Are Pathogenic Germline Variants in Metastatic Melanoma Associated with Resistance to Combined Immunotherapy?

 , , , ,
, , , , 

Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. DNA Extraction, Sequencing and Computational Analysis
2.2.1. Tumor Mutation Burden
2.2.2. Clinical and Biological Interpretation of Germline Alterations, Classification and Assessment of Potential Therapies
2.3. Statistical Methods
3. Results
3.1. Patient Characteristics
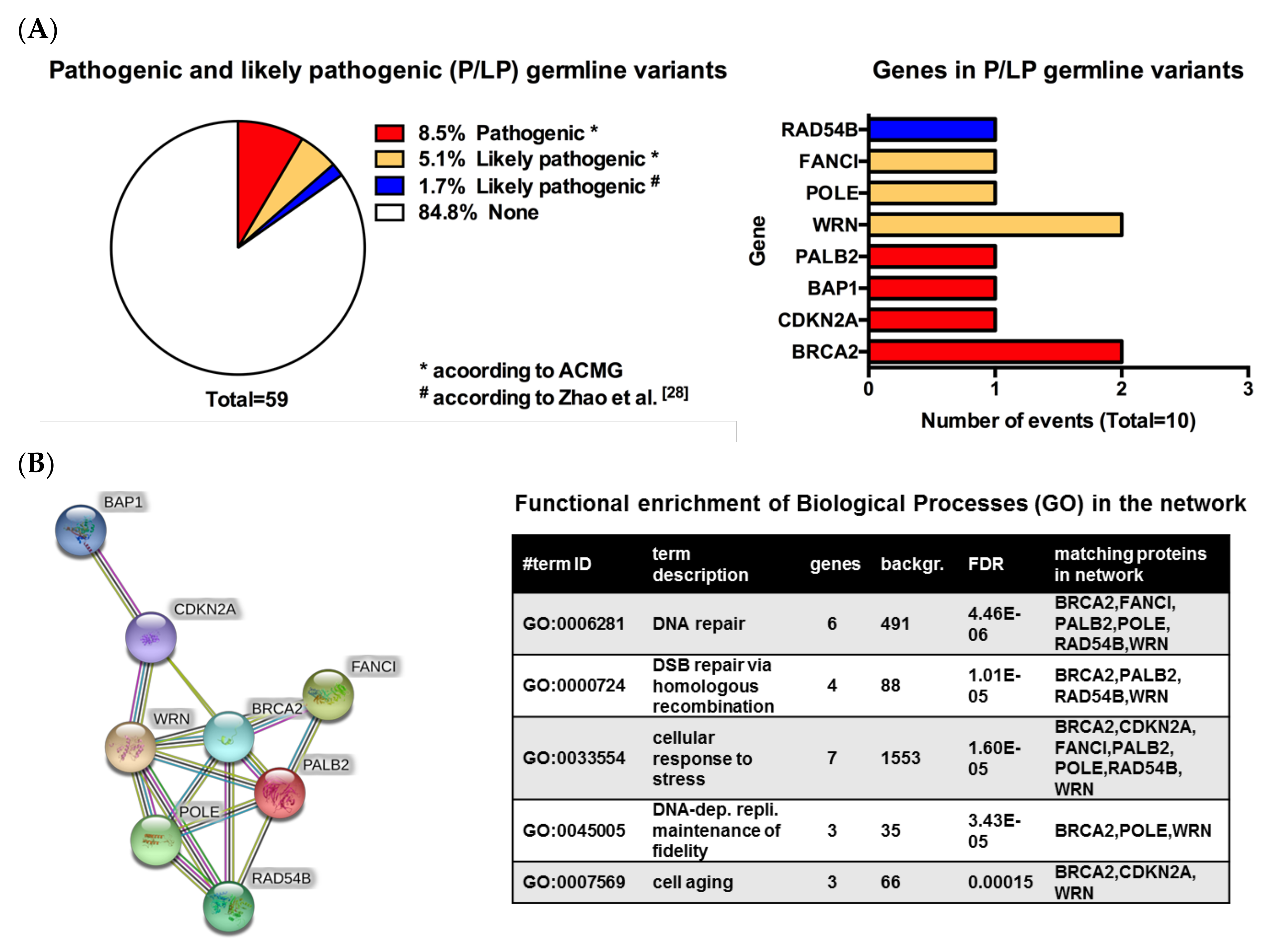
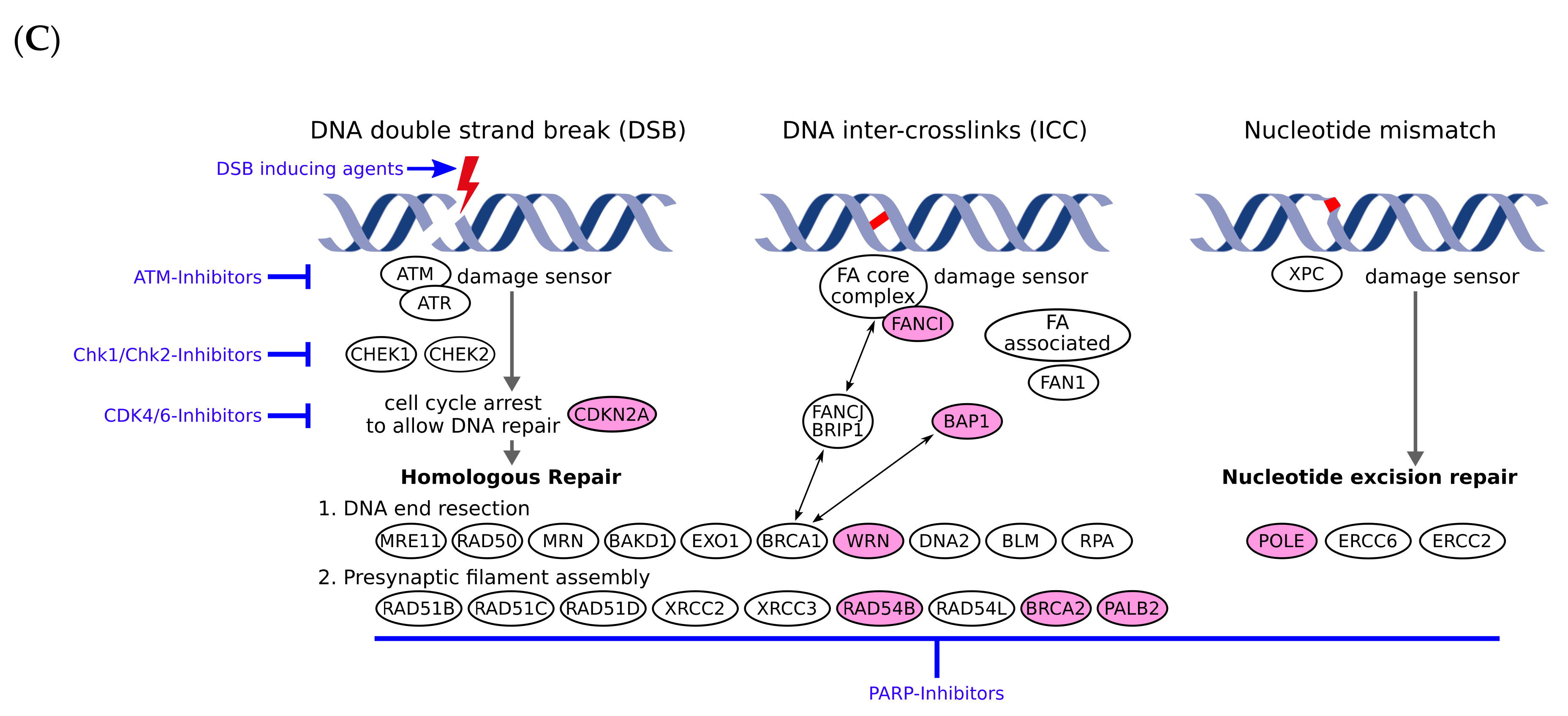
3.2. Pathogenic or Likely Pathogenic Germline Variants Identified
3.3. Response to Combined Immunotherapy
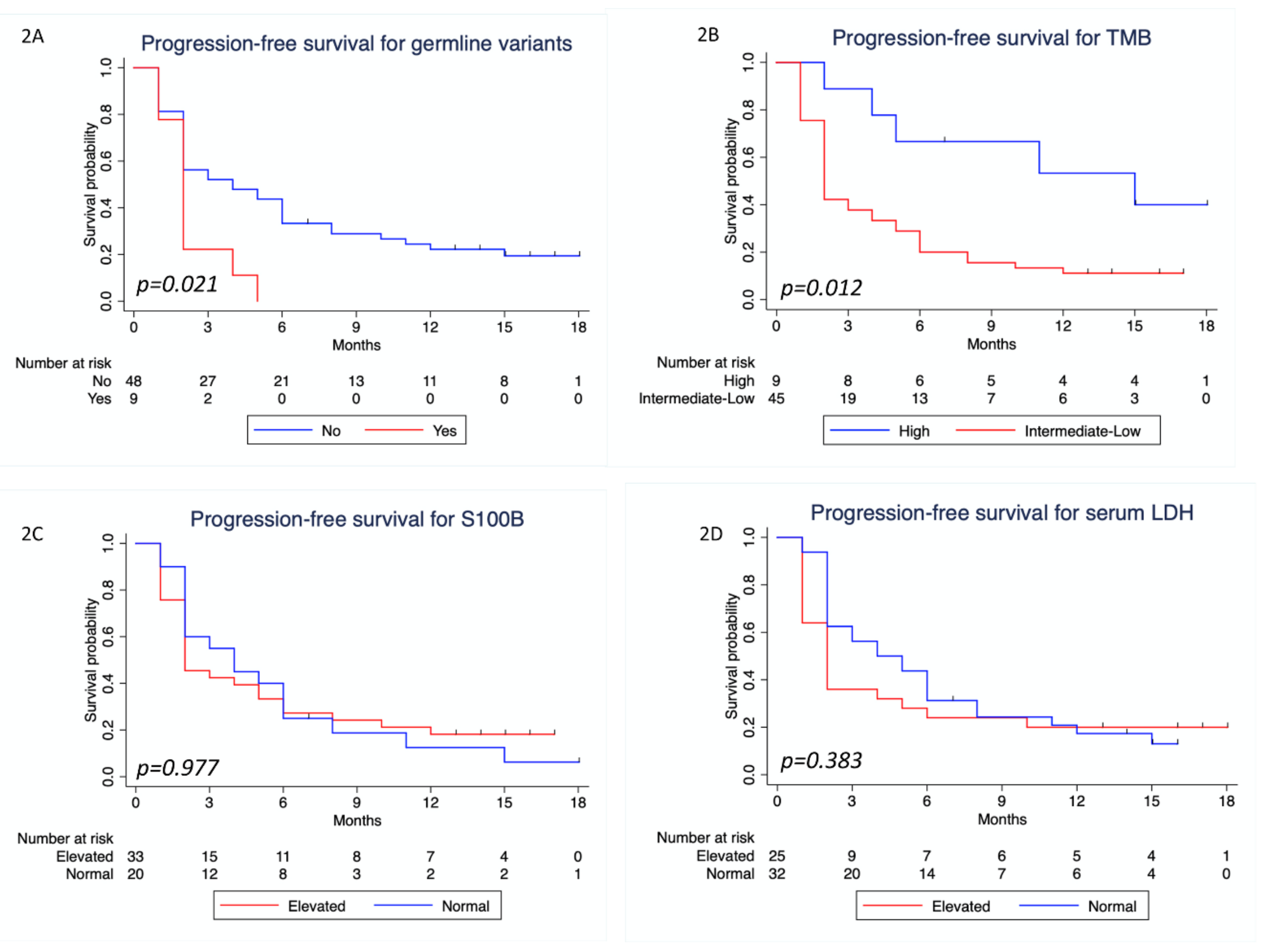
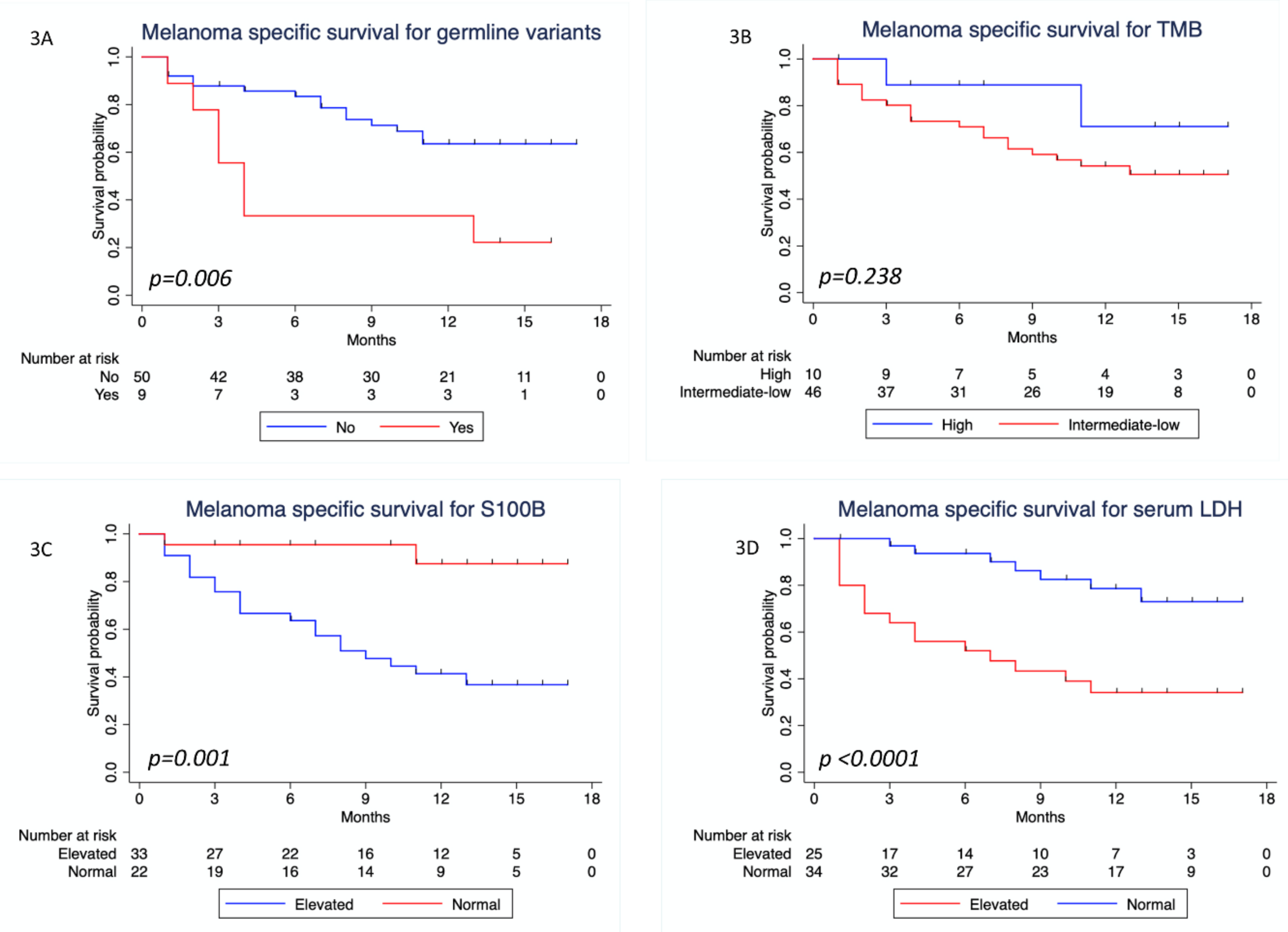
3.4. Survival Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science (New York, N.Y.) 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2018, 30, 44–56. [Google Scholar] [CrossRef]
- Ghazani, A.A.; Oliver, N.M.; St. Pierre, J.P.; Garofalo, A.; Rainville, I.R.; Hiller, E.; Treacy, D.J.; Rojas-Rudilla, V.; Wood, S.; Bair, E.; et al. Assigning clinical meaning to somatic and germ-line whole-exome sequencing data in a prospective cancer precision medicine study. Genet. Med. 2017, 19, 787. [Google Scholar] [CrossRef]
- Mandelker, D.; Donoghue, M.; Talukdar, S.; Bandlamudi, C.; Srinivasan, P.; Vivek, M.; Jezdic, S.; Hanson, H.; Snape, K.; Kulkarni, A.; et al. Germline-focussed analysis of tumour-only sequencing: Recommendations from the ESMO Precision Medicine Working Group. Ann. Oncol. 2019, 30, 1221–1231. [Google Scholar] [CrossRef]
