The Role of lncRNAs TAPIR-1 and -2 as Diagnostic Markers and Potential Therapeutic Targets in Prostate Cancer

 , , , , ,
, , , , ,  , ,
, , 

Abstract
1. Introduction
2. Results
2.1. TAPIR lncRNAs as a Potent Diagnostic Prostate Cancer Biomarker
2.2. TAPIR-1 and -2 Overexpression Is Restricted to Tumor Tissue
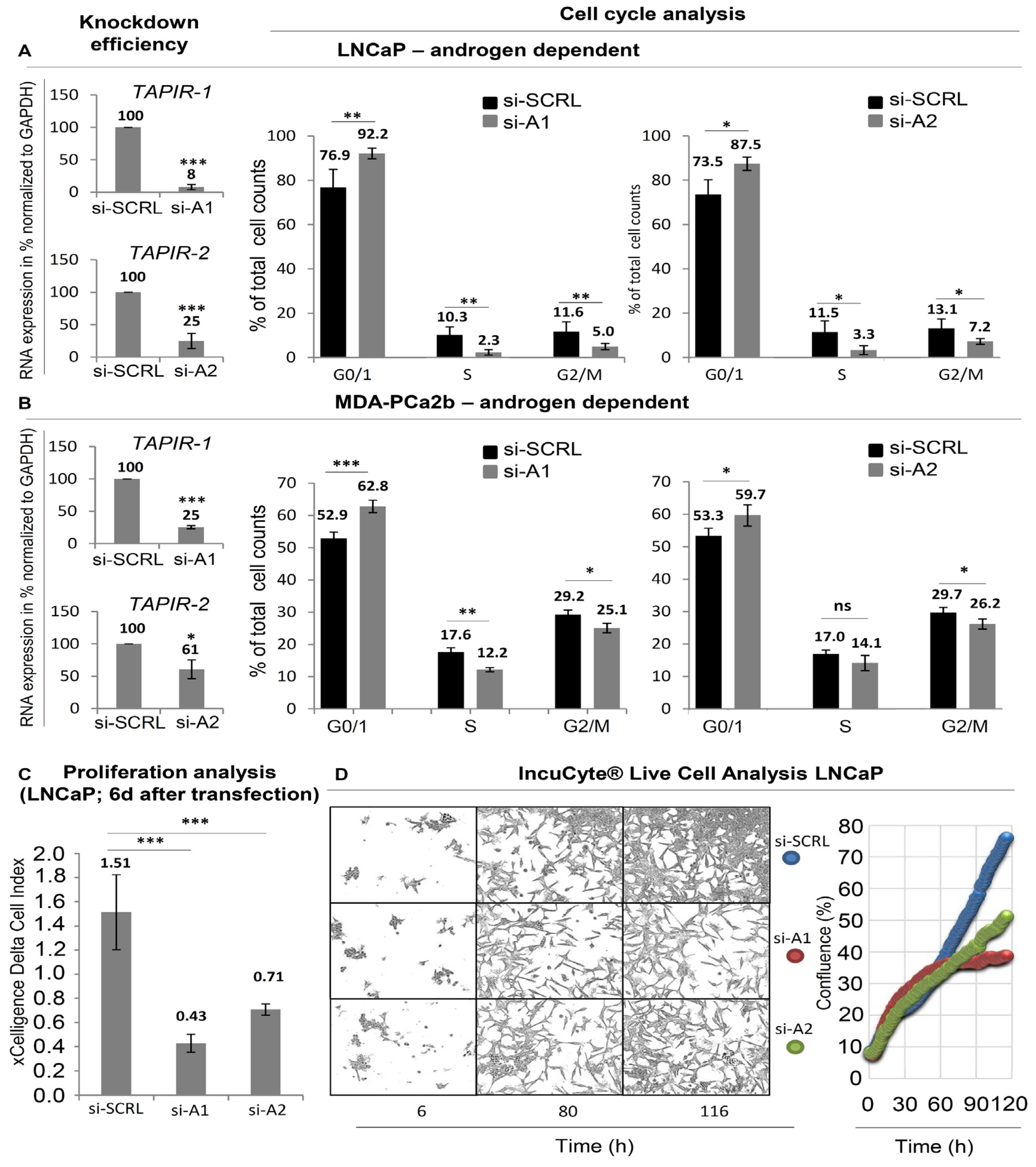
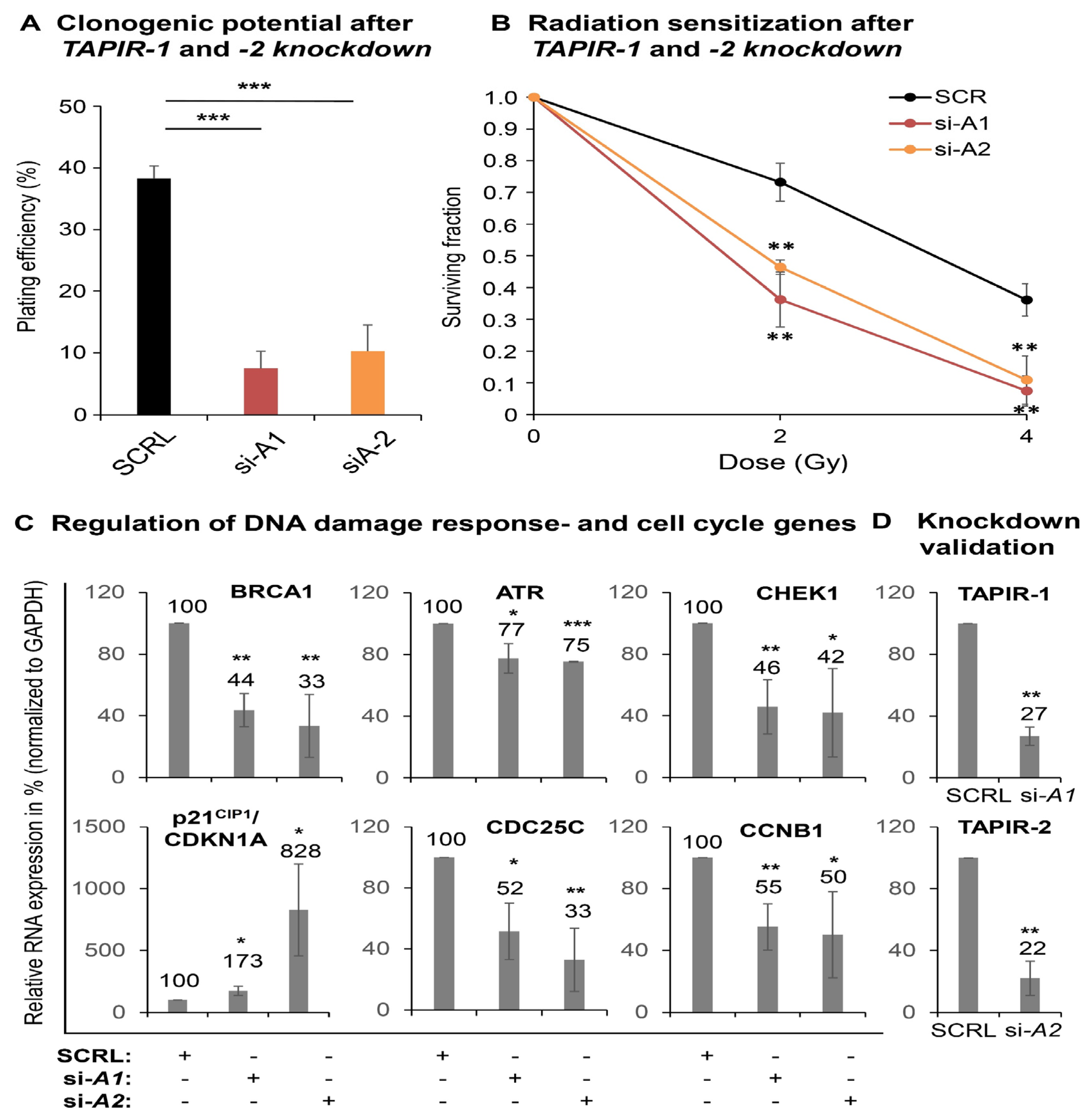
2.3. TAPIR Transcript Knockdown Causes Downregulation of Cell Cycle Genes and Growth Arrest in Prostate Tumor Cells
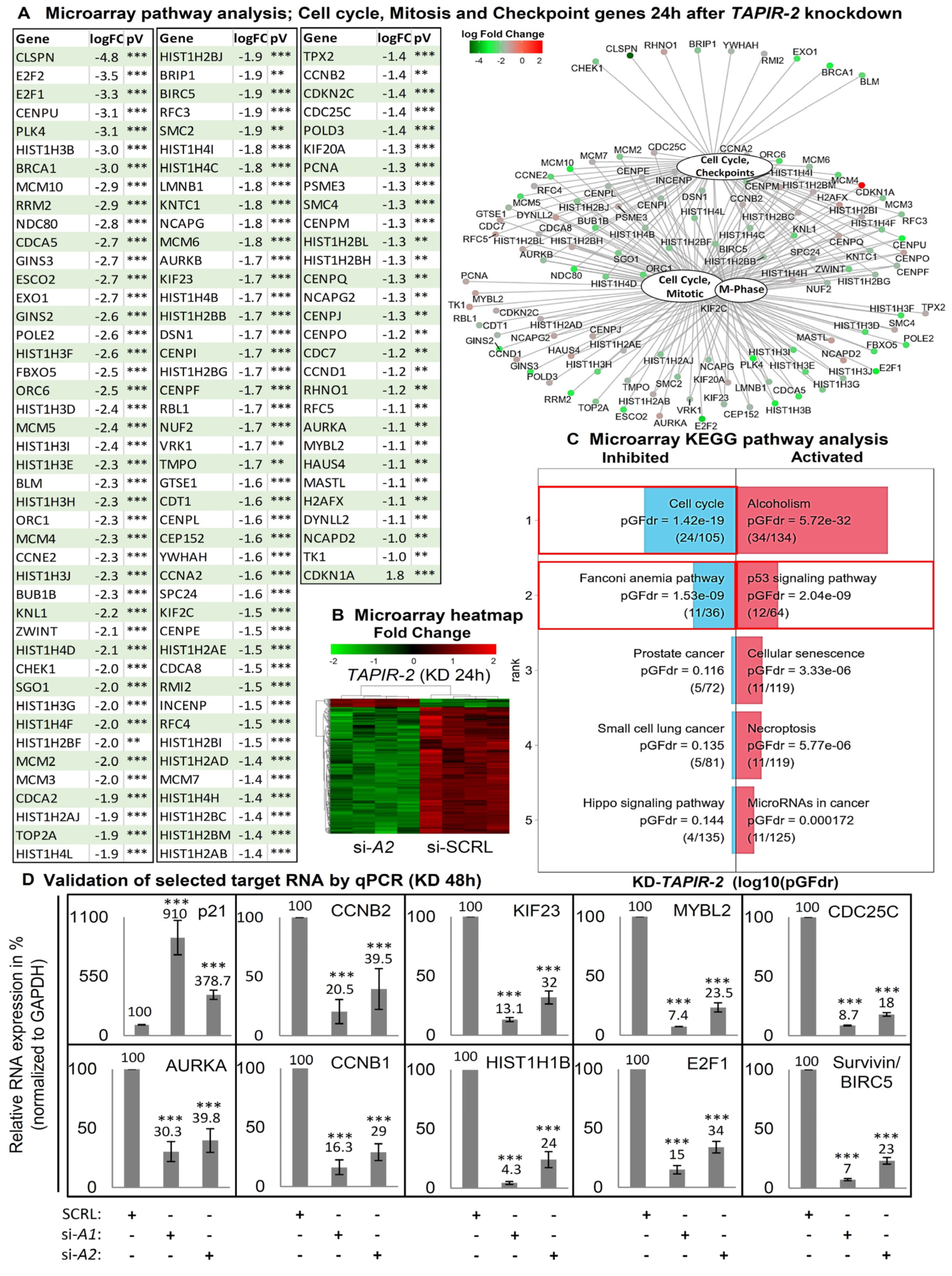
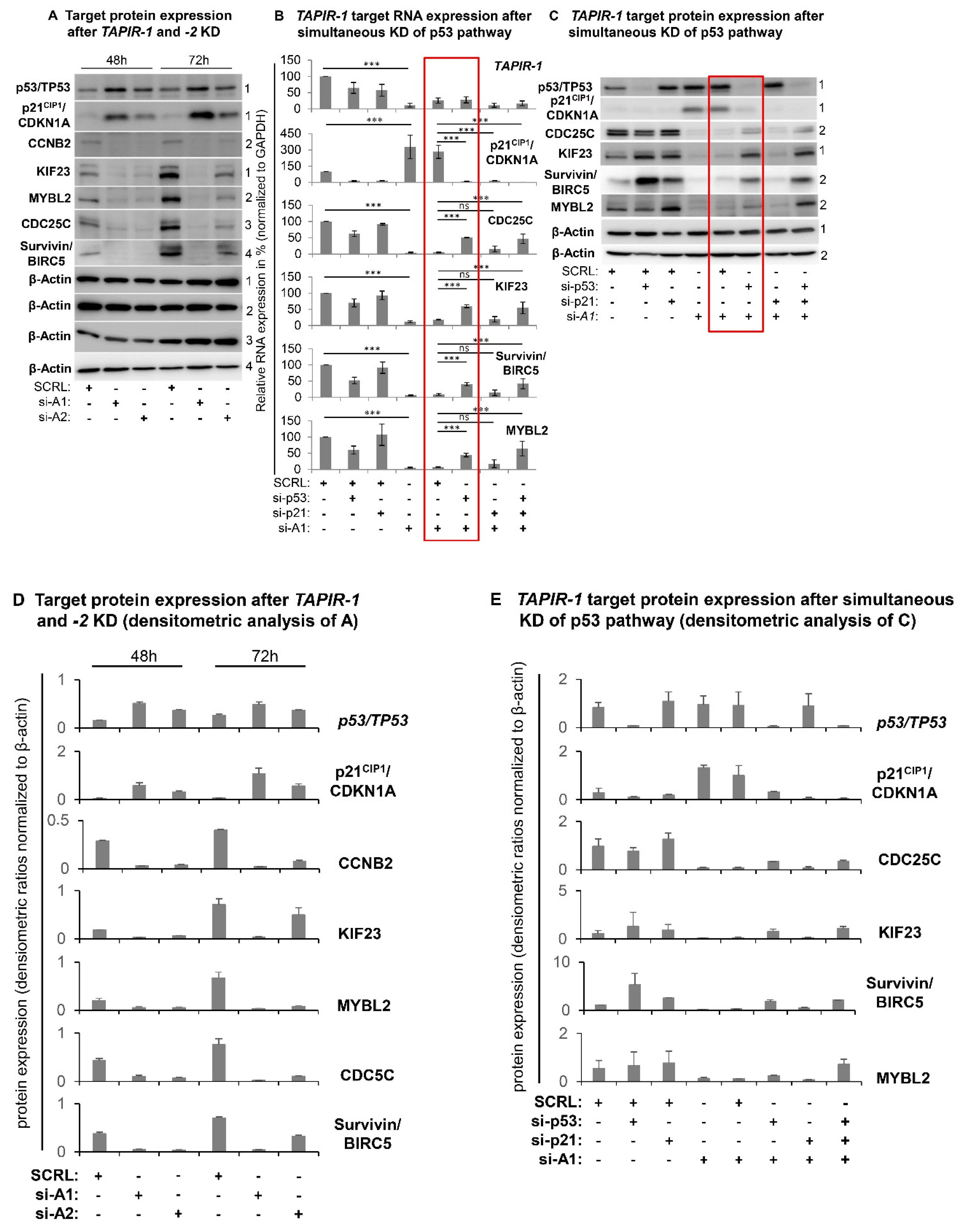
2.4. Increase in p53 Levels and Downregulation of Cell Cycle- and DNA-Repair Control Genes Following TAPIR Knockdown
2.5. TAPIR Knockdown Activates the p53-DREAM Tumor Suppressor Pathway
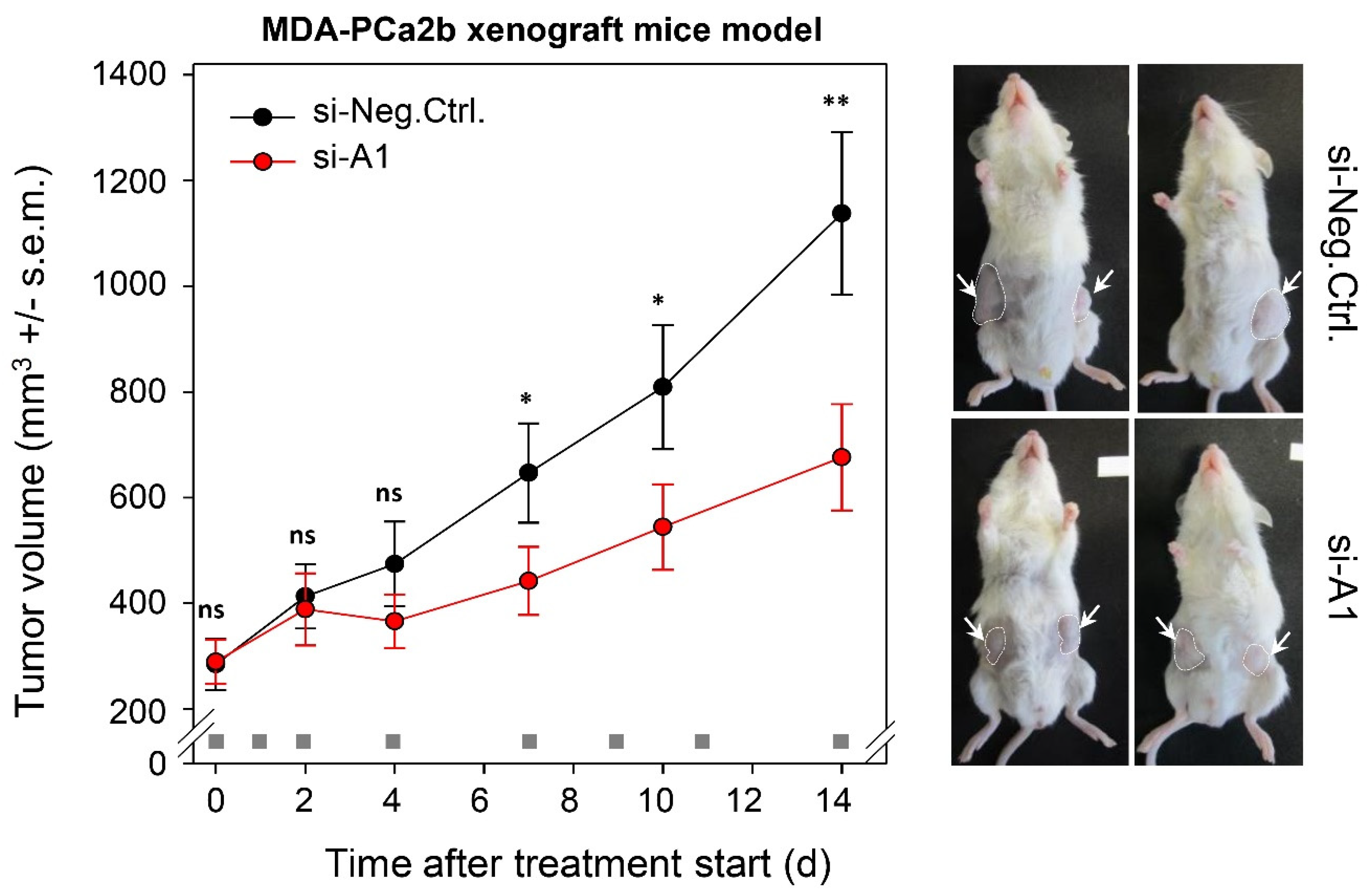
2.6. TAPIR Represents a Potential Therapeutic Target in Prostate Cancer
2.7. TAPIR-1 and -2 Knockdowns in Radiation-Resistant Prostate Cancer Cells Confer Renewed Sensitization to X-ray Treatment
3. Discussion
4. Materials and Methods
4.1. Patient Characteristics
4.2. Tissue Processing and RNA Isolation
4.3. Next-Generation Sequencing (NGS)
4.4. Agilent Custom Expression Microarray Processing
4.5. Coding Potential Prediction
4.6. Design and Synthesis of Small Interfering RNAs
4.7. Cell Lines, Cell Culture, and Tissue Samples
4.8. Microarray Analysis Following TAPIR Knockdown
4.9. Assessment of Pan-Tissue and Pan-Cancer TAPIR Expression
4.10. Isolation of RNA, cDNA Synthesis, and PCR Analysis
4.11. Xcelligence
4.12. IncuCyte ZOOM™
4.13. Cell Cycle Analysis
4.14. Westernblot
4.15. p53 Pathway Knockdown Experiments
4.16. ViewRNA™ In Situ Hybridization (ISH)
4.17. Mouse Xenograft Model and Therapeutic In Vivo siRNA Treatment
4.18. Clonogenic Cell Survival Assay
4.19. Statistical Analysis
4.20. Data Availability
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schröder, F.H.; Hugosson, J.; Roobol, M.J.; Tammela, T.L.J.; Zappa, M.; Nelen, V.; Kwiatkowski, M.; Lujan, M.; Määttänen, L.; Lilja, H.; et al. Screening and prostate cancer mortality: Results of the European Randomised Study of Screening for Prostate Cancer (ERSPC) at 13 years of follow-up. Lancet 2014, 384, 2027–2035. [Google Scholar] [CrossRef]
- Pinsky, P.F.; Parnes, H.L.; Andriole, G. Mortality and complications after prostate biopsy in the Prostate, Lung, Colorectal and Ovarian Cancer Screening (PLCO) trial. BJU Int. 2014, 113, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.M.; Donovan, J.L.; Turner, E.L.; Metcalfe, C.; Young, G.J.; Walsh, E.I.; Lane, J.A.; Noble, S.; Oliver, S.E.; Evans, S.; et al. Effect of a Low-Intensity PSA-Based Screening Intervention on Prostate Cancer Mortality: The CAP Randomized Clinical Trial. JAMA 2018, 319, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Bott, S.R.J. Prostate cancer management: (1) an update on localised disease. Postgrad. Med. J. 2003, 79, 575–580. