The Role of Deubiquitinating Enzymes in Hematopoiesis and Hematological Malignancies



Abstract
1. Introduction
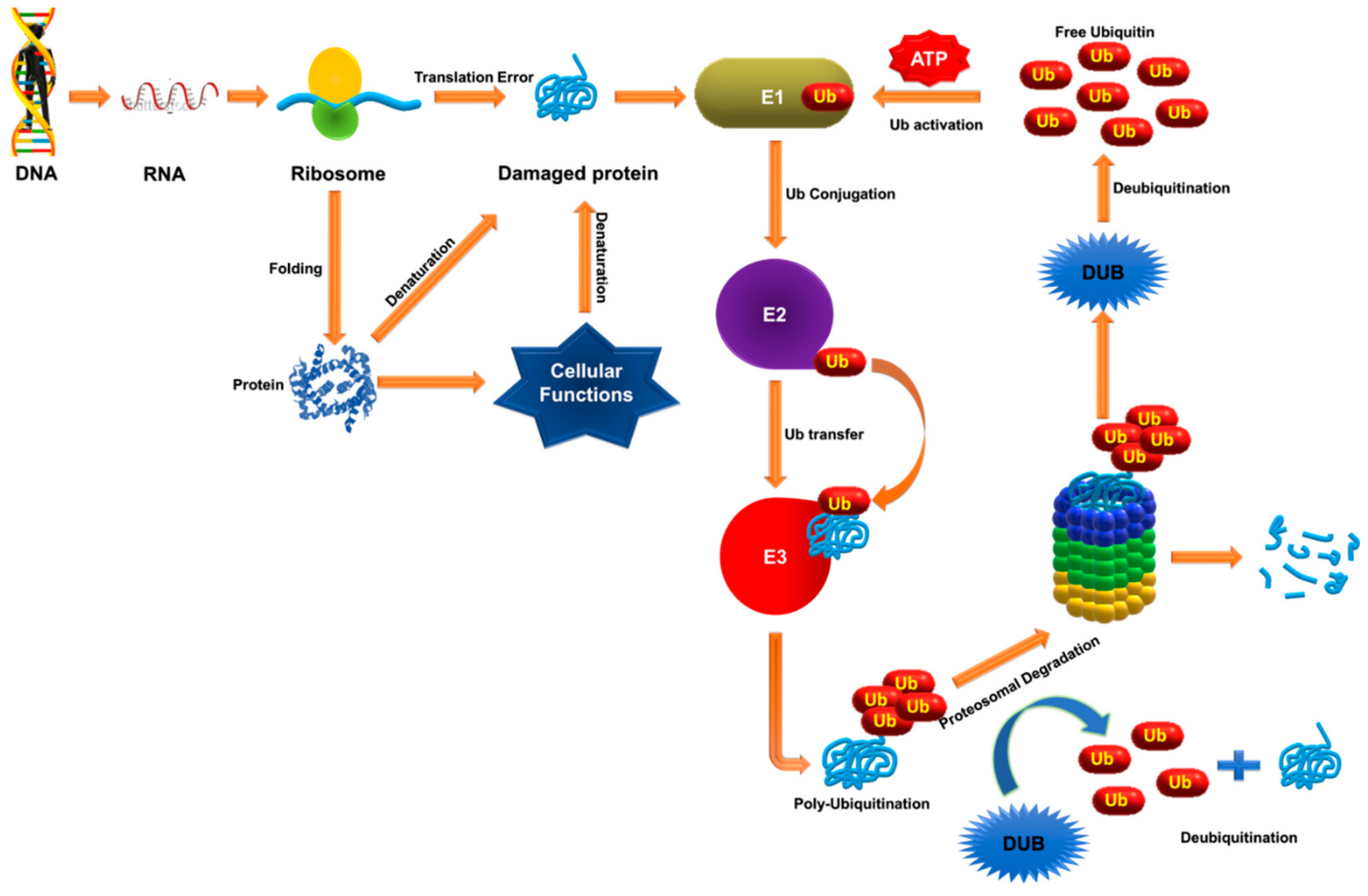
2. Ubiquitination and Deubiquitination
3. Importance of DUBs in Hematopoiesis
3.1. DUB-1, DUB-2A, and DUB-3
3.2. MYSM1
3.3. USP3
3.4. USP16
3.5. USP1 and USP10
3.6. USP15
3.7. Other DUBs
4. Importance of DUBs in Erythropoiesis and Angiogenesis
5. DUBs in Hematological Malignancies
5.1. USP1
5.2. A20
5.3. USP7
5.4. USP9X
5.5. USP14
5.6. USP24
5.7. CYLD
6. Deubiquitinases as Emerging Targets against Hematological Malignancies
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations:
| AML | acute myeloid leukemia |
| BM | bone marrow |
| CAM-DR | Cell adhesion mediated drug resistance |
| CDK | cyclin-dependent kinases |
| CFU-E | colony forming unit erythroid |
| DDR | DNA damage response |
| DUBs | Deubiquitinating enzymes |
| FA | Fanconi anemia |
| FL | fetal liver |
| IL | Interleukin |
| LUBAC | Linear ubiquitin chain assembly complex |
| MALT-1 | Mucosa-associated lymphoid tissue |
| MINDY | motif interacting with Ub-containing novel DUB family |
| MCPIP1 | monocyte chemotactic protein–induced protein 1 |
| MM | Multiple myeloma |
| NEMO | NF-kappa-B essential modulator |
| NK | natural killer |
| NF-kB | Nuclear factor-kB |
| OUT | Otu-domain ubiquitin aldehyde-binding proteins |
| PR-DUB | polycomb repressive deubiquitinase complex |
| PRC1 | polycomb repressive complex 1 |
| RBCs | red blood cells |
| SAC | spindle assembly checkpoint |
| TRAF | tumor necrosis factor receptor–associated factor |
| UPS | Ubiquitin proteasome system |
| USP | Ubiquitin-specific proteases |
| WM | Waldenstrom macroglobuliemia |
| XIAP | X-linked inhibitor of apoptotic protein |
References
- Moran-Crusio, K.; Reavie, L.B.; Aifantis, I. Regulation of hematopoietic stem cell fate by the ubiquitin proteasome system. Trends Immunol. 2012, 33, 357–363. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Orkin, S.H. Diversification of haematopoietic stem cells to specific lineages. Nat. Rev. Genet. 2000, 1, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Reavie, L.; Della Gatta, G.; Crusio, K.; Aranda-Orgilles, B.; Buckley, S.M.; Thompson, B.; Lee, E.; Gao, J.; Bredemeyer, A.L.; Helmink, B.A.; et al. Regulation of hematopoietic stem cell differentiation by a single ubiquitin ligase–substrate complex. Nat. Immunol. 2010, 11, 207. [Google Scholar] [CrossRef] [PubMed]
- Zivot, A.; Lipton, J.M.; Narla, A.; Blanc, L. Erythropoiesis: Insights into pathophysiology and treatments in 2017. Mol. Med. 2018, 24, 11. [Google Scholar] [CrossRef] [PubMed]
- Hattangadi, S.M.; Wong, P.; Zhang, L.; Flygare, J.; Lodish, H.F. From stem cell to red cell: Regulation of erythropoiesis at multiple levels by multiple proteins, RNAs, and chromatin modifications. Blood 2011, 118, 6258–6268. [Google Scholar] [CrossRef] [PubMed]
- Palis, J.; Robertson, S.; Kennedy, M.; Wall, C.; Keller, G. Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development 1999, 126, 5073–5084. [Google Scholar]
- Keerthivasan, G.; Wickrema, A.; Crispino, J.D. Erythroblast enucleation. Stem Cells Int. 2011, 2011, 1–9. [Google Scholar] [CrossRef]
- Cai, J.; Wei, J.; Schrott, V.; Zhao, J.; Bullock, G.; Zhao, Y. Induction of deubiquitinating enzyme USP50 during erythropoiesis and its potential role in the regulation of Ku70 stability. J. Investig. Med. 2018, 66, 1–6. [Google Scholar] [CrossRef]
- Adair, T.H.; Montani, J.-P. Angiogenesis; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010; pp. 1–8. [Google Scholar]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef]
- Ellis, L.M.; Liu, W.; Ahmad, S.A.; Fan, F.; Do Jung, Y.; Shaheen, R.M.; Reinmuth, N. Overview of angiogenesis: Biologic implications for antiangiogenic therapy. Semin. Oncol. 2001, 28, 94–104. [Google Scholar] [CrossRef]
- Rivkin, E.; Almeida, S.M.; Ceccarelli, D.F.; Juang, Y.-C.; MacLean, T.A.; Srikumar, T.; Huang, H.; Dunham, W.H.; Fukumura, R.; Xie, G.; et al. The linear ubiquitin-specific deubiquitinase gumby regulates angiogenesis. Nature 2013, 498, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Haq, S.; Ramakrishna, S. Deubiquitylation of deubiquitylases. Open Biol. 2017, 7, 170016. [Google Scholar] [CrossRef] [PubMed]
- Suresh, B.; Lee, J.; Kim, H.; Ramakrishna, S. Regulation of pluripotency and differentiation by deubiquitinating enzymes. Cell Death Differ. 2016, 23, 1257–1264. [Google Scholar] [CrossRef] [PubMed]
- Pfoh, R.; Lacdao, I.K.; Saridakis, V. Deubiquitinases and the new therapeutic opportunities offered to cancer. Endocr.-Relat. Cancer 2015, 22, T35–T54. [Google Scholar] [CrossRef]
- Hanpude, P.; Bhattacharya, S.; Dey, A.K.; Maiti, T.K. Deubiquitinating enzymes in cellular signaling and disease regulation. IUBMB Life 2015, 67, 544–555. [Google Scholar] [CrossRef]
- Amerik, A.Y.; Hochstrasser, M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta. 2004, 1695, 189–207. [Google Scholar] [CrossRef]
- Todi, S.V.; Paulson, H. Balancing act: Deubiquitinating enzymes in the nervous system. Trends Neurosci. 2011, 34, 370–382. [Google Scholar] [CrossRef]
- Clague, M.J.; Coulson, J.M.; Urbé, S. Cellular functions of the DUBs. J. Cell Sci. 2012, 125, 277–286. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Suresh, B.; Baek, K.-H. The role of deubiquitinating enzymes in apoptosis. Cell. Mol. Life Sci. 2011, 68, 15–26. [Google Scholar] [CrossRef]
- Pinto-Fernandez, A.; Kessler, B.M. DUBbing cancer: Deubiquitylating enzymes involved in epigenetics, DNA damage and the cell cycle as therapeutic targets. Front. Genet. 2016, 7, 133. [Google Scholar] [CrossRef]
