Usefulness of Two Independent DNA and RNA Tissue-Based Multiplex Assays for the Routine Care of Advanced NSCLC Patients


 ,
, 
 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Results
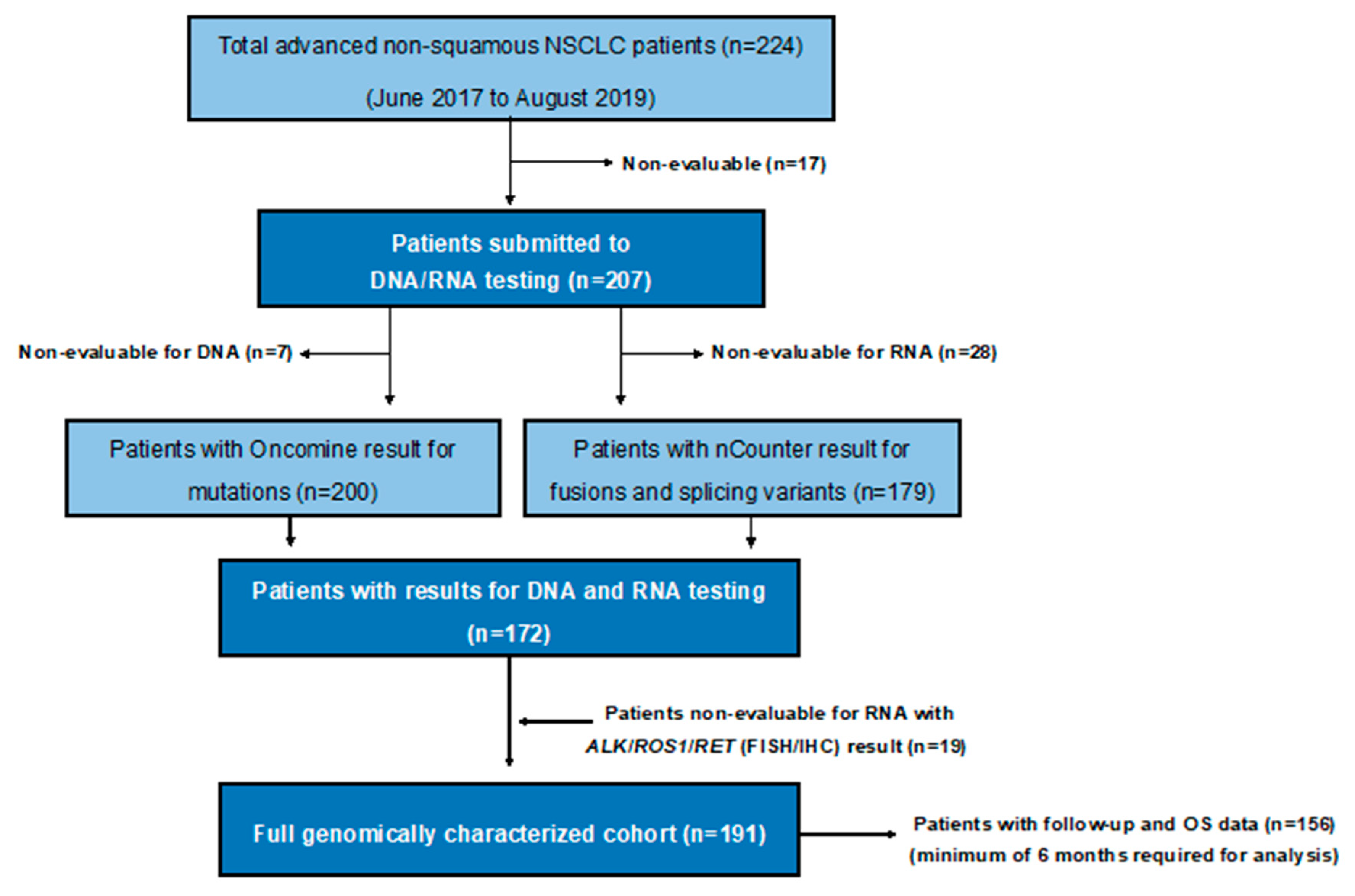
2.1. Patient Cohort and Clinical Data
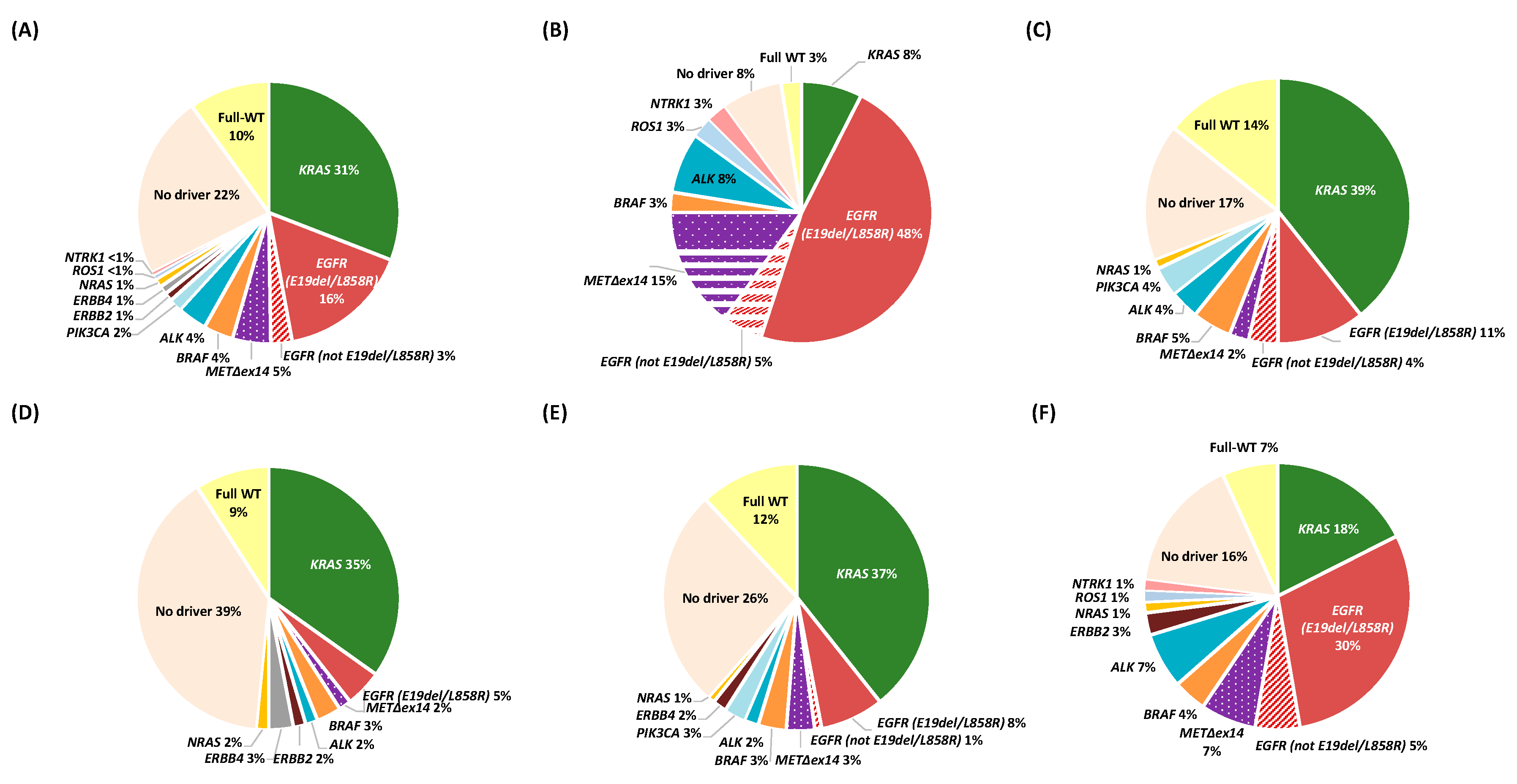
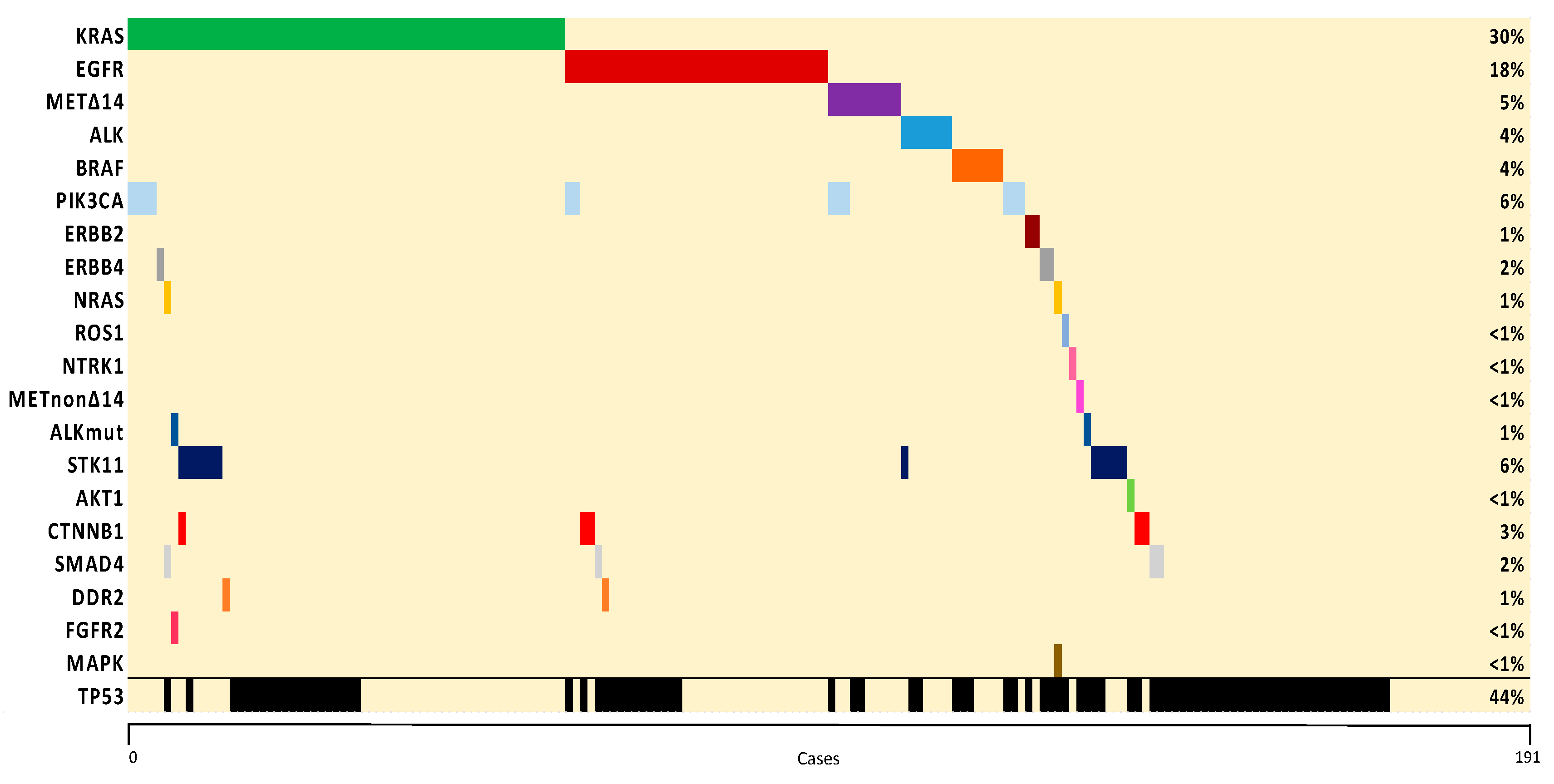
2.2. Molecular Characterization
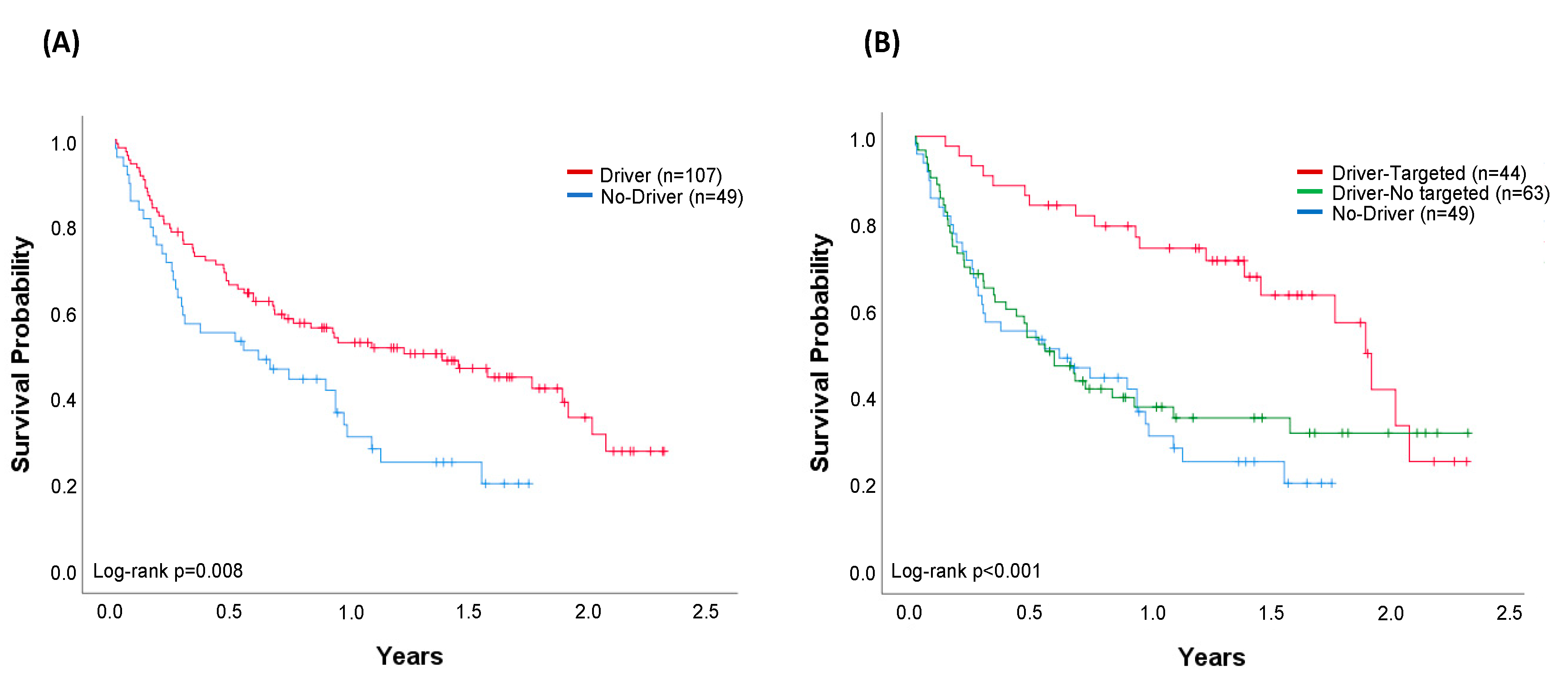
2.3. Treatment Strategies and Overall Survival (OS)
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Molecular Analyses
4.3. Molecular Subgroups
4.4. Statistical Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hunter, D.J. Uncertainty in the era of precision medicine. N. Engl. J. Med. 2016, 375, 711–713. [Google Scholar] [CrossRef]
- Pao, W.; Girard, N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011, 12, 175–180. [Google Scholar] [CrossRef]