- Chang, M.T.; Asthana, S.; Gao, S.P.; Lee, B.H.; Chapman, J.S.; Kandoth, C.; Gao, J.; Socci, N.D.; Solit, D.B.; Olshen, A.B.; et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 2015, 34, 155. [Google Scholar] [CrossRef]
- Parsons, D.W.; Roy, A.; Yang, Y.; Wang, T.; Scollon, S.; Bergstrom, K.; Kerstein, R.A.; Gutierrez, S.; Petersen, A.K.; Bavle, A.; et al. Diagnostic Yield of Clinical Tumor and Germline Whole-Exome Sequencing for Children With Solid TumorsDiagnostic Yield in Genetic Sequencing for Children With Solid TumorsDiagnostic Yield in Genetic Sequencing for Children With Solid Tumors. JAMA Oncol. 2016, 2, 616–624. [Google Scholar] [CrossRef]
- Schrader, K.A.; Cheng, D.T.; Joseph, V.; Prasad, M.; Walsh, M.; Zehir, A.; Ni, A.; Thomas, T.; Benayed, R.; Ashraf, A.; et al. Germline Variants in Targeted Tumor Sequencing Using Matched Normal DNAGermline Variants in Tumor Sequencing With Matched Normal DNAGermline Variants in Tumor Sequencing With Matched Normal DNA. JAMA Oncol. 2016, 2, 104–111. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Schachter, J.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.M.; Lotem, M.; et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): Post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet. Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2014, 372, 320–330. [Google Scholar] [CrossRef]