- Suresh, B.; Lee, J.; Kim, K.-S.; Ramakrishna, S. The importance of ubiquitination and deubiquitination in cellular reprogramming. Stem Cells Int. 2016, 2016, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, A.P.; Suresh, B.; Kim, H.; Kim, K.S.; Ramakrishna, S. Concise review: Fate determination of stem cells by deubiquitinating enzymes. Stem Cells 2017, 35, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Suresh, B.; Lee, J.; Hong, S.-H.; Kim, K.-S.; Ramakrishna, S. The role of deubiquitinating enzymes in spermatogenesis. Cell. Mol. Life Sci. 2015, 72, 4711–4720. [Google Scholar] [CrossRef] [PubMed]
- Clague, M.J.; Liu, H.; Urbé, S. Governance of endocytic trafficking and signaling by reversible ubiquitylation. Dev. Cell 2012, 23, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Farshi, P.; Deshmukh, R.R.; Nwankwo, J.O.; Arkwright, R.T.; Cvek, B.; Liu, J.; Dou, Q.P. Deubiquitinases (DUBs) and DUB inhibitors: A patent review. Expert Opin. Ther. Patents 2015, 25, 1191–1208. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.; Luna-Vargas, M.P.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.; Sixma, T.K.; Bernards, R. A genomic and functional inventory of deubiquitinating enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Saad, Y.; Lei, T.; Wang, J.; Qi, D.; Yang, Q.; Kolattukudy, P.E.; Fu, M. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-κB signaling. J. Exp. Med. 2010, 207, 2959–2973. [Google Scholar] [CrossRef]
- Rehman, S.A.A.; Kristariyanto, Y.A.; Choi, S.-Y.; Nkosi, P.J.; Weidlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 is a member of an evolutionarily conserved and structurally distinct new family of deubiquitinating enzymes. Mol. Cell 2016, 63, 146–155. [Google Scholar]
- Rice, K.L.; Hormaeche, I.; Licht, J.D. Epigenetic regulation of normal and malignant hematopoiesis. Oncogene 2007, 26, 6697–6714. [Google Scholar] [CrossRef]
- Higuchi, M.; Kawamura, H.; Matsuki, H.; Hara, T.; Takahashi, M.; Saito, S.; Saito, K.; Jiang, S.; Naito, M.; Kiyonari, H.; et al. USP10 is an essential deubiquitinase for hematopoiesis and inhibits apoptosis of long-term hematopoietic stem cells. Stem Cell Rep. 2016, 7, 1116–1129. [Google Scholar] [CrossRef]
- Zhu, Y.; Pless, M.; Inhorn, R.; Mathey-Prevot, B.; D’Andrea, A.D. The murine DUB-1 gene is specifically induced by the betac subunit of interleukin-3 receptor. Mol. Cell. Biol. 1996, 16, 4808–4817. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Lambert, K.; Corless, C.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; D’Andrea, A.D. DUB-2 is a member of a novel family of cytokine-inducible deubiquitinating enzymes. J. Biol. Chem. 1997, 272, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, J. Stat proteins and oncogenesis. J. Clin. Investig. 2002, 109, 1139–1142. [Google Scholar] [CrossRef]
- Wang, Y.; Yeung, Y.-G.; Langdon, W.Y.; Stanley, E.R. c-Cbl is transiently tyrosine-phosphorylated, ubiquitinated, and membrane-targeted following CSF-1 stimulation of macrophages. J. Biol. Chem. 1996, 271, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Verdier, F.; Chrétien, S.; Muller, O.; Varlet, P.; Yoshimura, A.; Gisselbrecht, S.; Lacombe, C.; Mayeux, P.J. Proteasomes regulate erythropoietin receptor and signal transducer and activator of transcription 5 (STAT5) activation Possible involvement of the ubiquitinated Cis protein. J. Biol. Chem. 1998, 273, 28185–28190. [Google Scholar] [CrossRef] [PubMed]
- Baek, K.-H.; Mondoux, M.A.; Jaster, R.; Fire-Levin, E.; D’Andrea, A.D. DUB-2A, a new member of the DUB subfamily of hematopoietic deubiquitinating enzymes. Blood 2001, 98, 636–642. [Google Scholar] [CrossRef]
- Strous, G.J.; Van Kerkhof, P.; Govers, R.; Rotwein, P.; Schwartz, A.L. Growth hormone-induced signal transduction depends on an intact ubiquitin system. J. Biol. Chem. 1997, 272, 40–43. [Google Scholar] [CrossRef][Green Version]
- Strous, G.J.; Van Kerkhof, P.; Govers, R.; Ciechanover, A.; Schwartz, A.L. The ubiquitin conjugation system is required for ligand-induced endocytosis and degradation of the growth hormone receptor. EMBO J. 1996, 15, 3806–3812. [Google Scholar] [CrossRef]
- Kim, T.K.; Maniatis, T. Regulation of interferon-γ-activated STAT1 by the ubiquitin-proteasome pathway. Science 1996, 273, 1717–1719. [Google Scholar] [CrossRef]
- Marteijn, J.; Jansen, J.; van der Reijden, B. Ubiquitylation in normal and malignant hematopoiesis: Novel therapeutic targets. Leukemia 2006, 20, 1511–1518. [Google Scholar] [CrossRef][Green Version]
- Burrows, J.F.; McGrattan, M.J.; Rascle, A.; Humbert, M.; Baek, K.-H.; Johnston, J.A. DUB-3, a cytokine-inducible deubiquitinating enzyme that blocks proliferation. J. Biol. Chem. 2004, 279, 13993–14000. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Carroll, M.; Papa, F.R.; Hochstrasser, M.; D’Andrea, A.D. DUB-1, a deubiquitinating enzyme with growth-suppressing activity. Proc. Natl. Acad. Sci. USA 1996, 93, 3275–3279. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Zhou, W.; Wang, J.; Puc, J.; Ohgi, K.A.; Erdjument-Bromage, H.; Tempst, P.; Glass, C.K.; Rosenfeld, M.G. A histone H2A deubiquitinase complex coordinating histone acetylation and H1 dissociation in transcriptional regulation. Mol. Cell 2007, 27, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.-X.; Nguyen, Q.; Chou, Y.; Wang, T.; Nandakumar, V.; Yates, P.; Jones, L.; Wang, L.; Won, H.; Lee, H.-R.; et al. Control of B cell development by the histone H2A deubiquitinase MYSM1. Immunity 2011, 35, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Grosschedl, R. Failure of B-cell differentiation in mice lacking the transcription factor EBF. Nature 1995, 376, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, H.; Gregory, S.C.; Yokota, T.; Sakaguchi, N.; Kincade, P.W. Transcription from the RAG1 locus marks the earliest lymphocyte progenitors in bone marrow. Immunity 2002, 17, 117–130. [Google Scholar] [CrossRef]