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zietman, A.L.; Bae, K.; Slater, J.D.; Shipley, W.U.; Efstathiou, J.A.; Coen, J.J.; Bush, D.A.; Lunt, M.; Spiegel, D.Y.; Skowronski, R.; et al. Randomized trial comparing conventional-dose with high-dose conformal radiation therapy in early-stage adenocarcinoma of the prostate: Long-term results from proton radiation oncology group/american college of radiology 95-09. J. Clin. Oncol. 2010, 28, 1106–1111. [Google Scholar] [CrossRef] [PubMed]
- Pahlajani, N.; Ruth, K.J.; Buyyounouski, M.K.; Chen, D.Y.T.; Horwitz, E.M.; Hanks, G.E.; Price, R.A.; Pollack, A. Radiotherapy doses of 80 Gy and higher are associated with lower mortality in men with Gleason score 8 to 10 prostate cancer. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 1949–1956. [Google Scholar] [CrossRef]
- Johansson, S.; Aström, L.; Sandin, F.; Isacsson, U.; Montelius, A.; Turesson, I. Hypofractionated proton boost combined with external beam radiotherapy for treatment of localized prostate cancer. Prostate Cancer 2012, 2012, 654861. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Consortium, E.P.; Bernstein, B.E.; Birney, E.; Dunham, I.; Green, E.D.; Gunter, C. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Rinn, J.; Guttman, M. RNA Function. RNA and dynamic nuclear organization. Science 2014, 345, 1240–1241. [Google Scholar] [CrossRef]
- Ramalho-Carvalho, J.; Fromm, B.; Henrique, R.; Jeronimo, C. Deciphering the function of non-coding RNAs in prostate cancer. Cancer Metastasis Rev. 2016, 35, 235–262. [Google Scholar] [CrossRef]
- Mattick, J.S.; Rinn, J.L. Discovery and annotation of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015, 22, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, evolution, and mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [PubMed]
- Kunej, T.; Obsteter, J.; Pogacar, Z.; Horvat, S.; Calin, G.A. The decalog of long non-coding RNA involvement in cancer diagnosis and monitoring. Crit. Rev. Clin. Lab. Sci. 2014, 51, 344–357. [Google Scholar] [CrossRef]
- Weber, D.G.; Johnen, G.; Casjens, S.; Bryk, O.; Pesch, B.; Jockel, K.-H.; Kollmeier, J.; Bruning, T. Evaluation of long noncoding RNA MALAT1 as a candidate blood-based biomarker for the diagnosis of non-small cell lung cancer. BMC Res. Notes 2013, 6, 518. [Google Scholar] [CrossRef]
- Di Gesualdo, F.; Capaccioli, S.; Lulli, M. A pathophysiological view of the long non-coding RNA world. Oncotarget 2014, 5, 10976–10996. [Google Scholar] [CrossRef]
