Breast Cancer. J. Biol. Chem. 2013, 288, 22993–23008. [Google Scholar] [CrossRef]
- Michaelis, C.; Ciosk, R.; Nasmyth, K. Cohesins: Chromosomal proteins that prevent premature separation of sister chromatids. Cell 1997, 91, 35–45. [Google Scholar] [CrossRef]
- Nasmyth, K.; Haering, C.H. Cohesin: Its roles and mechanisms. Annu. Rev. Genet. 2009, 43, 525–558. [Google Scholar] [CrossRef]
- Wendt, K.S.; Yoshida, K.; Itoh, T.; Bando, M.; Koch, B.; Schirghuber, E.; Tsutsumi, S.; Nagae, G.; Ishihara, K.; Mishiro, T.; et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature 2008, 451, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Stedman, W.; Kang, H.; Lin, S.; Kissil, J.L.; Bartolomei, M.S.; Lieberman, P.M. Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J. 2008, 27, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Cieply, B.; Riley, P.; Pifer, P.M.; Widmeyer, J.; Addison, J.B.; Ivanov, A.V.; Denvir, J.; Frisch, S.M. Suppression of the Epithelial–Mesenchymal Transition by Grainyhead-like-2. Cancer Res. 2012, 72, 2440–2453. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.F.; Liu, A.J.; Krishnakumar, R.; Freimer, J.W.; DeVeale, B.; Blelloch, R. GRHL2-Dependent Enhancer Switching Maintains a Pluripotent Stem Cell Transcriptional Subnetwork after Exit from Naive Pluripotency. Cell Stem Cell 2018, 23, 226–238.e4. [Google Scholar] [CrossRef]
- Chung, V.Y.; Tan, T.Z.; Ye, J.; Huang, R.-L.; Lai, H.-C.; Kappei, D.; Wollmann, H.; Guccione, E.; Huang, R.Y.-J. The role of GRHL2 and epigenetic remodeling in epithelial–mesenchymal plasticity in ovarian cancer cells. Commun. Biol. 2019, 2, s42003–s42019. [Google Scholar] [CrossRef]
- Chèneby, J.; Gheorghe, M.; Artufel, M.; Mathelier, A.; Ballester, B. ReMap 2018: An updated atlas of regulatory regions from an integrative analysis of DNA-binding ChIP-seq experiments. Nucleic Acids Res. 2018, 46, D267–D275. [Google Scholar] [CrossRef]
- Xiang, X.; Deng, Z.; Zhuang, X.; Ju, S.; Mu, J.; Jiang, H.; Zhang, L.; Yan, J.; Miller, D.; Zhang, H.-G. Grhl2 determines the epithelial phenotype of breast cancers and promotes tumor progression. PLoS ONE 2012, 7, e50781. [Google Scholar] [CrossRef]
- Goossens, S.; Vandamme, N.; Van Vlierberghe, P.; Berx, G. EMT transcription factors in cancer development re-evaluated: Beyond EMT and MET. Biochim. Biophys. Acta BBA Rev. Cancer 2017, 1868, 584–591. [Google Scholar] [CrossRef]
- Huang, R.Y.-J.; Guilford, P.; Thiery, J.P. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J. Cell Sci. 2012, 125, 4417–4422. [Google Scholar] [CrossRef]
- Guaita, S.; Puig, I.; Franci, C.; Garrido, M.; Dominguez, D.; Batlle, E.; Sancho, E.; Dedhar, S.; De Herreros, A.G.; Baulida, J. Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J. Biol. Chem. 2002, 277, 39209–39216. [Google Scholar] [CrossRef]
- Beltran, M.; Puig, I.; Peña, C.; García, J.M.; Álvarez, A.B.; Peña, R.; Bonilla, F.; Herreros, A.G. de A natural antisense transcript regulates Zeb2/Sip1 gene expression during Snail1-induced epithelial–mesenchymal transition. Genes Dev. 2008, 22, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Dave, N.; Guaita-Esteruelas, S.; Gutarra, S.; Frias, À.; Beltran, M.; Peiró, S.; de Herreros, A.G. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J. Biol. Chem. 2011, 286, 12024–12032. [Google Scholar] [CrossRef] [PubMed]