- Zandi, S.; Mansson, R.; Tsapogas, P.; Zetterblad, J.; Bryder, D.; Sigvardsson, M. EBF1 is essential for B-lineage priming and establishment of a transcription factor network in common lymphoid progenitors. J. Immunol. 2008, 181, 3364–3372. [Google Scholar] [CrossRef]
- Huo, Y.; Li, B.-Y.; Lin, Z.-F.; Wang, W.; Jiang, X.-X.; Chen, X.; Xi, W.-J.; Li, Y.-F.; Yang, A.-G.; Chen, S.-Y.; et al. MYSM1 is essential for maintaining hematopoietic stem cell (HSC) quiescence and survival. Med. Sci. Monit. 2018, 24, 2541. [Google Scholar] [CrossRef]
- Le Guen, T.; Touzot, F.; André-Schmutz, I.; Lagresle-Peyrou, C.; France, B.; Kermasson, L.; Lambert, N.; Picard, C.; Nitschke, P.; Carpentier, W.; et al. An in vivo genetic reversion highlights the crucial role of Myb-Like, SWIRM, and MPN domains 1 (MYSM1) in human hematopoiesis and lymphocyte differentiation. J. Allergy Clin. Immunol. 2015, 136, 1619–1626. [Google Scholar] [CrossRef]
- Nijnik, A.; Clare, S.; Hale, C.; Raisen, C.; McIntyre, R.E.; Yusa, K.; Everitt, A.R.; Mottram, L.; Podrini, C.; Lucas, M.; et al. The critical role of histone H2A-deubiquitinase Mysm1 in hematopoiesis and lymphocyte differentiation. Blood 2012, 119, 1370–1379. [Google Scholar] [CrossRef]
- Nandakumar, V.; Chou, Y.; Zang, L.; Huang, X.F.; Chen, S.-Y. Epigenetic control of natural killer cell maturation by histone H2A deubiquitinase, MYSM1. Proc. Natl. Acad. Sci. USA 2013, 110, E3927–E3936. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Nandakumar, V.; Jiang, X.-X.; Jones, L.; Yang, A.-G.; Huang, X.F.; Chen, S.-Y. The control of hematopoietic stem cell maintenance, self-renewal, and differentiation by Mysm1-mediated epigenetic regulation. Blood 2013, 122, 2812–2822. [Google Scholar] [CrossRef] [PubMed]
- Won, H.; Nandakumar, V.; Yates, P.; Sanchez, S.; Jones, L.; Huang, X.F.; Chen, S.-Y. Epigenetic control of dendritic cell development and fate determination of common myeloid progenitor by Mysm1. Blood 2014, 124, 2647–2656. [Google Scholar] [CrossRef] [PubMed]
- Opferman, J.T.; Iwasaki, H.; Ong, C.C.; Suh, H.; Mizuno, S.-I.; Akashi, K.; Korsmeyer, S.J. Obligate role of anti-apoptotic MCL-1 in the survival of Hematopoietic stem cells. Science 2005, 307, 1101–1104. [Google Scholar] [CrossRef]
- Nicassio, F.; Corrado, N.; Vissers, J.H.; Areces, L.B.; Bergink, S.; Marteijn, J.A.; Geverts, B.; Houtsmuller, A.B.; Vermeulen, W.; Di Fiore, P.P.; et al. Human USP3 is a chromatin modifier required for S phase progression and genome stability. Curr. Biol. 2007, 17, 1972–1977. [Google Scholar] [CrossRef]
- Lancini, C.; Gargiulo, G.; van den Berk, P.C.; Citterio, E. Quantitative analysis by next generation sequencing of hematopoietic stem and progenitor cells (LSK) and of splenic B cells transcriptomes from wild-type and Usp3-knockout mice. Data Brief 2016, 6, 556–561. [Google Scholar] [CrossRef][Green Version]
- Lancini, C.; van den Berk, P.C.; Vissers, J.H.; Gargiulo, G.; Song, J.-Y.; Hulsman, D.; Serresi, M.; Tanger, E.; Blom, M.; Vens, C.; et al. Tight regulation of ubiquitin-mediated DNA damage response by USP3 preserves the functional integrity of hematopoietic stem cells. J. Exp. Med. 2014, 211, 1759–1777. [Google Scholar] [CrossRef]
- Yang, S.; Wang, J.; Guo, S.; Huang, D.; Lorigados, I.B.; Nie, X.; Lou, D.; Li, Y.; Liu, M.; Kang, Y.; et al. Transcriptional activation of USP16 gene expression by NFκB signaling. Mol. Brain 2019, 12, 1–12. [Google Scholar] [CrossRef]
- Gu, Y.; Jones, A.E.; Yang, W.; Liu, S.; Dai, Q.; Liu, Y.; Swindle, C.S.; Zhou, D.; Zhang, Z.; Ryan, T.M.; et al. The histone H2A deubiquitinase Usp16 regulates hematopoiesis and hematopoietic stem cell function. Proc. Natl. Acad. Sci. USA 2016, 113, E51–E60. [Google Scholar] [CrossRef]
- Gu, Y.; Yang, W.; Jones, A.; Liu, S.; Dai, Q.; Swindle, C.S.; Ryan, T.; Townes, T.M.; Klug, C.; Wang, H. Regulation of Hematopoietic Stem Cell Function By the Histone H2A Deubiquitinase Usp16. Blood 2015, 126, 1177. [Google Scholar] [CrossRef]
- Yang, W.; Lee, Y.-H.; Jones, A.E.; Woolnough, J.L.; Zhou, D.; Dai, Q.; Wu, Q.; Giles, K.E.; Townes, T.M.; Wang, H. The histone H2A deubiquitinase Usp16 regulates embryonic stem cell gene expression and lineage commitment. Nat. Commun. 2014, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.; Huang, T.T.; Dirac, A.M.; Brummelkamp, T.R.; Kerkhoven, R.M.; D’Andrea, A.D.; Bernards, R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol. Cell 2005, 17, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Parmar, K.; Kim, J.; Sykes, S.M.; Shimamura, A.; Stuckert, P.; Zhu, K.; Hamilton, A.; Deloach, M.K.; Kutok, J.L.; Akashi, K.; et al. Hematopoietic stem cell defects in mice with deficiency of Fancd2 or Usp1. Stem Cells 2010, 28, 1186–1195. [Google Scholar] [CrossRef] [PubMed]
- Van den Berk, P.; Lancini, C.; Company, C.; Serresi, M.; Hulsman, D.; Pritchard, C.; Song, J.-Y.; Schmitt, M.J.; Tanger, E.; Huijbers, I.J.; et al. USP15 deubiquitinase safeguards hematopoiesis and genome integrity in hematopoietic stem cells and leukemia cells. Cell Rep. 2020, 20, 00384. [Google Scholar] [CrossRef]
- Niederkorn, M.; Hueneman, K.; Choi, K.; Varney, M.E.; Romano, L.; Pujato, M.A.; Greis, K.D.; Inoue, J.-i.; Meetei, R.; Starczynowski, D.T. TIFAB Regulates USP15-Mediated p53 Signaling during Stressed and Malignant Hematopoiesis. Cell Rep. 2020, 30, 2776–2790. [Google Scholar] [CrossRef]
- Sanchez-Pulido, L.; Kong, L.; Ponting, C.P. A common ancestry for BAP1 and Uch37 regulators. Bioinformatics 2012, 28, 1953–1956. [Google Scholar] [CrossRef]
- Paulsson, K.; Bekassy, A.; Olofsson, T.; Mitelman, F.; Johansson, B.; Panagopoulos, I. A novel and cytogenetically cryptic t (7; 21)(p22; q22) in acute myeloid leukemia results in fusion of RUNX1 with the ubiquitin-specific protease gene USP42. Leukemia 2006, 20, 224–229. [Google Scholar] [CrossRef]