- Planchard, D.; Hellmann, M.D.; Peters, S.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.F.; Faivre-Finn, C.; Mok, T.S.; Reck, M.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, 192–237. [Google Scholar] [CrossRef] [PubMed]
- Kris, M.G.; Johnson, B.E.; Berry, L.D.; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Varella-Garcia, M.; Franklin, W.A.; Aronson, S.L.; Su, P.-F.; et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014, 311, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Majem, M.; Juan, O.; Insa, A.; Reguart, N.; Trigo, J.M.; Carcereny, E.; García-Campelo, R.; García, Y.; Guirado, M.; Provencio, M. SEOM clinical guidelines for the treatment of non-small cell lung cancer (2018). Clin. Transl. Oncol. 2019, 21, 3–17. [Google Scholar] [CrossRef]
- Ettinger, D.S.; Aisner, D.L.; Wood, D.E.; Akerley, W.; Bauman, J.; Chang, J.Y.; Chirieac, L.R.; D’Amico, T.A.; Dilling, T.J.; Dobelbower, M.; et al. NCCN guidelines insights: Non-Small cell lung cancer, version 5.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Kalemkerian, G.P.; Narula, N.; Kennedy, E.B.; Biermann, W.A.; Donington, J.; Leighl, N.B.; Lew, M.; Pantelas, J.; Ramalingam, S.S.; Reck, M.; et al. Molecular testing guideline for the selection of patients with lung cancer for treatment with targeted tyrosine kinase inhibitors: American Society of Clinical Oncology endorsement of the College of American Pathologists/International Association for the study of lung cancer/Association for Molecular Pathology Clinical Practice guideline update. J. Clin. Oncol. 2018, 36, 911–919. [Google Scholar] [CrossRef]
- Pennell, N.A.; Arcila, M.E.; Gandara, D.R.; West, H. Biomarker testing for patients with advanced non–small cell lung cancer: Real-world issues and tough choices. Am. Soc. Clin. Oncol. Educ. Book 2019, 531–542. [Google Scholar] [CrossRef]
- Remon, J.; Lacroix, L.; Jovelet, C.; Caramella, C.; Howarth, K.; Plagnol, V.; Rosenfeld, N.; Morris, C.; Mezquita, L.; Pannet, C.; et al. Real-World utility of an amplicon-based next-generation sequencing liquid biopsy for broad molecular profiling in patients with advanced non-small-cell lung cancer. JCO Precis. Oncol. 2019, 1–14. [Google Scholar] [CrossRef]
- Europe LuCE. LuCE Report on Lung Cancer. Disparities in Diagnosis, Care and Treatment Access. 2017. Available online: https://www.lungcancereurope.eu/2017/11/07/disparities-in-diagnosis-care-and-500treatment-access/ (accessed on 23 December 2019).