- Ku, S.H.; Kim, K.; Choi, K.; Kim, S.H.; Kwon, I.C. Tumor-targeting multifunctional nanoparticles for siRNA delivery: Recent advances in cancer therapy. Adv. Healthc. Mater. 2014, 3, 1182–1193. [Google Scholar] [CrossRef]
- Dorsett, Y.; Tuschl, T. siRNAs: Applications in functional genomics and potential as therapeutics. Nat. Rev. Drug Discov. 2004, 3, 318–329. [Google Scholar] [CrossRef]
- Modarresi, F.; Faghihi, M.A.; Lopez-Toledano, M.A.; Fatemi, R.P.; Magistri, M.; Brothers, S.P.; van der Brug, M.P.; Wahlestedt, C. Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat. Biotechnol. 2012, 30, 453–459. [Google Scholar] [CrossRef]
- Lennox, K.A.; Behlke, M.A. Cellular localization of long non-coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic Acids Res. 2016, 44, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Pathak, K.; Vaidya, A. Molecular therapy using siRNA: Recent trends and advances of multi target inhibition of cancer growth. Int. J. Biol. Macromol. 2018, 116, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.-P.; Li, W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef]
- Fischer, M.; Quaas, M.; Steiner, L.; Engeland, K. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res. 2016, 44, 164–174. [Google Scholar] [CrossRef]
- Muller, G.A.; Stangner, K.; Schmitt, T.; Wintsche, A.; Engeland, K. Timing of transcription during the cell cycle: Protein complexes binding to E2F, E2F/CLE, CDE/CHR, or CHR promoter elements define early and late cell cycle gene expression. Oncotarget 2017, 8, 97736–97748. [Google Scholar] [CrossRef]
- Müller, G.A.; Wintsche, A.; Stangner, K.; Prohaska, S.J.; Stadler, P.F.; Engeland, K. The CHR site: Definition and genome-wide identification of a cell cycle transcriptional element. Nucleic Acids Res. 2014, 42, 10331–10350. [Google Scholar] [CrossRef]
- Quaas, M.; Müller, G.A.; Engeland, K. p53 can repress transcription of cell cycle genes through a p21(WAF1/CIP1)-dependent switch from MMB to DREAM protein complex binding at CHR promoter elements. Cell Cycle 2012, 11, 4661–4672. [Google Scholar] [CrossRef]
- Fischer, M.; Quaas, M.; Nickel, A.; Engeland, K. Indirect p53-dependent transcriptional repression of Survivin, CDC25C, and PLK1 genes requires the cyclin-dependent kinase inhibitor p21/CDKN1A and CDE/CHR promoter sites binding the DREAM complex. Oncotarget 2015, 6, 41402–41417. [Google Scholar] [CrossRef]
- Werth, S.; Urban-Klein, B.; Dai, L.; Höbel, S.; Grzelinski, M.; Bakowsky, U.; Czubayko, F.; Aigner, A. A low molecular weight fraction of polyethylenimine (PEI) displays increased transfection efficiency of DNA and siRNA in fresh or lyophilized complexes. J. Control. Release 2006, 112, 257–270. [Google Scholar] [CrossRef]