- Wefes, I.; Mastrandrea, L.D.; Haldeman, M.; Koury, S.T.; Tamburlin, J.; Pickart, C.M.; Finley, D. Induction of ubiquitin-conjugating enzymes during terminal erythroid differentiation. Proc. Natl. Acad. Sci. USA 1995, 92, 4982–4986. [Google Scholar] [CrossRef]
- Aressy, B.; Jullien, D.; Cazales, M.; Marcellin, M.; Bugler, B.; Burlet-Schiltz, O.; Ducommun, B. A screen for deubiquitinating enzymes involved in the G2/M checkpoint identifies USP50 as a regulator of HSP90-dependent Wee1 stability. Cell Cycle 2010, 9, 3839–3846. [Google Scholar] [CrossRef]
- Lee, J.Y.; Seo, D.; You, J.; Chung, S.; Park, J.S.; Lee, J.-H.; Jung, S.M.; Lee, Y.S.; Park, S.H. The deubiquitinating enzyme, ubiquitin-specific peptidase 50, regulates inflammasome activation by targeting the ASC adaptor protein. FEBS Lett. 2017, 591, 479–490. [Google Scholar] [CrossRef]
- Forster, M.; Belle, J.; Petrov, J.C.; Ryder, E.; Clare, S.; Nijnik, A. Deubiquitinase MYSM1 Is Essential for Normal Fetal Liver Hematopoiesis and for the Maintenance of Hematopoietic Stem Cells in Adult Bone Marrow. Stem Cells Dev. 2015, 24, 1865–1877. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liang, L.; Ye, M.; Zhang, J.; Zhang, Y.; Kuang, Y.; Zhu, Z.; Peng, Y.; An, X. Deubiquitinase USP7 Regulates Erythroid Development Via Deubiquinating GATA1. Blood 2017, 130, 10. [Google Scholar]
- Damgaard, R.B.; Walker, J.A.; Marco-Casanova, P.; Morgan, N.V.; Titheradge, H.L.; Elliott, P.; McHale, D.; Maher, E.R.; McKenzie, A.N.; Komander, D. The Deubiquitinase OTULIN Is an Essential Negative Regulator of Inflammation and Autoimmunity. Cell 2016, 166, 1215–1230. [Google Scholar] [CrossRef] [PubMed]
- Paleolog, E.M.; Hunt, M.; Elliott, M.J.; Feldmann, M.; Maini, R.N.; Woody, J.N. Deactivation of vascular endothelium by monoclonal anti–tumor necrosis factor α antibody in rheumatoid arthritis. Arthritis Rheum. 1996, 39, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Chng, H.W.; Camplejohn, R.S.; Stone, M.G.; Hart, I.R.; Nicholson, L.J. A new role for the anti-apoptotic gene A20 in angiogenesis. Exp. Cell Res. 2006, 312, 2897–2907. [Google Scholar] [CrossRef]
- Yoo, S.Y.; Kwon, S.-M. Angiogenesis and Its Therapeutic Opportunities. Mediat. Inflamm. 2013, 2013, 1–11. [Google Scholar] [CrossRef]
- Lim, R.; Sugino, T.; Nolte, H.; Andrade, J.; Zimmermann, B.; Shi, C.; Doddaballapur, A.; Ong, Y.T.; Wilhelm, K.; Fasse, J.W.D.; et al. Deubiquitinase USP10 regulates Notch signaling in the endothelium. Science 2019, 364, 188–193. [Google Scholar]
- Jura, J.; Skalniak, L.; Koj, A. Monocyte chemotactic protein-1-induced protein-1 (MCPIP1) is a novel multifunctional modulator of inflammatory reactions. Biochim. Biophys. Acta 2012, 1823, 1905–1913. [Google Scholar] [CrossRef]
- Roy, A.; Zhang, M.; Saad, Y.; Kolattukudy, P.E. Antidicer RNAse activity of monocyte chemotactic protein-induced protein-1 is critical for inducing angiogenesis. Am. J. Physiol. Cell Physiol. 2013, 305, C1021–C1032. [Google Scholar] [CrossRef]
- Wang, B.; Cai, W.; Ai, D.; Zhang, X.; Yao, L. The Role of Deubiquitinases in Vascular Diseases. J. Cardiovasc. Transl. Res. 2019, 13, 1–11. [Google Scholar] [CrossRef]
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Frescas, D.; Pagano, M. Deregulated proteolysis by the F-box proteins SKP2 and β-TrCP: Tipping the scales of cancer. Nat. Rev. Cancer 2008, 8, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Corn, P.G. Role of the ubiquitin proteasome system in renal cell carcinoma. BMC Biochem. 2007, 8, S4. [Google Scholar] [CrossRef]
- Crawford, L.J.; Irvine, A.E. Targeting the ubiquitin proteasome system in haematological malignancies. Blood Rev. 2013, 27, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Kleeff, J.; Wildi, S.; Friess, H.; Büchler, M.W.; Israel, M.A.; Korc, M. Id-1 and Id-2 Are Overexpressed in Pancreatic Cancer and in Dysplastic Lesions in Chronic Pancreatitis. Am. J. Pathol. 1999, 155, 815–822. [Google Scholar] [CrossRef]
- Lin, C.Q.; Singh, J.; Murata, K.; Itahana, Y.; Parrinello, S.; Liang, S.H.; Gillett, C.E.; Campisi, J.; Desprez, P.-Y. A role for Id-1 in the aggressive phenotype and steroid hormone response of human breast cancer cells. Cancer Res. 2000, 60, 1332–1340. [Google Scholar]
- O’Brien, C.A.; Kreso, A.; Ryan, P.; Hermans, K.G.; Gibson, L.; Wang, Y.; Tsatsanis, A.; Gallinger, S.; Dick, J.E. ID1 and ID3 regulate the self-renewal capacity of human colon cancer-initiating cells through p21. Cancer Cell 2012, 21, 777–792. [Google Scholar] [CrossRef]
- Soroceanu, L.; Murase, R.; Limbad, C.; Singer, E.; Allison, J.; Adrados, I.; Kawamura, R.; Pakdel, A.; Fukuyo, Y.; Nguyen, D.; et al. Id-1 is a key transcriptional regulator of glioblastoma aggressiveness and a novel therapeutic target. Cancer Res. 2013, 73, 1559–1569. [Google Scholar] [CrossRef]
- Tang, R.; Hirsch, P.; Fava, F.; Lapusan, S.; Marzac, C.; Teyssandier, I.; Pardo, J.; Marie, J.-P.; Legrand, O. High Id1 expression is associated with poor prognosis in 237 patients with acute myeloid leukemia. Blood 2009, 114, 2993–3000. [Google Scholar] [CrossRef]
- Mistry, H.; Hsieh, G.; Buhrlage, S.J.; Huang, M.; Park, E.; Cuny, G.D.; Galinsky, I.; Stone, R.M.; Gray, N.S.; D’Andrea, A.D. Small-molecule inhibitors of USP1 target ID1 degradation in leukemic cells. Mol. Cancer Ther. 2013, 12, 2651–2662. [Google Scholar] [CrossRef]
- Das, D.S.; Das, A.; Ray, A.; Song, Y.; Samur, M.K.; Munshi, N.C.; Chauhan, D.; Anderson, K.C. Blockade of deubiquitylating enzyme USP1 inhibits DNA repair and triggers apoptosis in multiple myeloma cells. Clin. Cancer Res. 2017, 23, 4280–4289. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, L.; Wang, X.; Zhou, L.; Liao, Z.; Xu, L.; Wu, H.; Ren, J.; Li, Z.; Yang, L.; et al. Characteristics of A20 gene polymorphisms and clinical significance in patients with rheumatoid arthritis. J. Transl. Med. 2015, 13, 215. [Google Scholar] [CrossRef]