- Hiley, C.T.; Le Quesne, J.; Santis, G.; Sharpe, R.; de Castro, D.G.; Middleton, G.; Swanton, C. Challenges in molecular testing in non-small-cell lung cancer patients with advanced disease. Lancet 2016, 388, 1002–1011. [Google Scholar] [CrossRef]
- Sholl, L.M.; Aisner, D.L.; Varella-Garcia, M.; Berry, L.D.; Dias-Santagata, D.; Wistuba, I.I.; Chen, H.; Fujimoto, J.; Kugler, K.; Franklin, W.A.; et al. Multi-Institutional oncogenic driver mutation analysis in lung adenocarcinoma: The lung cancer mutation consortium experience. J. Thorac. Oncol. 2015, 10, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Barlesi, F.; Mazieres, J.; Merlio, J.-P.; Debieuvre, D.; Mosser, J.; Lena, H.; Ouafik, L.H.; Besse, B.; Rouquette, I.; Westeel, V.; et al. Routine molecular profiling of patients with advanced non-small-cell lung cancer: Results of a 1-year nationwide programme of the French Cooperative Thoracic Intergroup (IFCT). Lancet 2016, 387, 1415–1426. [Google Scholar] [CrossRef]
- Volckmar, A.L.; Leichsenring, J.; Kirchner, M.; Christopoulos, P.; Neumann, O.; Budczies, J.; Morais de Oliveira, C.M.; Rempel, E.; Buchhalter, I.; Brandt, R.; et al. Combined targeted DNA and RNA sequencing of advanced NSCLC in routine molecular diagnostics: Analysis of the first 3000 Heidelberg cases. Int. J. Cancer 2019, 145, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, L.; Zhu, Y.; Huang, C.; Qin, Y.; Liu, H.; Ren-Heidenreich, L.; Shi, B.; Ren, H.; Chu, X.; et al. Coexistence of EGFR with KRAS, or BRAF, or PIK3CA somatic mutations in lung cancer: A comprehensive mutation profiling from 5125 Chinese cohorts. Br. J. Cancer 2014, 110, 2812–2820. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.H.; Johnson, A.; Albacker, L.; Wang, K.; Chmielecki, J.; Frampton, G.; Gay, L.; Elvin, J.A.; Vergilio, J.-A.; Ali, S.; et al. Comprehensive genomic profiling facilitates implementation of the national comprehensive cancer network guidelines for lung cancer biomarker testing and identifies patients who may benefit from enrollment in mechanism-driven clinical trials. Oncologist 2016, 21, 684–691. [Google Scholar] [CrossRef]
- Chatziandreou, I.; Tsioli, P.; Sakellariou, S.; Mourkioti, I.; Giannopoulou, I.; Levidou, G.; Korkolopoulou, P.; Patsouris, E.; Saetta, A.A. Comprehensive molecular analysis of NSCLC: Clinicopathological Associations. PLoS ONE 2015, 10, e0133859. [Google Scholar] [CrossRef]
- Tsoulos, N.; Papadopoulou, E.; Metaxa-Mariatou, V.; Tsaousis, G.; Efstathiadou, C.; Tounta, G.; Scapeti, A.; Bourkoula, E.; Zarogoulidis, P.; Pentheroudakis, G.; et al. Tumor molecular profiling of NSCLC patients using next generation sequencing. Oncol. Rep. 2017, 38, 3419–3429. [Google Scholar] [CrossRef][Green Version]
- Martín Martorell, P.; Huerta, M.; Compañ Quilis, A.; Abellán, R.; Seda, E.; Blesa, S.; Chaves, F.J.; Dualde Beltrán, D.; Roselló Keränen, S.; Franco, J.; et al. Coexistence of EGFR, KRAS, BRAF, and PIK3CA mutations and ALK rearrangement in a comprehensive cohort of 326 consecutive Spanish nonsquamous NSCLC patients. Clin. Lung Cancer 2017, 18, e395–e402. [Google Scholar] [CrossRef]