- Schwaederle, M.; Zhao, M.; Lee, J.J.; Lazar, V.; Leyland-Jones, B.; Schilsky, R.L.; Mendelsohn, J.; Kurzrock, R. Association of Biomarker-Based Treatment Strategies With Response Rates and Progression-Free Survival in Refractory Malignant Neoplasms: A Meta-analysisOutcomes for Biomarker-Based Treatment Strategies in Refractory Malignant NeoplasmsOutcomes for Biomarker-Based Treatment Strategies in Refractory Malignant Neoplasms. JAMA Oncol. 2016, 2, 1452–1459. [Google Scholar] [CrossRef]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef]
- Lopez-Chavez, A.; Thomas, A.; Rajan, A.; Raffeld, M.; Morrow, B.; Kelly, R.; Carter, C.A.; Guha, U.; Killian, K.; Lau, C.C.; et al. Molecular profiling and targeted therapy for advanced thoracic malignancies: A biomarker-derived, multiarm, multihistology phase II basket trial. J. Clin. Oncol. 2015, 33, 1000–1007. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Robert, C.; Hersey, P.; Nathan, P.; Garbe, C.; Milhem, M.; Demidov, L.V.; Hassel, J.C.; Rutkowski, P.; Mohr, P.; et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 2012, 367, 107–114. [Google Scholar] [CrossRef]
- Benafif, S.; Hall, M. An update on PARP inhibitors for the treatment of cancer. OncoTargets Ther. 2015, 8, 519–528. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib Monotherapy in Patients With Advanced Cancer and a Germline BRCA1/2 Mutation. J. Clin. Oncol. 2014, 33, 244–250. [Google Scholar] [CrossRef]