- Höbel, S.; Prinz, R.; Malek, A.; Urban-Klein, B.; Sitterberg, J.; Bakowsky, U.; Czubayko, F.; Aigner, A. Polyethylenimine PEI F25-LMW allows the long-term storage of frozen complexes as fully active reagents in siRNA-mediated gene targeting and DNA delivery. Eur. J. Pharm. Biopharm. 2008, 70, 29–41. [Google Scholar] [CrossRef]
- Höbel, S.; Aigner, A. Polyethylenimine (PEI)/siRNA-mediated gene knockdown in vitro and in vivo. Methods Mol. Biol. 2010, 623, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Hendruschk, S.; Wiedemuth, R.; Aigner, A.; Töpfer, K.; Cartellieri, M.; Martin, D.; Kirsch, M.; Ikonomidou, C.; Schackert, G.; Temme, A. RNA interference targeting survivin exerts antitumoral effects in vitro and in established glioma xenografts in vivo. Neuro-Oncology 2011, 13, 1074–1089. [Google Scholar] [CrossRef] [PubMed]
- Hampl, V.; Martin, C.; Aigner, A.; Hoebel, S.; Singer, S.; Frank, N.; Sarikas, A.; Ebert, O.; Prywes, R.; Gudermann, T.; et al. Depletion of the transcriptional coactivators megakaryoblastic leukaemia 1 and 2 abolishes hepatocellular carcinoma xenograft growth by inducing oncogene-induced senescence. EMBO Mol. Med. 2013, 5, 1367–1382. [Google Scholar] [CrossRef] [PubMed]
- Frömberg, A.; Rabe, M.; Aigner, A. Multiple effects of the special AT-rich binding protein 1 (SATB1) in colon carcinoma. Int. J. Cancer 2014, 135, 2537–2546. [Google Scholar] [CrossRef]
- Frömberg, A.; Rabe, M.; Oppermann, H.; Gaunitz, F.; Aigner, A. Analysis of cellular and molecular antitumor effects upon inhibition of SATB1 in glioblastoma cells. BMC Cancer 2017, 17, 3. [Google Scholar] [CrossRef][Green Version]
- Ibrahim, A.F.; Weirauch, U.; Thomas, M.; Grünweller, A.; Hartmann, R.K.; Aigner, A. MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Cancer Res. 2011, 71, 5214–5224. [Google Scholar] [CrossRef]
- Höbel, S.; Koburger, I.; John, M.; Czubayko, F.; Hadwiger, P.; Vornlocher, H.-P.; Aigner, A. Polyethylenimine/small interfering RNA-mediated knockdown of vascular endothelial growth factor in vivo exerts anti-tumor effects synergistically with Bevacizumab. J. Gene Med. 2010, 12, 287–300. [Google Scholar] [CrossRef]
- Peitzsch, C.; Cojoc, M.; Hein, L.; Kurth, I.; Mäbert, K.; Trautmann, F.; Klink, B.; Schröck, E.; Wirth, M.P.; Krause, M.; et al. An Epigenetic Reprogramming Strategy to Resensitize Radioresistant Prostate Cancer Cells. Cancer Res. 2016, 76, 2637–2651. [Google Scholar] [CrossRef]
- Cojoc, M.; Peitzsch, C.; Kurth, I.; Trautmann, F.; Kunz-Schughart, L.A.; Telegeev, G.D.; Stakhovsky, E.A.; Walker, J.R.; Simin, K.; Lyle, S.; et al. Aldehyde Dehydrogenase Is Regulated by β-Catenin/TCF and Promotes Radioresistance in Prostate Cancer Progenitor Cells. Cancer Res. 2015, 75, 1482–1494. [Google Scholar] [CrossRef]