- Serizawa, M.; Koh, Y.; Kenmotsu, H.; Isaka, M.; Murakami, H.; Akamatsu, H.; Mori, K.; Abe, M.; Hayashi, I.; Taira, T.; et al. Assessment of mutational profile of Japanese lung adenocarcinoma patients by multitarget assays: A prospective, single-institute study. Cancer 2014, 120, 1471–1481. [Google Scholar] [CrossRef]
- Bast, E.; Morrissey, L.; Tammireddy, S.; Shaw, A.T.; Borger, D.R.; Lennes, I.T.; Baselga, J.; Engelman, J.A.; Temel, J.S.; Sequist, L.V.; et al. Implementing multiplexed genotyping of non-small-cell lung cancers into routine clinical practice. Ann. Oncol. 2011, 22, 2616–2624. [Google Scholar] [CrossRef]
- Scheffler, M.; Ihle, M.A.; Hein, R.; Merkelbach-Bruse, S.; Scheel, A.H.; Siemanowski, J.; Brägelmann, J.; Kron, A.; Abedpour, N.; Ueckeroth, F.; et al. K-ras mutation subtypes in NSCLC and associated co-occuring mutations in other oncogenic pathways. J. Thorac. Oncol. 2019, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Reguart, N.; Teixidó, C.; Giménez-Capitán, A.; Paré, L.; Galván, P.; Viteri, S.; Rodríguez, S.; Peg, V.; Aldeguer, E.; Viñolas, N.; et al. Identification of ALK, ROS1, and RET fusions by a multiplexed mRNA-based assay in formalin-fixed, paraffin-embedded samples from advanced non-small-cell lung cancer patients. Clin. Chem. 2017, 63, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Simarro, J.; Murria, R.; Pérez-Simó, G.; Llop, M.; Mancheño, N.; Ramos, D.; de Juan, I.; Barragán, E.; Laiz, B.; Cases, E.; et al. Development, implementation and assessment of molecular diagnostics by next generation sequencing in personalized treatment of cancer: Experience of a public reference healthcare hospital. Cancers 2019, 1196. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Peled, N.; Greer, J.; Wu, W.; Choi, P.; Berger, A.H.; Wong, S.; Jen, K.Y.; Seo, Y.; Hann, B.; et al. Exon 14 mutation encodes an actionable therapeutic target in lung adenocarcinoma. Cancer Res. 2017, 77, 4498–4505. [Google Scholar] [CrossRef] [PubMed]
- Reungwetwattana, T.; Liang, Y.; Zhu, V.; Ou, S.-H.I. The race to target MET exon 14 skipping alterations in non-small cell lung cancer: The why, the how, the who, the unknown, and the inevitable. Lung Cancer 2017, 103, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Poirot, B.; Doucet, L.; Benhenda, S.; Champ, J.; Meignin, V.; Lehmann-Che, J. MET exon 14 alterations and new resistance mutations to tyrosine kinase inhibitors: Risk of inadequate detection with current amplicon-based NGS panels. J. Thorac. Oncol. 2017, 12, 1582–1587. [Google Scholar] [CrossRef]
- Tong, J.H.; Yeung, S.F.; Chan, A.W.H.; Chung, L.Y.; Chau, S.L.; Lung, R.W.M.; Tong, C.Y.; Chow, C.; Tin, E.K.Y.; Yu, Y.H.; et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non-small cell lung carcinoma with poor prognosis. Clin. Cancer Res. 2016, 22, 3048–3056. [Google Scholar] [CrossRef]
- Mazza, V. New molecular drivers in NSCLC: The role of MET. Oncol. Res. Rev. 2019, 2, 1–7. [Google Scholar] [CrossRef]
- Novartis. Novartis Investigational Lung Cancer Therapy Capmatinib (INC280) Granted FDA Breakthrough Therapy Designation for Patients with MET-Mutated Advanced Non-Small Cell Lung Cancer (news release). Available online: https://www.novartis.com/news/media-releases/novartis-investigational-lung-cancer-therapy-capmatinib-inc280granted-fda-breakthrough-therapy-designation-patients-met-mutated-advanced-non-small-cell-lung (accessed on 21 December 2019).