- Song, H.Y.; Rothe, M.; Goeddel, D.V. The tumor necrosis factor-inducible zinc finger protein A20 interacts with TRAF1/TRAF2 and inhibits NF-kappaB activation. Proc. Natl. Acad. Sci. USA 1996, 93, 6721–6725. [Google Scholar] [CrossRef] [PubMed]
- Hymowitz, S.G.; Wertz, I.E. A20: From ubiquitin editing to tumour suppression. Nat. Rev. Cancer 2010, 10, 332–341. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, L.; Liao, Z.; Zhang, F.; Xu, L.; Xu, Y.; Chen, S.; Yang, L.; Zhou, Y.; Li, Y. Alternative Expression Pattern of MALT1-A20-NF-B in Patients with Rheumatoid Arthritis. J. Immunol. Res. 2014, 2014, 1–7. [Google Scholar] [CrossRef]
- Lu, T.T.; Onizawa, M.; Hammer, G.E.; Turer, E.E.; Yin, Q.; Damko, E.; Agelidis, A.; Shifrin, N.; Advincula, R.; Barrera, J.; et al. Dimerization and ubiquitin mediated recruitment of A20, a complex deubiquitinating enzyme. Immunity 2013, 38, 896–905. [Google Scholar] [CrossRef]
- Kato, M.; Sanada, M.; Kato, I.; Sato, Y.; Takita, J.; Takeuchi, K.; Niwa, A.; Chen, Y.; Nakazaki, K.; Nomoto, J.; et al. Frequent inactivation of A20 in B-cell lymphomas. Nature 2009, 459, 712–716. [Google Scholar] [CrossRef]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-κB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef]
- Chanudet, E.; Huang, Y.; Ichimura, K.; Dong, G.; Hamoudi, R.A.; Radford, J.; Wotherspoon, A.C.; Isaacson, P.G.; Ferry, J.; Du, M.Q. A20 is targeted by promoter methylation, deletion and inactivating mutation in MALT lymphoma. Leukemia 2010, 24, 483–487. [Google Scholar] [CrossRef]
- Honma, S. The mammalian circadian system: A hierarchical multi-oscillator structure for generating circadian rhythm. J. Physiol. Sci. 2018, 68, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Novak, U.; Rinaldi, A.; Kwee, I.; Nandula, S.V.; Rancoita, P.M.V.; Compagno, M.; Cerri, M.; Rossi, D.; Murty, V.V.; Zucca, E.; et al. The NF-κB negative regulator TNFAIP3 (A20) is inactivated by somatic mutations and genomic deletions in marginal zone lymphomas. Blood 2009, 113, 4918–4921. [Google Scholar] [CrossRef] [PubMed]
- Honma, K.; Tsuzuki, S.; Nakagawa, M.; Karnan, S.; Aizawa, Y.; Kim, W.S.; Kim, Y.D.; Ko, Y.H.; Seto, M. TNFAIP3 is the target gene of chromosome band 6q23. 3–q24. 1 loss in ocular adnexal marginal zone B cell lymphoma. Genes Chromosomes Cancer 2008, 47, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.A.; Schmitz, R.; Brune, V.; Tiacci, E.; Döring, C.; Hansmann, M.-L.; Siebert, R.; Küppers, R. Mutations in the genes coding for the NF-κB regulating factors IκBα and A20 are uncommon in nodular lymphocyte-predominant Hodgkin’s lymphoma. Haematologica 2010, 95, 153–157. [Google Scholar] [CrossRef]
- Schmitz, R.; Hansmann, M.-L.; Bohle, V.; Martin-Subero, J.I.; Hartmann, S.; Mechtersheimer, G.; Klapper, W.; Vater, I.; Giefing, M.; Gesk, S.; et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J. Exp. Med. 2009, 206, 981–989. [Google Scholar] [CrossRef]
- Nomoto, J.; Hiramoto, N.; Kato, M.; Sanada, M.; Maeshima, A.M.; Taniguchi, H.; Hosoda, F.; Asakura, Y.; Munakata, W.; Sekiguchi, N.; et al. Deletion of the TNFAIP3/A20 gene detected by FICTION analysis in classical Hodgkin lymphoma. BMC Cancer 2012, 12, 457. [Google Scholar] [CrossRef]
- Weiner, G.J. Rituximab: Mechanism of action. Semin. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef]
- Zhou, X.; Hu, W.; Qin, X. The role of complement in the mechanism of action of rituximab for B-cell lymphoma: Implications for therapy. Oncologist 2008, 13, 954–966. [Google Scholar] [CrossRef]
- Hu, S.; Liang, S.; Guo, H.; Zhang, D.; Li, H.; Wang, X.; Yang, W.; Qian, W.; Hou, S.; Wang, H.; et al. Comparison of the inhibition mechanisms of adalimumab and infliximab in treating tumor necrosis factor α-associated diseases from a molecular view. J. Biol. Chem. 2013, 288, 27059–27067. [Google Scholar] [CrossRef]
- Miller, S.C.; Huang, R.; Sakamuru, S.; Shukla, S.J.; Attene-Ramos, M.S.; Shinn, P.; Van Leer, D.; Leister, W.; Austin, C.P.; Xia, M. Identification of known drugs that act as inhibitors of NF-κB signaling and their mechanism of action. Biochem. Pharmacol. 2010, 79, 1272–1280. [Google Scholar] [CrossRef]
- Colland, F. The Therapeutic Potential of Deubiquitinating Enzyme Inhibitors. Biochem. Soc. Trans. 2010, 38, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Boutell, C.; Canning, M.; Orr, A.; Everett, R. Reciprocal activities between herpes simplex virus type 1 regulatory protein ICP0, a ubiquitin E3 ligase, and ubiquitin-specific protease USP7. J. Virol. 2005, 79, 12342–12354. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Jensen, J.P.; Ludwig, R.L.; Vousden, K.H.; Weissman, A.M. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J. Biol. Chem. 2000, 275, 8945–8951. [Google Scholar] [CrossRef]
- Li, M.; Brooks, C.L.; Kon, N.; Gu, W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 2004, 13, 879–886. [Google Scholar] [CrossRef]
- Anderson, K.C. Targeted therapy of multiple myeloma based upon tumor-microenvironmental interactions. Exp. Hematol. 2007, 35, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Waller, E.K.; Richardson, P.G.; Jagannath, S.; Orlowski, R.Z.; Giver, C.R.; Jaye, D.L.; Francis, D.; Giusti, S.; Torre, C.; et al. Risk factors and kinetics of thrombocytopenia associated with bortezomib for relapsed, refractory multiple myeloma. Blood 2005, 106, 3777–3784. [Google Scholar] [CrossRef]
- Chauhan, D.; Tian, Z.; Nicholson, B.; Kumar, K.S.; Zhou, B.; Carrasco, R.; McDermott, J.L.; Leach, C.A.; Fulcinniti, M.; Kodrasov, M.; et al. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer cell 2012, 22, 345–358. [Google Scholar] [CrossRef]
- Lim, M.S.; Elenitoba-Johnson, K.S.J. Ubiquitin ligases in malignant lymphoma. Leuk. Lymphoma 2004, 45, 1329–1339. [Google Scholar] [CrossRef]