- Schalken, J.A.; Hessels, D.; Verhaegh, G. New targets for therapy in prostate cancer: Differential display code 3 (DD3(PCA3)), a highly prostate cancer-specific gene. Urology 2003, 62, 34–43. [Google Scholar] [CrossRef]
- Fischer, M.; Müller, G.A. Cell cycle transcription control: DREAM/MuvB and RB-E2F complexes. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 638–662. [Google Scholar] [CrossRef] [PubMed]
- Mages, C.F.; Wintsche, A.; Bernhart, S.H.; Müller, G.A. The DREAM complex through its subunit Lin37 cooperates with Rb to initiate quiescence. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Steiner, L.; Engeland, K. The transcription factor p53: Not a repressor, solely an activator. Cell Cycle 2014, 13, 3037–3058. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 2013, 10, 925–933. [Google Scholar] [CrossRef]
- van Heesch, S.; van Iterson, M.; Jacobi, J.; Boymans, S.; Essers, P.B.; de Bruijn, E.; Hao, W.; MacInnes, A.W.; Cuppen, E.; Simonis, M. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome Biol. 2014, 15, R6. [Google Scholar] [CrossRef]
- Peitzsch, C.; Kurth, I.; Kunz-Schughart, L.; Baumann, M.; Dubrovska, A. Discovery of the cancer stem cell related determinants of radioresistance. Radiother. Oncol. 2013, 108, 378–387. [Google Scholar] [CrossRef]
- Baumann, M.; Krause, M.; Hill, R. Exploring the role of cancer stem cells in radioresistance. Nat. Rev. Cancer 2008, 8, 545–554. [Google Scholar] [CrossRef]
- Gorodetska, I.; Kozeretska, I.; Dubrovska, A. BRCA Genes: The Role in Genome Stability, Cancer Stemness and Therapy Resistance. J. Cancer 2019, 10, 2109–2127. [Google Scholar] [CrossRef]
- Gorodetska, I.; Lukiyanchuk, V.; Peitzsch, C.; Kozeretska, I.; Dubrovska, A. BRCA1 and EZH2 cooperate in regulation of prostate cancer stem cell phenotype. Int. J. Cancer 2019, 145, 2974–2985. [Google Scholar] [CrossRef]
- Yang, C.-C.; Suzuki, M.; Yamakawa, S.; Uno, S.; Ishii, A.; Yamazaki, S.; Fukatsu, R.; Fujisawa, R.; Sakimura, K.; Tsurimoto, T.; et al. Claspin recruits Cdc7 kinase for initiation of DNA replication in human cells. Nat. Commun. 2016, 7, 12135. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.; D’Andrea, A. Fanconi anemia pathway. Curr. Biol. 2017, 27, R986–R988. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, C.; Cui, Y.; Nadiminty, N.; Lou, W.; Gao, A.C. Interleukin-6 induces neuroendocrine differentiation (NED) through suppression of RE-1 silencing transcription factor (REST). Prostate 2014, 74, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Pernicová, Z.; Slabáková, E.; Fedr, R.; Šimečková, Š.; Jaroš, J.; Suchánková, T.; Bouchal, J.; Kharaishvili, G.; Král, M.; Kozubík, A.; et al. The role of high cell density in the promotion of neuroendocrine transdifferentiation of prostate cancer cells. Mol. Cancer 2014, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- Farach, A.; Ding, Y.; Lee, M.; Creighton, C.; Delk, N.A.; Ittmann, M.; Miles, B.; Rowley, D.; Farach-Carson, M.C.; Ayala, G.E. Neuronal Trans-Differentiation in Prostate Cancer Cells. Prostate 2016, 76, 1312–1325. [Google Scholar] [CrossRef] [PubMed]