- Wolf, J.; Seto, T.; Han, J.-Y.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.-W.; Hida, T.; De Jonge, M.J.; Orlov, S.V.; et al. Capmatinib (INC280) in METΔex14-mutated advanced non-small cell lung cancer (NSCLC): Efficacy data from the phase II GEOMETRY mono-1 study. J. Clin. Oncol. 2019, 37, 9004. [Google Scholar] [CrossRef]
- Hames, M.L.; Chen, H.; Iams, W.; Aston, J.; Lovly, C.M.; Horn, L. Correlation between KRAS mutation status and response to chemotherapy in patients with advanced non-small cell lung cancer☆. Lung Cancer 2016, 92, 29–34. [Google Scholar] [CrossRef]
- Yang, H.; Liang, S.-Q.; Schmid, R.A.; Peng, R.-W. New horizons in KRAS-mutant lung cancer: Dawn after darkness. Front. Oncol. 2019, 9, 953. [Google Scholar] [CrossRef] [PubMed]
- Govindan, R.; Fakih, M.G.; Price, T.J.; Falchook, G.S.; Desai, J.; Kuo, J.C.; Strickler, J.H.; Krauss, J.C.; Li, B.T.; Denlinger, C.S.; et al. 446PD phase I study of AMG 510, a novel molecule targeting KRAS G12C mutant solid tumours. Ann. Oncol. 2019, 30, v159–v193. [Google Scholar] [CrossRef]
- Goldstraw, P.; Crowley, J.; Chansky, K.; Giroux, D.J.; Groome, P.A.; Rami-Porta, R.; Postmus, P.E.; Rusch, V.; Sobin, L. The IASLC lung cancer staging project: Proposals for the revision of the TNM stage groupings in the forthcoming (seventh) edition of the TNM classification of malignant tumours. J. Thorac. Oncol. 2007, 2, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.G.; Zhang, S.M.; Ding, X.X.; He, B.; Zhang, H.Q. Driver genes in non-small cell lung cancer: Characteristics, detection methods, and targeted therapies. Oncotarget 2017, 8, 57680–57692. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Hanker, A.B.; Garrett, J.T. Genomic alterations of ERBB receptors in cancer: Clinical implications. Oncotarget 2017, 8, 114371–114392. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, K.; Sequist, L.V.; Arcila, M.E.; Lovly, C.M.; Chen, X.; Rudin, C.M.; Moran, T.; Camidge, D.R.; Vnencak-Jones, C.L.; Berry, L.; et al. Characteristics of lung cancers harboring NRAS mutations. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 2584–2591. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Total, N. (%) (n = 224) | Targeted Therapy, N. (%) (n = 45) | No Targeted Therapy, N. (%) (n = 179) |
|---|---|---|---|
| Age at metastatic disease, median (IQR) | 70 (60–77) | 70 (59–77) | 69 (60–76) |
| Sex | |||
| Women | 82 (37) | 30 (67) | 52 (29) |
| Men | 142 (63) | 15 (33) | 127 (71) |
| Histology | |||
| Adenocarcinoma | 206 (92) | 44 (98) | 162 (91) |
| Other 1 | 18 (8) | 1 (2) | 17 (9) |
| Performance Status 2 | |||
| 0 | 32 (14) | 11 (24) | 21 (12) |
| 1 | 96 (43) | 23 (51) | 73 (41) |
| 2 | 40 (18) | 7 (16) | 33 (18) |
| 3 | 48 (22) | 4 (9) | 44 (25) |
| 4 | 5 (2) | 0 (0) | 5 (3) |
| Unknown | 3 (1) | 0 (0) | 3 (1) |
| Smoking history | |||
| Never | 42 (19) | 26 (58) | 16 (9) |
| Former 3 | 99 (44) | 15 (33) | 84 (47) |
| Current | 82 (37) | 4 (9) | 78 (43) |
| Unknown | 1 (<1%) | 0 (0) | 1 (< 1) |
| Stage at diagnose | |||
| I | 8 (3) | 2 (4) | 6 (3) |