- Low, S.-K.; Nakamura, Y. The road map of cancer precision medicine with the innovation of advanced cancer detection technology and personalized immunotherapy. Jpn. J. Clin. Oncol. 2019, 49, 596–603. [Google Scholar] [CrossRef]
- Mateo, J.; Chakravarty, D.; Dienstmann, R.; Jezdic, S.; Gonzalez-Perez, A.; Lopez-Bigas, N.; Ng, C.K.Y.; Bedard, P.L.; Tortora, G.; Douillard, J.Y.; et al. A framework to rank genomic alterations as targets for cancer precision medicine: The ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann. Oncol. 2018, 29, 1895–1902. [Google Scholar] [CrossRef]
- Thavaneswaran, S.; Rath, E.; Tucker, K.; Joshua, A.M.; Hess, D.; Pinese, M.; Ballinger, M.L.; Thomas, D.M. Therapeutic implications of germline genetic findings in cancer. Nat. Rev. Clin. Oncol. 2019, 16, 386–396. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Brusco, L.; Daniels, M.; Wathoo, C.; Bailey, A.M.; Strong, L.; Shaw, K.; Lu, K.; Qi, Y.; Zhao, H.; et al. Incidental germline variants in 1000 advanced cancers on a prospective somatic genomic profiling protocol. Ann. Oncol. 2016, 27, 795–800. [Google Scholar] [CrossRef]
- Gershenwald, J.E.; Scolyer, R.A. Melanoma Staging: American Joint Committee on Cancer (AJCC) 8th Edition and Beyond. Ann. Surg. Oncol. 2018, 25, 2105–2110. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Zhao, Q.; Yang, J.; Li, L.; Cao, D.; Yu, M.; Shen, K. Germline and somatic mutations in homologous recombination genes among Chinese ovarian cancer patients detected using next-generation sequencing. J. Gynecol. Oncol. 2017, 28, e39. [Google Scholar] [CrossRef]