- Agathanggelou, A.; Smith, E.; Davies, N.J.; Kwok, M.; Zlatanou, A.; Oldreive, C.E.; Mao, J.; Da Costa, D.; Yadollahi, S.; Perry, T.; et al. USP7 inhibition alters homologous recombination repair and targets CLL cells independently of ATM/p53 functional status. Blood 2017, 130, 156–166. [Google Scholar] [CrossRef]
- Kategaya, L.; Di Lello, P.; Rougé, L.; Pastor, R.; Clark, K.R.; Drummond, J.; Kleinheinz, T.; Lin, E.; Upton, J.-P.; Prakash, S.; et al. USP7 small-molecule inhibitors interfere with ubiquitin binding. Nature 2017, 550, 534–538. [Google Scholar] [CrossRef]
- Sahasrabuddhe, A.A.; Elenitoba-Johnson, K.S.J. Role of the ubiquitin proteasome system in hematologic malignancies. Immunol. Rev. 2015, 263, 224–239. [Google Scholar] [CrossRef] [PubMed]
- Schwickart, M.; Huang, X.; Lill, J.R.; Liu, J.; Ferrando, R.; French, D.M.; Maecker, H.; O’Rourke, K.; Bazan, F.; Eastham-Anderson, J.; et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 2010, 463, 103–107. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Karp, J.E.; Svingen, P.A.; Krajewski, S.; Burke, P.J.; Gore, S.D.; Reed, J.C. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood 1998, 91, 991–1000. [Google Scholar] [CrossRef]
- Robillard, N.; Pellat-Deceunynck, C.; Bataille, R. Phenotypic characterization of the human myeloma cell growth fraction. Blood 2005, 105, 4845–4848. [Google Scholar] [CrossRef]
- Sun, H.; Kapuria, V.; Peterson, L.F.; Fang, D.; Bornmann, W.G.; Bartholomeusz, G.; Talpaz, M.; Donato, N.J. Bcr-Abl ubiquitination and Usp9x inhibition block kinase signaling and promote CML cell apoptosis. Blood 2011, 117, 3151–3162. [Google Scholar] [CrossRef] [PubMed]
- Peterson, L.F.; Sun, H.; Liu, Y.; Potu, H.; Kandarpa, M.; Ermann, M.; Courtney, S.M.; Young, M.; Showalter, H.D.; Sun, D.; et al. Targeting deubiquitinase activity with a novel small-molecule inhibitor as therapy for B-cell malignancies. Blood 2015, 125, 3588–3597. [Google Scholar] [CrossRef] [PubMed]
- Vong, Q.P.; Cao, K.; Li, H.Y.; Iglesias, P.A.; Zheng, Y. Chromosome alignment and segregation regulated by ubiquitination of survivin. Science 2005, 310, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Engel, K.; Rudelius, M.; Slawska, J.; Jacobs, L.; Abhari, B.A.; Altmann, B.; Kurutz, J.; Rathakrishnan, A.; Fernández-Sáiz, V.; Brunner, A.; et al. USP9X stabilizes XIAP to regulate mitotic cell death and chemoresistance in aggressive B-cell lymphoma. EMBO Mol. Med. 2016, 8, 851–862. [Google Scholar] [CrossRef]
- D’arcy, P.; Brnjic, S.; Olofsson, M.H.; Fryknäs, M.; Lindsten, K.; De Cesare, M.; Perego, P.; Sadeghi, B.; Hassan, M.; Larsson, R.; et al. Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat. Med. 2011, 17, 1636. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata, S.; Katayama, J.; Shindo, H.; Ozawa, Y.; Itoh, K.; Mizugaki, M. Increased expression of queuosine synthesizing enzyme, tRNA-guanine transglycosylase, and queuosine levels in tRNA of leukemic cells. J. Biochem. 2001, 129, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Dick, L.R.; Fleming, P.E. Building on bortezomib: Second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov. Today 2010, 15, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Diefenbach, C.; Zain, J.; O’Connor, O.A. Emerging role of carfilzomib in treatment of relapsed and refractory lymphoid neoplasms and multiple myeloma. Core Evid. 2011, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Kupperman, E.; Lee, E.C.; Cao, Y.; Bannerman, B.; Fitzgerald, M.; Berger, A.; Yu, J.; Yang, Y.; Hales, P.; Bruzzese, F.; et al. Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 2010, 70, 1970–1980. [Google Scholar] [CrossRef]
- Hazlehurst, L.A.; Argilagos, R.F.; Emmons, M.; Boulware, D.; Beam, C.A.; Sullivan, D.M.; Dalton, W.S. Cell adhesion to fibronectin (CAM-DR) influences acquired mitoxantrone resistance in U937 cells. Cancer Res. 2006, 66, 2338–2345. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, J.; Shen, C.; Ding, L.; Zhong, F.; Ouyang, Y.; Wang, Y.; He, S. The role of ubiquitin-specific protease 14 (USP 14) in cell adhesion-mediated drug resistance (CAM-DR) of multiple myeloma cells. Eur. J. Haematol. 2017, 98, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Paulus, A.; Akhtar, S.; Caulfield, T.; Samuel, K.; Yousaf, H.; Bashir, Y.; Paulus, S.; Tran, D.; Hudec, R.; Cogen, D.; et al. Coinhibition of the deubiquitinating enzymes, USP14 and UCHL5, with VLX1570 is lethal to ibrutinib-or bortezomib-resistant Waldenstrom macroglobulinemia tumor cells. Blood Cancer J. 2016, 6, e492. [Google Scholar] [CrossRef] [PubMed]
- Chitta, K.; Paulus, A.; Akhtar, S.; Blake, M.K.K.; Caulfield, T.R.; Novak, A.J.; Ansell, S.M.; Advani, P.; Ailawadhi, S.; Sher, T.; et al. Targeted inhibition of the deubiquitinating enzymes, USP 14 and UCHL 5, induces proteotoxic stress and apoptosis in W aldenström macroglobulinaemia tumour cells. Br. J. Haematol. 2015, 169, 377–390. [Google Scholar] [CrossRef]
- D’Arcy, P.; Brnjic, S.; Olofsson, M.H.; Fryknas, M.; Larsson, R.; Linder, S.T. Inhibition of Proteasome Deubiquitinating Activity as a Novel Cancer Therapy. Cancer Res. 2012, 72, 2941. [Google Scholar]
- Tian, Z.; D’Arcy, P.; Wang, X.; Ray, A.; Tai, Y.-T.; Hu, Y.; Carrasco, R.D.; Richardson, P.; Linder, S.; Chauhan, D.; et al. A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 2014, 123, 706–716. [Google Scholar] [CrossRef]
- Lee, B.-H.; Lee, M.J.; Park, S.; Oh, D.-C.; Elsasser, S.; Chen, P.-C.; Gartner, C.; Dimova, N.; Hanna, J.; Gygi, S.P.; et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 2010, 467, 179–184. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Wu, Y.-S.; Hung, C.-Y.; Wang, S.-A.; Young, M.-J.; Hsu, T.-I.; Hung, J.-J. USP24 induces IL-6 in tumor-associated microenvironment by stabilizing p300 and β-TrCP and promotes cancer malignancy. Nat. Commun. 2018, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Lub, S.; Maes, K.; Menu, E.; De Bruyne, E.; Vanderkerken, K.; Van Valckenborgh, E. Novel strategies to target the ubiquitin proteasome system in multiple myeloma. Oncotarget 2016, 7, 6521. [Google Scholar] [CrossRef]