| II | 22 (10) | 3 (7) | 19 (11) |
| III | 33 (15) | 6 (13) | 27 (15) |
| IV | 161 (72) | 34 (76) | 127 (71) |
| Treatment setting at molecular diagnosis | |||
| Treatment naïve | 188 (84) | 35 (78) | 153 (85) |
| Previously treated | 36 (16) | 10 (22) | 26 (15) |
| Molecular Alteration | Genotyping, N. (%) 1 (n = 191) | Targeted therapy, N. (%) 2 (n = 45) |
|---|---|---|
| Driver oncogene 3 | 129 (68) | 45 (35) |
| EGFR | 36 (19) | 27 (75) |
| EGFR E21 | 1 (<1) | 0 (0) |
| EGFR E19del | 25 (13) | 22 (88) |
| EGFR E20 | 4 (2) | 1 (25) |
| EGFR L858R | 6 (3) | 5 (83) |
| BRAF | 7 (4) | 3 (43) |
| BRAF V600E | 2 (1) | 1 (50) |
| BRAF non-V600E | 5 (3) | 2 (40) |
| KRAS | 59 (31) | 0 (0) |
| KRAS G12C | 23 (12) | 0 (0) |
| KRAS G12V | 14 (7) | 0 (0) |
| KRAS G12 (various) | 17 (9) | 0 (0) |
| KRAS G13 | 2 (1) | 0 (0) |
| KRAS (other) | 3 (2) | 0 (0) |
| MET∆ex14 | 9 (5) | 7 (78) |
| ALK | 7 (4) | 7 (100) |
| ERBB2 (HER2) | 2 (1) | 0 (0) |
| ERBB4 | 2 (1) | 0 (0) |
| PIK3CA | 3 (2) | 0 (0) |
| NTRK1 | 1 (<1) | 1 (100) |
| ROS1 | 1 (<1) | 0 (0) |
| NRAS | 2 (1) | 0 (0) |
| Non-Driver oncogene 4 | 43 (22) | 0 (0) |
| TP53 | 31 (16) | 0 (0) |
| STK11 | 3 (2) | 0 (0) |
| CTNNB1 | 1 (<1) | 0 (0) |
| >1 gene alteration | 8 (4) | 0 (0) |
| Full-WT | 19 (10) | 0 (0) |
| Variable | Multivariable | ||
|---|---|---|---|
| HR | 95% CI | p | |
| Age | 1.00 | 0.98–1.02 | 0.851 |
| Gender | 0.98 | 0.61–1.60 | 0.951 |
| ECOG | 2.47 | 1.98–3.09 | <0.001 |
| Targeted Therapy | 0.40 | 0.21–0.71 | 0.002 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marin, E.; Teixido, C.; Carmona-Rocha, E.; Reyes, R.; Arcocha, A.; Viñolas, N.; Rodríguez-Mues, M.; Cabrera, C.; Sánchez, M.; Vollmer, I.; et al. Usefulness of Two Independent DNA and RNA Tissue-Based Multiplex Assays for the Routine Care of Advanced NSCLC Patients. Cancers 2020, 12, 1124. https://doi.org/10.3390/cancers12051124
Marin E, Teixido C, Carmona-Rocha E, Reyes R, Arcocha A, Viñolas N, Rodríguez-Mues M, Cabrera C, Sánchez M, Vollmer I, et al. Usefulness of Two Independent DNA and RNA Tissue-Based Multiplex Assays for the Routine Care of Advanced NSCLC Patients. Cancers. 2020; 12(5):1124. https://doi.org/10.3390/cancers12051124
Chicago/Turabian StyleMarin, Elba, Cristina Teixido, Elena Carmona-Rocha, Roxana Reyes, Ainara Arcocha, Nuria Viñolas, MªCarmen Rodríguez-Mues, Carlos Cabrera, Marcelo Sánchez, Ivan Vollmer, and et al. 2020. "Usefulness of Two Independent DNA and RNA Tissue-Based Multiplex Assays for the Routine Care of Advanced NSCLC Patients" Cancers 12, no. 5: 1124. https://doi.org/10.3390/cancers12051124
APA StyleMarin, E., Teixido, C., Carmona-Rocha, E., Reyes, R., Arcocha, A., Viñolas, N., Rodríguez-Mues, M., Cabrera, C., Sánchez, M., Vollmer, I., Castillo, S., Muñoz, S., Sullivan, I. G., Rodriguez, A., Garcia, M., Alos, S., Jares, P., Martinez, A., Prat, A., ... Reguart, N. (2020). Usefulness of Two Independent DNA and RNA Tissue-Based Multiplex Assays for the Routine Care of Advanced NSCLC Patients. Cancers, 12(5), 1124. https://doi.org/10.3390/cancers12051124