- Jiang, H.; Lei, R.; Ding, S.W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shaw, K.; Cooper, D.N. The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Curr. Protoc. Bioinform. 2012. [Google Scholar] [CrossRef]
- Forschner, A.; Battke, F.; Hadaschik, D.; Schulze, M.; Weißgraeber, S.; Han, C.-T.; Kopp, M.; Frick, M.; Klumpp, B.; Tietze, N.; et al. Tumor mutation burden and circulating tumor DNA in combined CTLA-4 and PD-1 antibody therapy in metastatic melanoma–results of a prospective biomarker study. J. Immunother. Cancer 2019, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; Yusko, E.; Xu, Y.; Guo, X.; Ennis, R.C.; Fabrizio, D.; Chalmers, Z.R.; Greenbowe, J.; et al. Targeted Next Generation Sequencing Identifies Markers of Response to PD-1 Blockade. Cancer Immunol. Res. 2016, 4, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Furumichi, M.; Morishima, K.; Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019, 47, D590–D595. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci 2019, 28, 1947–1951. [Google Scholar] [CrossRef]
- Chakravarty, D.; Gao, J.; Phillips, S.M.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef]
- STRING. Protein-Protein Interaction Networks Functional Enrichment Analysis. Available online: https://string-db.org (accessed on 20 March 2020).