- Pagan, J.; Seto, T.; Pagano, M.; Cittadini, A. Role of the ubiquitin proteasome system in the heart. Circ. Res. 2013, 112, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Lork, M.; Verhelst, K.; Beyaert, R. CYLD, A20 and OTULIN deubiquitinases in NF-κ B signaling and cell death: So similar, yet so different. Cell Death Differ. 2017, 24, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Massoumi, R.; Paus, R. Cylindromatosis and the CYLD gene: New lessons on the molecular principles of epithelial growth control. Bioessays 2007, 29, 1203–1214. [Google Scholar] [CrossRef]
- Kovalenko, A.; Chable-Bessia, C.; Cantarella, G.; Israël, A.; Wallach, D.; Courtois, G. The tumour suppressor CYLD negatively regulates NF-κB signalling by deubiquitination. Nature 2003, 424, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Arora, M.; Kaul, D.; Varma, N.; Marwaha, R. Cellular proteolytic modification of tumor-suppressor CYLD is critical for the initiation of human T-cell acute lymphoblastic leukemia. Blood Cells Mol. Dis. 2015, 54, 132–138. [Google Scholar] [CrossRef]
- Jeon, H.-M.; Jin, X.; Lee, J.-S.; Oh, S.-Y.; Sohn, Y.-W.; Park, H.-J.; Joo, K.M.; Park, W.-Y.; Nam, D.-H.; DePinho, R.A.; et al. Inhibitor of differentiation 4 drives brain tumor-initiating cell genesis through cyclin E and notch signaling. Genes Dev. 2008, 22, 2028–2033. [Google Scholar] [CrossRef]
- Screpanti, I.; Bellavia, D.; Campese, A.F.; Frati, L.; Gulino, A. Notch, a unifying target in T-cell acute lymphoblastic leukemia? Trends Mol. Med. 2003, 9, 30–35. [Google Scholar] [CrossRef]
- Vilimas, T.; Mascarenhas, J.; Palomero, T.; Mandal, M.; Buonamici, S.; Meng, F.; Thompson, B.; Spaulding, C.; Macaroun, S.; Alegre, M.-L.; et al. Targeting the NF-κB signaling pathway in Notch1-induced T-cell leukemia. Nat. Med. 2007, 13, 70–77. [Google Scholar] [CrossRef]
- Espinosa, L.; Cathelin, S.; D’Altri, T.; Trimarchi, T.; Statnikov, A.; Guiu, J.; Rodilla, V.; Inglés-Esteve, J.; Nomdedeu, J.; Bellosillo, B.; et al. The Notch/Hes1 pathway sustains NF-κB activation through CYLD repression in T cell leukemia. Cancer cell 2010, 18, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Wickström, S.A.; Masoumi, K.C.; Khochbin, S.; Fässler, R.; Massoumi, R. CYLD negatively regulates cell-cycle progression by inactivating HDAC6 and increasing the levels of acetylated tubulin. EMBO J. 2010, 29, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Stegmeier, F.; Sowa, M.E.; Nalepa, G.; Gygi, S.P.; Harper, J.W.; Elledge, S.J. The tumor suppressor CYLD regulates entry into mitosis. Proc. Natl. Acad. Sci. USA 2007, 104, 8869–8874. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Huo, L.; Sun, X.; Liu, M.; Li, D.; Dong, J.-T.; Zhou, J. The tumor suppressor CYLD regulates microtubule dynamics and plays a role in cell migration. J. Biol. Chem. 2008, 283, 8802–8809. [Google Scholar] [CrossRef]
- Li, D.; Gao, J.; Yang, Y.; Sun, L.; Suo, S.; Luo, Y.; Shui, W.; Zhou, J.; Liu, M. CYLD coordinates with EB1 to regulate microtubule dynamics and cell migration. Cell Cycle 2014, 13, 974–983. [Google Scholar] [CrossRef]
- Niu, J.; Shi, Y.; Xue, J.; Miao, R.; Huang, S.; Wang, T.; Wu, J.; Fu, M.; Wu, Z.H. USP10 inhibits genotoxic NF-κB activation by MCPIP1-facilitated deubiquitination of NEMO. EMBO J. 2013, 32, 3206–3219. [Google Scholar] [CrossRef]
- Sun, W.; Tan, X.; Shi, Y.; Xu, G.; Mao, R.; Gu, X.; Fan, Y.; Yu, Y.; Burlingame, S.; Zhang, H.; et al. USP11 negatively regulates TNFα-induced NF-κB activation by targeting on IκBα. Cell Signal. 2010, 22, 386–394. [Google Scholar] [CrossRef]
- Xu, G.; Tan, X.; Wang, H.; Sun, W.; Shi, Y.; Burlingame, S.; Gu, X.; Cao, G.; Zhang, T.; Qin, J.; et al. Ubiquitin-specific peptidase 21 inhibits tumor necrosis factor α-induced nuclear factor κB activation via binding to and deubiquitinating receptor-interacting protein 1. J. Biol. Chem. 2010, 285, 969–978. [Google Scholar] [CrossRef]
- Harhaj, E.W.; Dixit, V.M. Deubiquitinases in the regulation of NF-κB signaling. Cell Res. 2011, 21, 22–39. [Google Scholar] [CrossRef]
- Keusekotten, K.; Elliott, P.R.; Glockner, L.; Fiil, B.K.; Damgaard, R.B.; Kulathu, Y.; Wauer, T.; Hospenthal, M.K.; Gyrd-Hansen, M.; Krappmann, D.; et al. OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell 2013, 153, 1312–1326. [Google Scholar] [CrossRef]
- Hussain, S.; Zhang, Y.; Galardy, P. DUBs and cancer: The role of deubiquitinating enzymes as oncogenes, non-oncogenes and tumor suppressors. Cell Cycle 2009, 8, 1688–1697. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Young, M.A.; Donato, N. Emerging potential of therapeutic targeting of ubiquitin-specific proteases in the treatment of cancer. Cancer Res. 2014, 74, 4955–4966. [Google Scholar] [CrossRef]
- Gutierrez-Diaz, B.T.; Gu, W.; Ntziachristos, P. Deubiquitinases: Pro-oncogenic Activity and Therapeutic Targeting in Blood Malignancies. Trends Immunol. 2020, 41, 327–340. [Google Scholar] [CrossRef]
- Wuilleme-Toumi, S.; Robillard, N.; Gomez, P.; Moreau, P.; Le Gouill, S.; Avet-Loiseau, H.; Harousseau, J.; Amiot, M.; Bataille, R. Mcl-1 is overexpressed in multiple myeloma and associated with relapse and shorter survival. Leukemia 2005, 19, 1248–1252. [Google Scholar] [CrossRef]
- Kapuria, V.; Peterson, L.F.; Fang, D.; Bornmann, W.G.; Talpaz, M.; Donato, N. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 2010, 70, 9265–9276. [Google Scholar] [CrossRef]
- Boise, L.H. DUB-ling down on B-cell malignancies. Blood 2015, 125, 3522–3523. [Google Scholar] [CrossRef] [PubMed]