- Ecke, T.H.; Schlechte, H.H.; Schiemenz, K.; Sachs, M.D.; Lenk, S.V.; Rudolph, B.D.; Loening, S.A. TP53 gene mutations in prostate cancer progression. Anticancer Res. 2010, 30, 1579–1586. [Google Scholar]
- Kämpf, C.; Specht, M.; Scholz, A.; Puppel, S.-H.; Doose, G.; Reiche, K.; Schor, J.; Hackermüller, J. uap: Reproducible and robust HTS data analysis. BMC Bioinform. 2019, 20, 1–9. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Hoffmann, S.; Otto, C.; Kurtz, S.; Sharma, C.M.; Khaitovich, P.; Vogel, J.; Stadler, P.F.; Hackermüller, J. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput. Biol. 2009, 5, e1000502. [Google Scholar] [CrossRef]
- Hoffmann, S.; Otto, C.; Doose, G.; Tanzer, A.; Langenberger, D.; Christ, S.; Kunz, M.; Holdt, L.M.; Teupser, D.; Hackermüller, J.; et al. A multi-split mapping algorithm for circular RNA, splicing, trans-splicing and fusion detection. Genome Biol. 2014, 15, R34. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2008. [Google Scholar]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Di, W.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Matthews, L.; Gopinath, G.; Gillespie, M.; Caudy, M.; Croft, D.; de Bono, B.; Garapati, P.; Hemish, J.; Hermjakob, H.; Jassal, B.; et al. Reactome knowledgebase of human biological pathways and processes. Nucleic Acids Res. 2009, 37, D619–D622. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology. R package version 2.38.1. 2019. Available online: http://www.bioconductor.org/packages/release/bioc/html/topGO.html (accessed on 24 April 2020).
- Sales, G.; Calura, E.; Cavalieri, D.; Romualdi, C. graphite—A Bioconductor package to convert pathway topology to gene network. BMC Bioinform. 2012, 13, 20. [Google Scholar] [CrossRef]
- Tarca, A.L.; Draghici, S.; Khatri, P.; Hassan, S.S.; Mittal, P.; Kim, J.S.; Kim, C.J.; Kusanovic, J.P.; Romero, R. A novel signaling pathway impact analysis. Bioinformatics. 2009, 1, 75–82. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Yu, G.; He, Q.-Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Ligtenberg, W. reactome.db: A set of annotation maps for reactome. R package version 1.68.0. 2019. Available online: https://www.bioconductor.org/packages/release/data/annotation/html/reactome.db.html (accessed on 24 April 2020).
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; García, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Kirschner, R.D.; Sänger, K.; Müller, G.A.; Engeland, K. Transcriptional activation of the tumor suppressor and differentiation gene S100A2 by a novel p63-binding site. Nucleic Acids Res. 2008, 36, 2969–2980. [Google Scholar] [CrossRef]