- Schwartz, L.H.; Litière, S.; de Vries, E.; Ford, R.; Gwyther, S.; Mandrekar, S.; Shankar, L.; Bogaerts, J.; Chen, A.; Dancey, J.; et al. RECIST 1.1-Update and clarification: From the RECIST committee. Eur. J. Cancer 2016, 62, 132–137. [Google Scholar] [CrossRef]
- Mandelker, D.; Zhang, L.; Kemel, Y.; Stadler, Z.K.; Joseph, V.; Zehir, A.; Pradhan, N.; Arnold, A.; Walsh, M.F.; Li, Y.; et al. Mutation Detection in Patients With Advanced Cancer by Universal Sequencing of Cancer-Related Genes in Tumor and Normal DNA vs Guideline-Based Germline TestingMutation Detection in Patients With Advanced CancerMutation Detection in Patients With Advanced Cancer. JAMA 2017, 318, 825–835. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Rulli, E.; Legramandi, L.; Salvati, L.; Mandala, M. The impact of targeted therapies and immunotherapy in melanoma brain metastases: A systematic review and meta-analysis. Cancer 2019, 125, 3776–3789. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Maillard, A.; Penel, N.; Baciarello, G.; Allouache, D.; Daugaard, G.; Van De Wouw, A.; Soler, G.; Vauleon, E.; Chaigneau, L.; et al. Adjuvant immunotherapy with nivolumab (NIVO) alone or in combination with ipilimumab (IPI) versus placebo in stage IV melanoma patients with no evidence of disease (NED): A randomized, double-blind phase 2 trial (IMMUNED). Ann. Oncol. 2019, 30, v851. [Google Scholar] [CrossRef]
- Bednarski, J.J.; Sleckman, B.P. At the intersection of DNA damage and immune responses. Nat. Immunol. 2019, 19, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Mak, T.W.; Hakem, A.; McPherson, J.P.; Shehabeldin, A.; Zablocki, E.; Migon, E.; Duncan, G.S.; Bouchard, D.; Wakeham, A.; Cheung, A.; et al. Brca1 required for T cell lineage development but not TCR loci rearrangement. Nat. Immunol. 2000, 1, 77. [Google Scholar] [CrossRef] [PubMed]
- Galgano, A.; Barinov, A.; Vasseur, F.; de Villartay, J.-P.; Rocha, B. CD8 Memory Cells Develop Unique DNA Repair Mechanisms Favoring Productive Division. PLoS ONE 2015, 10, e0140849. [Google Scholar] [CrossRef]
- Bednarski, J.J.; Sleckman, B.P. Lymphocyte development: Integration of DNA damage response signaling. Adv. Immunol. 2012, 116, 175–204. [Google Scholar] [CrossRef]
- Bredemeyer, A.L.; Helmink, B.A.; Innes, C.L.; Calderon, B.; McGinnis, L.M.; Mahowald, G.K.; Gapud, E.J.; Walker, L.M.; Collins, J.B.; Weaver, B.K.; et al. DNA double-strand breaks activate a multi-functional genetic program in developing lymphocytes. Nature 2008, 456, 819–823. [Google Scholar] [CrossRef]
- Bredemeyer, A.L.; Sharma, G.G.; Huang, C.Y.; Helmink, B.A.; Walker, L.M.; Khor, K.C.; Nuskey, B.; Sullivan, K.E.; Pandita, T.K.; Bassing, C.H.; et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature 2006, 442, 466–470. [Google Scholar] [CrossRef]
- Caddle, L.B.; Hasham, M.G.; Schott, W.H.; Shirley, B.-J.; Mills, K.D. Homologous recombination is necessary for normal lymphocyte development. Mol. Cell. Boil. 2008, 28, 2295–2303. [Google Scholar] [CrossRef]
- Beerman, I.; Seita, J.; Inlay, M.A.; Weissman, I.L.; Rossi, D.J. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 2014, 15, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Kraya, A.A.; Maxwell, K.N.; Wubbenhorst, B.; Wenz, B.M.; Pluta, J.; Rech, A.J.; Dorfman, L.M.; Lunceford, N.; Barrett, A.; Mitra, N.; et al. Genomic Signatures Predict the Immunogenicity of BRCA-Deficient Breast Cancer. Clin. Cancer Res. 2019, 25, 4363. [Google Scholar] [CrossRef] [PubMed]