- Colland, F.; Formstecher, E.; Jacq, X.; Reverdy, C.; Planquette, C.; Conrath, S.; Trouplin, V.; Bianchi, J.; Aushev, V.N.; Camonis, J.; et al. Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol. Cancer Ther. 2009, 8, 2286–2295. [Google Scholar] [CrossRef] [PubMed]
- Reverdy, C.; Conrath, S.; Lopez, R.; Planquette, C.; Atmanene, C.; Collura, V.; Harpon, J.; Battaglia, V.; Vivat, V.; Sippl, W.; et al. Discovery of specific inhibitors of human USP7/HAUSP deubiquitinating enzyme. Chem. Biol. 2012, 19, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, D.; Shiloh, A.; Luo, J.; Nikolaev, A.Y.; Qin, J.; Gu, W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002, 416, 648–653. [Google Scholar] [CrossRef]
- Dar, A.; Shibata, E.; Dutta, A. Deubiquitination of Tip60 by USP7 determines the activity of the p53-dependent apoptotic pathway. Mol. Cell Biol. 2013, 33, 3309–3320. [Google Scholar] [CrossRef]
- Yuan, T.; Yan, F.; Ying, M.; Cao, J.; He, Q.; Zhu, H.; Yang, B. Inhibition of ubiquitin-specific proteases as a novel anticancer therapeutic strategy. Front. Pharmacol. 2018, 9, 1080. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, L.; Wu, J.; Sokirniy, I.; Nguyen, P.; Bregnard, T.; Weinstock, J.; Mattern, M.; Bezsonova, I.; Hancock, W.; et al. Active site-targeted covalent irreversible inhibitors of USP7 impair the functions of Foxp3+ T-regulatory cells by promoting ubiquitination of Tip60. PLoS ONE 2017, 12, e0189744. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.L.; Schauer, N.J.; Yang, J.; Lamberto, I.; Doherty, L.; Bhatt, S.; Nonami, A.; Meng, C.; Letai, A.; Wright, R.; et al. Inhibition of USP10 induces degradation of oncogenic FLT3. Nat. Chem. Biol. 2017, 13, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Van Twest, S.; Murphy, V.J.; Hodson, C.; Tan, W.; Swuec, P.; O’Rourke, J.J.; Heierhorst, J.; Crismani, W.; Deans, A. J Mechanism of ubiquitination and deubiquitination in the Fanconi anemia pathway. Mol. Cell 2017, 65, 247–259. [Google Scholar] [CrossRef]
- Meledin, R.; Mali, S.M.; Kleifeld, O.; Brik, A. Activity-Based Probes Developed by Applying a Sequential Dehydroalanine Formation Strategy to Expressed Proteins Reveal a Potential α-Globin-Modulating Deubiquitinase. Angew. Chem. Int. Ed. Engl. 2018, 130, 5747–5751. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Tefferi, A.; Levine, R. Role of TET2 and ASXL1 mutations in the pathogenesis of myeloproliferative neoplasms. Hematol. Oncol. Clin. N. Am. 2012, 26, 1053–1064. [Google Scholar] [CrossRef]
- Dey, A.; Seshasayee, D.; Noubade, R.; French, D.M.; Liu, J.; Chaurushiya, M.S.; Kirkpatrick, D.S.; Pham, V.C.; Lill, J.R.; Bakalarski, C.E.; et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science 2012, 337, 1541–1546. [Google Scholar] [CrossRef]
- Paulus, A.; Akhtar, S.; Kuranz-Blake, M.; Novak, A.J.; Ansell, S.; Gertz, M.A.; Kyle, R.A.; Martin, P.; Coleman, M.; Ailawadhi, S.; et al. Targeted Disruption of USP14 and UCHL5 with the Novel Deubiquitinase Enzyme (DUB) Inhibitor, VLX1570, Induces Immense Proteotoxicity and Cell Death in Malignant Plasma Cells. Blood 2014, 124, 3116. [Google Scholar] [CrossRef]
- Chitta, K.S.; Paulus, A.; Akhtar, S.; Kuranz, M.; Roy, V.; Ansell, S.M.; Novak, A.J.; Martin, P.; Furman, R.R.; Coleman, M. Inhibition Of The Deubiquitinating Enzymes UCHL5 and USP14 Is Lethal To Waldenströms Macroglobulinemia Cells. Blood 2013, 122, 1823. [Google Scholar] [CrossRef]
- Bahrami, E.; Witzel, M.; Racek, T.; Puchałka, J.; Hollizeck, S.; Greif-Kohistani, N.; Kotlarz, D.; Horny, H.-P.; Feederle, R.; Schmidt, H.; et al. Myb-like, SWIRM, and MPN domains 1 (MYSM1) deficiency: Genotoxic stress-associated bone marrow failure and developmental aberrations. J. Allergy Clin. Immunol. 2017, 140, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Schmid, U.; Stenzel, W.; Koschel, J.; Raptaki, M.; Wang, X.; Naumann, M.; Matuschewski, K.; Schlüter, D.; Nishanth, G. The Deubiquitinating Enzyme Cylindromatosis Dampens CD8+ T Cell Responses and Is a Critical Factor for Experimental Cerebral Malaria and Blood–Brain Barrier Damage. Front. Immunol. 2017, 8, 27. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
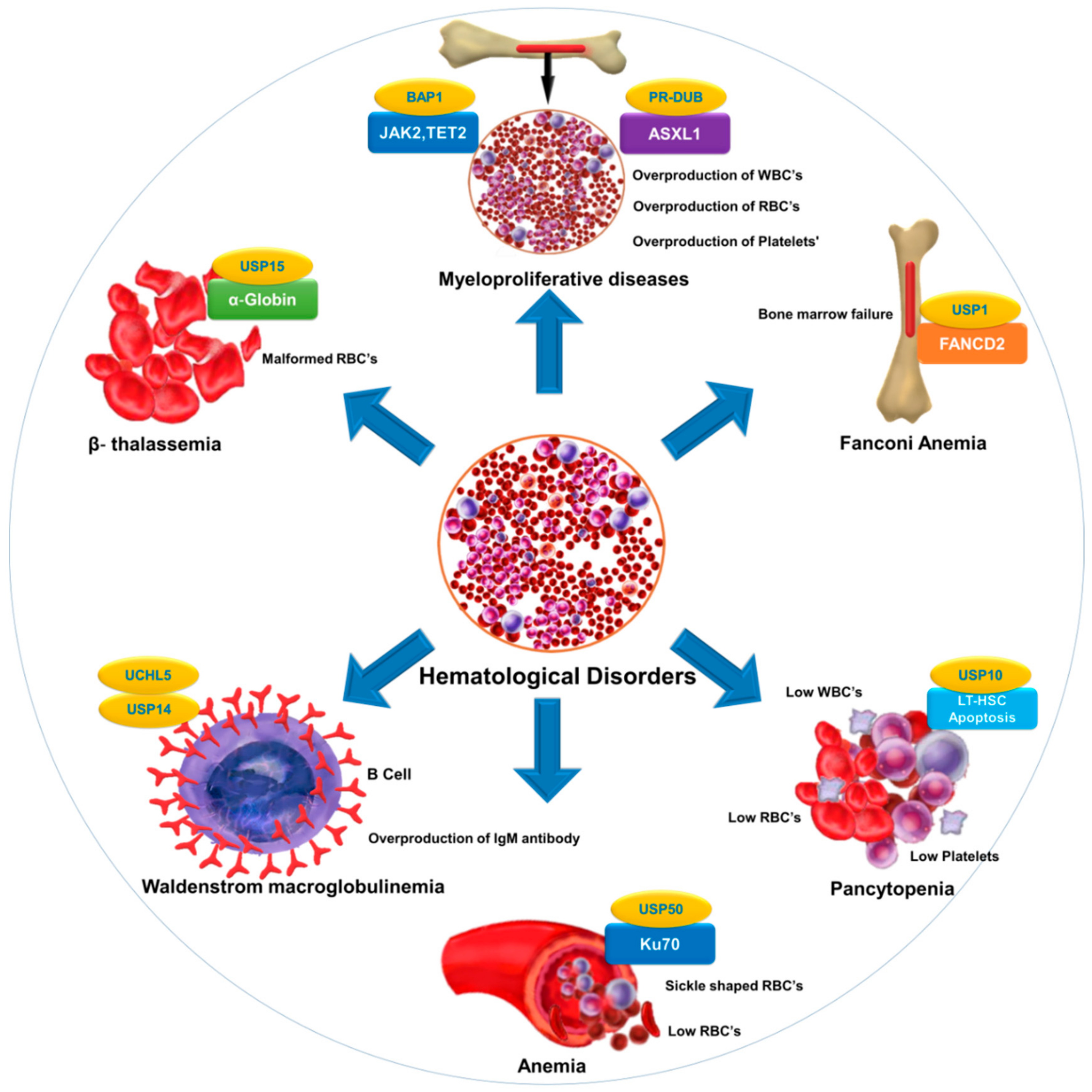
| Disorder | Associated Substrate | Regulatory DUB | Reference |
|---|---|---|---|
| Fanconi anemia | FANCD2 | USP1 | [63,176] |
| Anemia | Ku70 | USP50 | [8] |
| β-thalassemia | α-globin | USP15 | [177] |
| Pancytopenia | Reduction in LT-HSC | USP10 | [31] |
| Myeloproliferative diseases | ASXL1, EZH2,JAK2,TET2 | PR-DUB,BAP1 | [178,179] |
| Waldenstrom macroglobulinemia (WM) | Overexpression of USP14 and UCHL5 in drug-resistant WM-tumor cells | USP14 and UCHL5 | [137,180,181] |
| Bone marrow failure | B-cell factor 1 (Ebf1), paired box 5 (Pax5), and other B-lymphoid genes | MYSM1 | [45,182] |
| Malaria | CD8+ T cells | CYLD | [162,183] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarodaya, N.; Karapurkar, J.; Kim, K.-S.; Hong, S.-H.; Ramakrishna, S. The Role of Deubiquitinating Enzymes in Hematopoiesis and Hematological Malignancies. Cancers 2020, 12, 1103. https://doi.org/10.3390/cancers12051103
Sarodaya N, Karapurkar J, Kim K-S, Hong S-H, Ramakrishna S. The Role of Deubiquitinating Enzymes in Hematopoiesis and Hematological Malignancies. Cancers. 2020; 12(5):1103. https://doi.org/10.3390/cancers12051103
Chicago/Turabian StyleSarodaya, Neha, Janardhan Karapurkar, Kye-Seong Kim, Seok-Ho Hong, and Suresh Ramakrishna. 2020. "The Role of Deubiquitinating Enzymes in Hematopoiesis and Hematological Malignancies" Cancers 12, no. 5: 1103. https://doi.org/10.3390/cancers12051103
APA StyleSarodaya, N., Karapurkar, J., Kim, K.-S., Hong, S.-H., & Ramakrishna, S. (2020). The Role of Deubiquitinating Enzymes in Hematopoiesis and Hematological Malignancies. Cancers, 12(5), 1103. https://doi.org/10.3390/cancers12051103