- Montserrat, N.; Mozos, A.; Llobet, D.; Dolcet, X.; Pons, C.; de Herreros, A.G.; Matias-Guiu, X.; Prat, J. Epithelial to mesenchymal transition in early stage endometrioid endometrial carcinoma. Hum. Pathol. 2012, 43, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Yan, M.; Patra, J.; Natrajan, R.; Yan, Y.; Swagemakers, S.; Tomaszewski, J.M.; Verschoor, S.; Millar, E.K.; van der Spek, P.; et al. Enhanced RAD21 cohesin expression confers poor prognosis and resistance to chemotherapy in high grade luminal, basal and HER2 breast cancers. Breast Cancer Res. 2011, 13, R9. [Google Scholar] [CrossRef] [PubMed]
- Deb, S.; Xu, H.; Tuynman, J.; George, J.; Yan, Y.; Li, J.; Ward, R.L.; Mortensen, N.; Hawkins, N.J.; McKay, M.J.; et al. RAD21 cohesin overexpression is a prognostic and predictive marker exacerbating poor prognosis in KRAS mutant colorectal carcinomas. Br. J. Cancer 2014, 110, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; Lajoie, B.R.; Jain, G.; Dekker, J. The long-range interaction landscape of gene promoters. Nature 2012, 489, 109–113. [Google Scholar] [CrossRef]
- Qiao, Y.; Shiue, C.-N.; Zhu, J.; Zhuang, T.; Jonsson, P.; Wright, A.P.H.; Zhao, C.; Dahlman-Wright, K. AP-1-mediated chromatin looping regulates ZEB2 transcription: New insights into TNFα-induced epithelial–mesenchymal transition in triple-negative breast cancer. Oncotarget 2015, 6, 7804–7814. [Google Scholar] [CrossRef]
- Jacobs, J.; Atkins, M.; Davie, K.; Imrichova, H.; Romanelli, L.; Christiaens, V.; Hulselmans, G.; Potier, D.; Wouters, J.; Taskiran, I.I.; et al. The transcription factor Grainy head primes epithelial enhancers for spatiotemporal activation by displacing nucleosomes. Nat. Genet. 2018, 50, 1011–1020. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sundararajan, V.; Tan, M.; Zea Tan, T.; Pang, Q.Y.; Ye, J.; Chung, V.Y.; Huang, R.Y.-J. SNAI1-Driven Sequential EMT Changes Attributed by Selective Chromatin Enrichment of RAD21 and GRHL2. Cancers 2020, 12, 1140. https://doi.org/10.3390/cancers12051140
Sundararajan V, Tan M, Zea Tan T, Pang QY, Ye J, Chung VY, Huang RY-J. SNAI1-Driven Sequential EMT Changes Attributed by Selective Chromatin Enrichment of RAD21 and GRHL2. Cancers. 2020; 12(5):1140. https://doi.org/10.3390/cancers12051140
Chicago/Turabian StyleSundararajan, Vignesh, Ming Tan, Tuan Zea Tan, Qing You Pang, Jieru Ye, Vin Yee Chung, and Ruby Yun-Ju Huang. 2020. "SNAI1-Driven Sequential EMT Changes Attributed by Selective Chromatin Enrichment of RAD21 and GRHL2" Cancers 12, no. 5: 1140. https://doi.org/10.3390/cancers12051140
APA StyleSundararajan, V., Tan, M., Zea Tan, T., Pang, Q. Y., Ye, J., Chung, V. Y., & Huang, R. Y.-J. (2020). SNAI1-Driven Sequential EMT Changes Attributed by Selective Chromatin Enrichment of RAD21 and GRHL2. Cancers, 12(5), 1140. https://doi.org/10.3390/cancers12051140