- Tavner, F.; Frampton, J.; Watson, R.J. Targeting an E2F site in the mouse genome prevents promoter silencing in quiescent and post-mitotic cells. Oncogene 2007, 26, 2727–2735. [Google Scholar] [CrossRef]
- Bach, F.C.; Rutten, K.; Hendriks, K.; Riemers, F.M.; Cornelissen, P.; de Bruin, A.; Arkesteijn, G.J.; Wubbolts, R.; Horton, W.A.; Penning, L.C.; et al. The paracrine feedback loop between vitamin D₃ (1,25(OH)₂D₃) and PTHrP in prehypertrophic chondrocytes. J. Cell. Physiol. 2014, 229, 1999–2014. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNA Stealth siOligos and siRNA | Application | Target | ||
|---|---|---|---|---|
| TAPIR-1 | si-A1 | 5′-CGTCTGTCTGGAGTTTCCGCTTCAT-3′ | TAPIR-1 knockdown | Exon 3 |
| TAPIR-1 | si-B1 | 5′-CCAGGGAGCACACTTTATTAGGAAA-3′ | TAPIR-1 knockdown | Exon 3 |
| TAPIR-2 | si-A2 | 5′-GGTCAGCGTCTCGTTCACCTCTTTA-3′ | TAPIR-2 knockdown | Exon 2 |
| TAPIR-2 | si-B2 | 5′-CCGTGACAGTTTGAGTTGAGGACAT-3′ | TAPIR-2 knockdown | Exon 2 |
| p53/TP53 | si-p53 | Invitrogen cat# 1299001 VHS40366 | p53 knockdown | n.d. |
| p21CIP1/CDKN1A | si-p21CIP1 | Invitrogen, cat# 1299001 VHS40209 | p21CIP1 knockdown | n.d. |
| AR | si-AR | Dharmacon, cat# L-003400-00-0005 | AR knockdown | n.d. |
| siRNA control (medium GC) | si-SCRL | Invitrogen, cat# 12935-300 | negative control | none |
| Target | Strand | Sequence | Application | Product Size (bp) |
|---|---|---|---|---|
| GAPDH | For Rev | GTCAGTGGTGGACCTGACCT AGGGGAGATTCAGTGTGGTG | housekeeper, qPCR, normalization | 395 |
| ß-actin | For Rev | TCGTGCGTGACATTAAGGAGAA AGCAGCCGTGGCCATCT | housekeeper, qPCR, normalization | 71 |
| TAPIR-1 (Ex3) | For Rev | TTCATAGACCGTGCTTCCGT TGTGAACCTGGAGCTTTCCT | qPCR TAPIR-1, RNA expression | 271 |
| TAPIR-1 (Ex2-3) | For Rev | GGGACAACTCGAAGCAAAGG CTACAGAATGCGTGGTGTGG | qPCR TAPIR-1, RNA expression | 389 |
| TAPIR-1 (Ex1-2) | For Rev | TCTCAAAGCAGGCTCCATGA CGTCTGCTTCGAGTCTTCCT | qPCR TAPIR-1, RNA expression | 249 |
| TAPIR-2 (Ex1-2) | For Rev | GACTTCACCACTCACCAGGA AGGGACCCGTCTTATGTGAC | qPCR TAPIR-2, RNA expression | 237 |
| AURKA | For Rev | TTGAACACCCCTGGATCACA AGCGTTCTAGATTGAGGGCA | Validation of microarray, qPCR, RNA expression | 280 |
| CCNB1 | For Rev | TGGTGCACTTTCCTCCTTCT TTAGCATGCTTCGATGTGGC | Validation of microarray, qPCR, RNA expression | 420 |
| CCNB2 | For Rev | GCTGAACTCAAAAGCCGTCA ACTGGTCTGAGAAGAGGTTTCA | Validation of microarray, qPCR, RNA expression | 306 |
| p21CIP1/ CDKN1A | For Rev | TGTCTTGTACCCTTGTGCCT AAGATGTAGAGCGGGCCTTT | Validation of microarray, qPCR, RNA expression | 480 |
| E2F1 | For Rev | CTCTAACTGCACTTTCGGCC CCTCCTCCCCTTTGCTGATT | Validation of microarray, qPCR, RNA expression | 360 |