- Ock, C.-Y.; Hwang, J.-E.; Keam, B.; Kim, S.-B.; Shim, J.-J.; Jang, H.-J.; Park, S.; Sohn, B.H.; Cha, M.; Ajani, J.A.; et al. Genomic landscape associated with potential response to anti-CTLA-4 treatment in cancers. Nat. Commun. 2017, 8, 1050. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.A.; Zhao, F.; Khare, S.; Roszik, J.; Woodman, S.E.; Andrea, K.; Wubbenhorst, B.; Rimm, D.L.; Kirkwood, J.M.; Kluger, H.M.; et al. Copy Number Changes Are Associated with Response to Treatment with Carboplatin, Paclitaxel, and Sorafenib in Melanoma. Clin. Cancer Res. 2016, 22, 374. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.J.; Thomson, A.J. The Mechanism of Action of Antitumor Platinum Compounds. In Progress in Nucleic Acid Research and Molecular Biology; Cohn, W.E., Ed.; Academic Press: Cambridge, MA, USA, 1979; Volume 22, pp. 71–133. [Google Scholar]
- Stewart, R.A.; Pilie, P.G.; Yap, T.A. Development of PARP and Immune-Checkpoint Inhibitor Combinations. Cancer Res. 2018, 78, 6717–6725. [Google Scholar] [CrossRef]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, S.N.; et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J. Mol. Diagn. 2015, 17, 251–264. [Google Scholar] [CrossRef]
- FoundationOne CDx™. Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf17/P170019C.pdf (accessed on 15 August 2019).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient No. | Melanoma Subtype | Gene Name | Germline Variant | ACMG Classifiable [1] | Rationale | TMB (mut/Mb) | Pathway Involved | Potential Therapy | LOE$ |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Cutaneous (nodular) | BRCA2 | c.6446_6449delTTAA; p.Ile2149Lysfs*18 (het.), NM_000059.3 | Class 5 | Pathogenic germline variant | 40.9 | DNA repair, homologous recombination | PARPi | 2A |
| 2 | Cutaneous (nodular) | POLE | c.1270C>T; p.Leu424Phe (het.), NM_006231.3 | Class 4 | Likely pathogenic germline variant | 21.4 | DNA repair, homologous recombination | PARPi | 2B |
| Immunotherapy | 1B | ||||||||
| WRN | c.2893_2899del; p.Lys965Cysfs*7 (het.), NM_000553.4 | Class 4 | Likely pathogenic germline variant | Homologous recombination | PARPi | 2B | |||
| 3 | Acral | FANCI | CNV coding exon 9 (het. Deletion), NM_001113378.1 | Class 4 | Likely pathogenic germline variant | 1.6 | DNA repair, homologous recombination | PARPi | 4 |
| 4 | Cutaneous (superficial) | WRN | c.348delC; p.Met117Cysfs*8 (het.), NM_000553.4 | Class 4 | Likely pathogenic germline variant | 7.9 | Homologous recombination | PARPi | 2B |
| 5 | Cutaneous (polypoid) | CDKN2A | c.301G>T; p.Gly101Trp (het.), NM_000077.4 | Class 5 | Pathogenic germline variant | 5.8 | Cell cycle | CDK 4/6 inhibitors | 2B |
| 6 | Uveal | BAP1 | c.37+1G>T; p.? (het.), NM_004656.3 | Class 5 | Pathogenic germline variant | 1.0 | Homologous recombination, epigenetics | PARPi | 2B |
| EZH2i | 3 | ||||||||
| HDACi | 4 | ||||||||
| 7 | Cutaneous (superficial) | PALB2 | c.757_758delCT; p.Leu253Ilefs*3 (het.), NM_024675.3 | Class 5 | Pathogenic germline variant | 3.1 | Homologous recombination | PARPi | 2B |
| 8 | Cutaneous (nodular) | BRCA2 | c.1888dupA; p.Thr630Asnfs*6 (het.), NM_000059.3 | Class 5 | Pathogenic germline variant | 12.0 | DNA repair, homologous recombination | PARPi | 2A |
| 9 | Cutaneous (nodular) | RAD54B | c.889C>T; p.Arg297* (het.), NM_012415.3 | No | Likely pathogenic germline variant/ Potentially therapy relevant | 13.1 | DNA repair, homologous recombination | PARPi | 4 |
| Characteristic | N (%) |
|---|---|
| Sex | |
| Male | 36 (61) |
| Female | 23 (39) |
| Age | |
| Primary disease diagnosis (median years; IQR) | 58; (49–70) |
| Initiation of combined immunotherapy (median years; IQR) | 61; (51–74) |
| Melanoma subtype | |
| Cutaneous melanoma | 35 (59.3) |
| Acral melanoma | 6 (10.2) |
| Mucosal melanoma | 4 (6.7) |
| Melanoma of unknown primary | 5 (8.5) |
| Uveal melanoma | 9 (15.3) |
| Tumor mutation burden* (n = 56) | |
| TMB (mut/Mb median; IQR) | 4.7; (1.7–13.97) |
| Intermediate-low (≤23.1 mut/Mb) | 46 (82.1) |
| High (>23.1 mut/Mb) | 10 (17.9) |
| Serum biomarkers | |
| LDH | |
| Normal | 34 (57.6) |
| Elevated | 25 (42.4) |
| S100B* (n = 55) | |
| Normal | 22 (40) |
| Elevated | 33 (60) |
| BRAF mutation* (n = 57) | |
| Yes | 29 (50.9) |
| No | 28 (49.1) |
| Metastasis at start of nivolumab plus ipilimumab | |
| Presence of cerebral metastasis | 24 (40.7) |
| Presence of liver metastasis | 17 (28.8) |
| Presence of lung metastasis | 32 (54.2) |
| Nivolumab plus ipilimumab | |
| First line | 29 (49.2) |
| Second line or more | 30 (50.8) |
| Response to nivolumab and ipilimumab at first staging* (n = 55) | |
| Progressive disease | 34 (61.8) |
| Disease control | 21 (38.2) |
| Category | Disease Control N = 21 | Progressive Disease N = 34 | p | Odds Ratio (95% CI) |
|---|---|---|---|---|
| N (%) | ||||
| Pathogenic and likely pathogenic germline variants | ||||
| Present | 0 | 9 (26.5) | 0.010 | NA* |
| Not present | 21 (100) | 25 (73.5) | ||
| Tumor mutation burden (n = 52) | ||||
| Low /Intermediate | 11 (57.9) | 32 (97) | <0.0001 | 23.27 (2.61–207.7) |
| High | 8 (42.1) | 1 (3) | ||
| Serum biomarkers | ||||
| S100B (n = 51) | ||||
| Normal | 9 (47.4) | 11 (34.4) | 0.36 | |
| Elevated | 10 (52.6) | 21 (65.6) | 1.72 (0.54–5.47) | |
| LDH | ||||
| Normal | 15 (71.4) | 17 (50) | 0.118 | |
| Elevated | 6 (28.6) | 17 (50) | 2.50 (0.78–7.98) | |
| Category | Progression-Free Survival | Melanoma Specific Survival | |||||||
|---|---|---|---|---|---|---|---|---|---|
| HR (95% CI)1 | p1 | HR (95% CI)2 | p2 | HR (95% CI)1 | p1 | HR (95% CI)2 | p2 | ||
| Pathogenic and likely pathogenic germline variants | |||||||||
| Present vs. not present | 2.16 (1.01–4.64) | 0.048 | 1.93 (0.89–4.15) | 0.095 | 3.21 (1.31–7.87) | 0.011 | 2.93 (1.07–8.0) | 0.036 | |
| Tumor mutation burden | |||||||||
| Low/intermediate vs. high | 2.88 (1.12–7.38) | 0.028 | 2.75 (1.07–7.09) | 0.036 | 2.31 (0.54–9.85) | 0.258 | NA | ||
| S100B | |||||||||
| Elevated vs. normal | 0.992 (0.55–1.81) | 0.979 | NA | 7.45 (1.74–31.91) | 0.007 | 4.65 (1.04–20.76) | 0.044 | ||
| LDH | |||||||||
| Elevated vs. normal | 1.26 (0.71–2.26) | 0.433 | NA | 4.33 (1.77–10.56) | 0.001 | 5.16 (1.90–13.96) | 0.001 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amaral, T.; Schulze, M.; Sinnberg, T.; Nieser, M.; Martus, P.; Battke, F.; Garbe, C.; Biskup, S.; Forschner, A. Are Pathogenic Germline Variants in Metastatic Melanoma Associated with Resistance to Combined Immunotherapy? Cancers 2020, 12, 1101. https://doi.org/10.3390/cancers12051101
Amaral T, Schulze M, Sinnberg T, Nieser M, Martus P, Battke F, Garbe C, Biskup S, Forschner A. Are Pathogenic Germline Variants in Metastatic Melanoma Associated with Resistance to Combined Immunotherapy? Cancers. 2020; 12(5):1101. https://doi.org/10.3390/cancers12051101
Chicago/Turabian StyleAmaral, Teresa, Martin Schulze, Tobias Sinnberg, Maike Nieser, Peter Martus, Florian Battke, Claus Garbe, Saskia Biskup, and Andrea Forschner. 2020. "Are Pathogenic Germline Variants in Metastatic Melanoma Associated with Resistance to Combined Immunotherapy?" Cancers 12, no. 5: 1101. https://doi.org/10.3390/cancers12051101
APA StyleAmaral, T., Schulze, M., Sinnberg, T., Nieser, M., Martus, P., Battke, F., Garbe, C., Biskup, S., & Forschner, A. (2020). Are Pathogenic Germline Variants in Metastatic Melanoma Associated with Resistance to Combined Immunotherapy? Cancers, 12(5), 1101. https://doi.org/10.3390/cancers12051101