| HIST1H1B | For Rev | ACTCCGAAGAAGGCGAAGAA GCTTTAGGTTTTGCTGCTTTGG | Validation of microarray, qPCR, RNA expression | 321 |
| Survivin/ BIRC5 | For Rev | TGATGAGAGAATGGAGACAGAG ACAGCAGTGGCAAAAGGAG | Validation of microarray, qPCR, RNA expression | 223 |
| MYBL2 | For Rev | CAGCAATGCCAGTACAGGTG CTCAGGGTTGAGGTGGTTGT | Validation of microarray qPCR | 198 |
| CCNE1 | For Rev | GCAGGATCCAGATGAAGAAATG GGGTCTGCACAGACTGCAT | Validation of microarray qPCR | 96 |
| CDC25C | For Rev | CTCAGATGCTGGAGGAAGAATC GGGACGATGGGCTTCTTCAG | Validation of microarray qPCR | 283 |
| FOXM1 | For Rev | CGTGGATTGAGGACCACTTT GGCTTAAACACCTGGTCCAA | Validation of microarray qPCR | 187 |
| MKI67 | For Rev | CCAGCTTCCTGTTGTGTCAA AGCCGTACAGGCTCATCAAT | Validation of microarray qPCR | 129 |
| KIF23 | For Rev | GTAGAGTGGCAGCCAAACAGC CTGATCAGGTTGAAAGAGTAAAGGC | Validation of microarray qPCR | 197 |
| AR | For Rev | GCCAGCAGAAATGATTG AGCCTCTCCTTCCTCCTG | qPCR AR, RNA expression | 150 |
| BRCA1 | For Rev | CAGCTTGACACAGGTTTG GGATTTTCGGGTTCACTC | qPCR, RNA expression | 155 |
| ATR | For Rev | AACCTCCGTGATGTTGCTTG AATGACAGGAGGGAGTTGCT | qPCR, RNA expression | 164 |
| CHEK1 | For Rev | TCAGACTTTGGCTTGGCAAC GGTTGGTCCCATGGCAATTC | qPCR, RNA expression | 194 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friedrich, M.; Wiedemann, K.; Reiche, K.; Puppel, S.-H.; Pfeifer, G.; Zipfel, I.; Binder, S.; Köhl, U.; Müller, G.A.; Engeland, K.; et al. The Role of lncRNAs TAPIR-1 and -2 as Diagnostic Markers and Potential Therapeutic Targets in Prostate Cancer. Cancers 2020, 12, 1122. https://doi.org/10.3390/cancers12051122
Friedrich M, Wiedemann K, Reiche K, Puppel S-H, Pfeifer G, Zipfel I, Binder S, Köhl U, Müller GA, Engeland K, et al. The Role of lncRNAs TAPIR-1 and -2 as Diagnostic Markers and Potential Therapeutic Targets in Prostate Cancer. Cancers. 2020; 12(5):1122. https://doi.org/10.3390/cancers12051122
Chicago/Turabian StyleFriedrich, Maik, Karolin Wiedemann, Kristin Reiche, Sven-Holger Puppel, Gabriele Pfeifer, Ivonne Zipfel, Stefanie Binder, Ulrike Köhl, Gerd A. Müller, Kurt Engeland, and et al. 2020. "The Role of lncRNAs TAPIR-1 and -2 as Diagnostic Markers and Potential Therapeutic Targets in Prostate Cancer" Cancers 12, no. 5: 1122. https://doi.org/10.3390/cancers12051122
APA StyleFriedrich, M., Wiedemann, K., Reiche, K., Puppel, S.-H., Pfeifer, G., Zipfel, I., Binder, S., Köhl, U., Müller, G. A., Engeland, K., Aigner, A., Füssel, S., Fröhner, M., Peitzsch, C., Dubrovska, A., Rade, M., Christ, S., Schreiber, S., Hackermüller, J., ... Horn, F. (2020). The Role of lncRNAs TAPIR-1 and -2 as Diagnostic Markers and Potential Therapeutic Targets in Prostate Cancer. Cancers, 12(5), 1122. https://doi.org/10.3390/cancers12051122