YM155-Adapted Cancer Cell Lines Reveal Drug-Induced Heterogeneity and Enable the Identification of Biomarker Candidates for the Acquired Resistance Setting

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Results
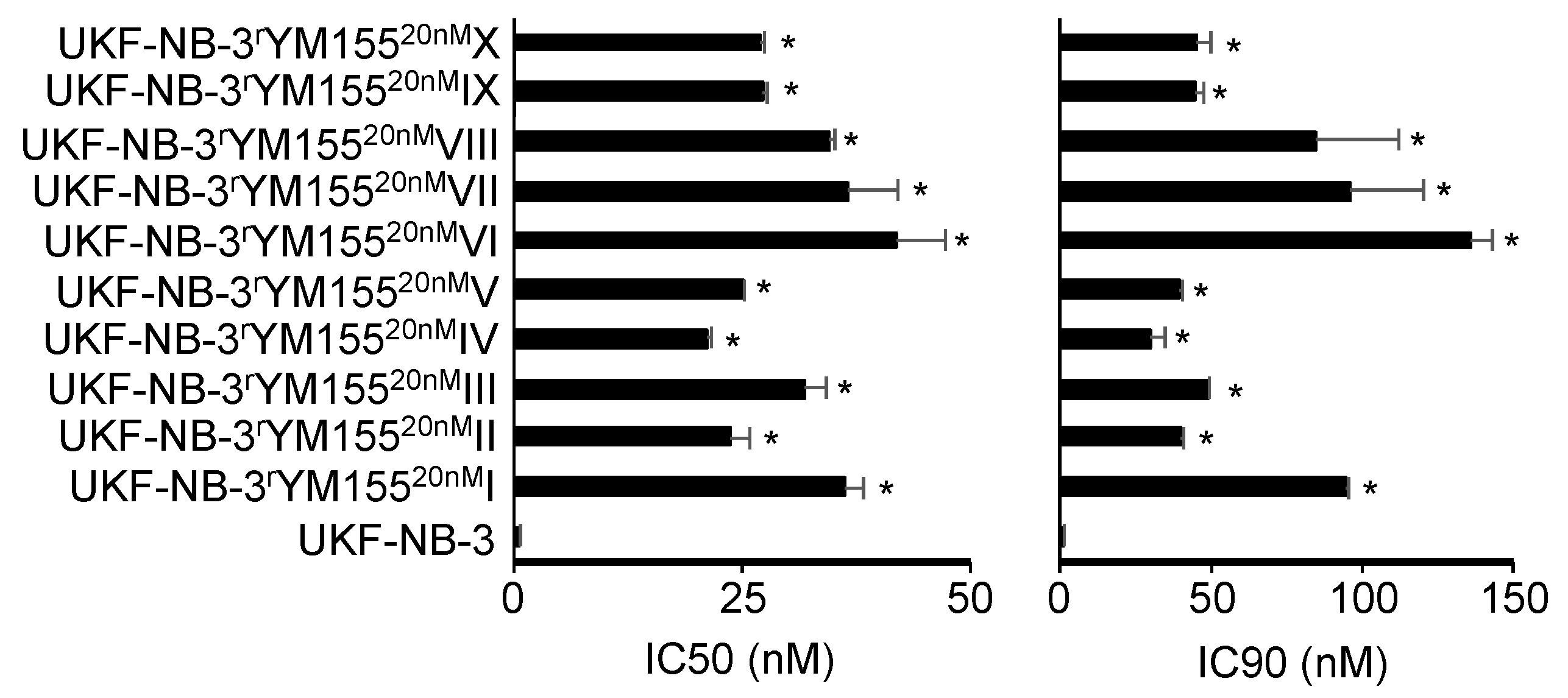
2.1. YM155-Adapted UKF-NB-3 Sublines Display Pronounced YM155 Resistance
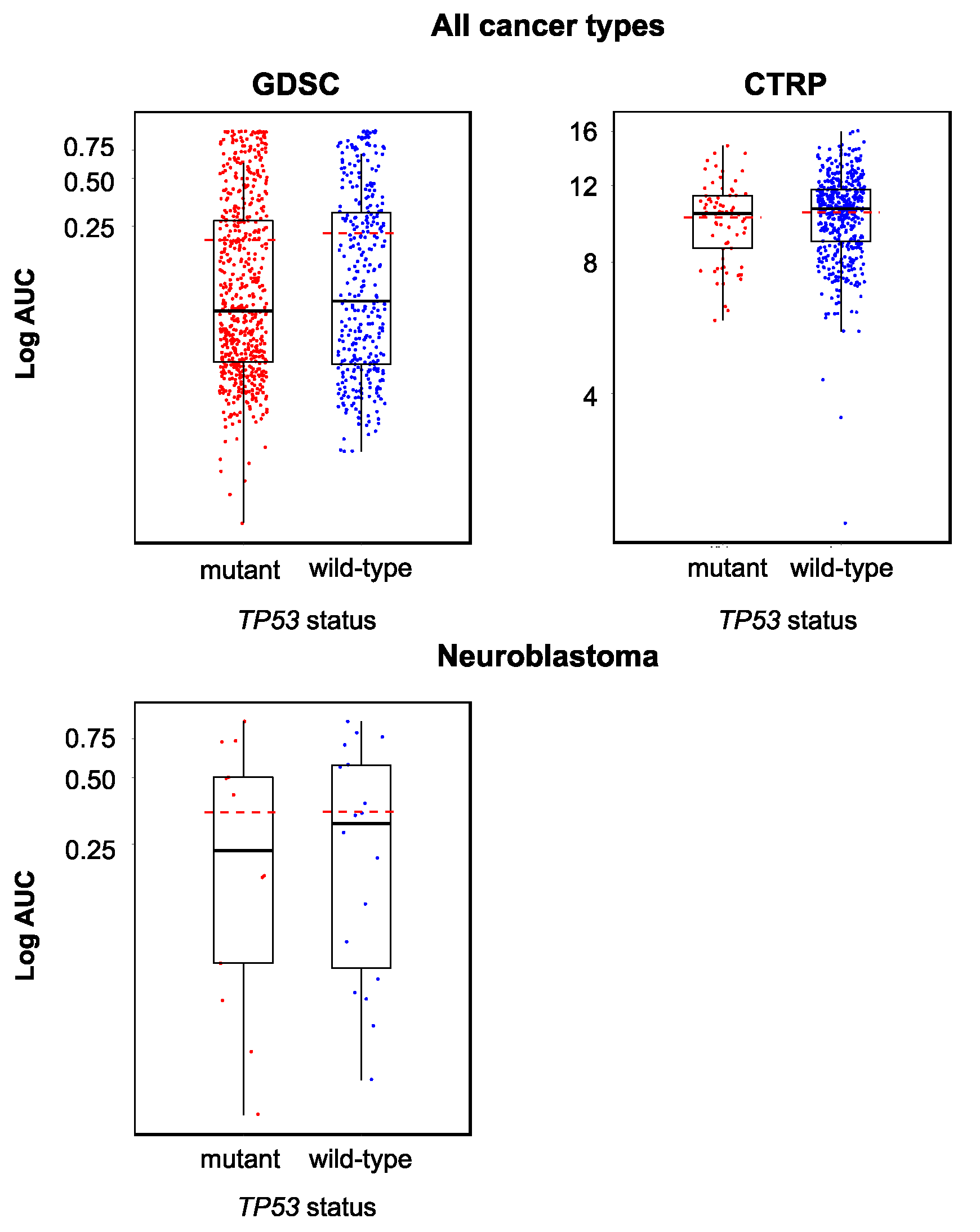
2.2. The Cellular TP53 Status is Not a Reliable Indicator of YM155 Sensitivity
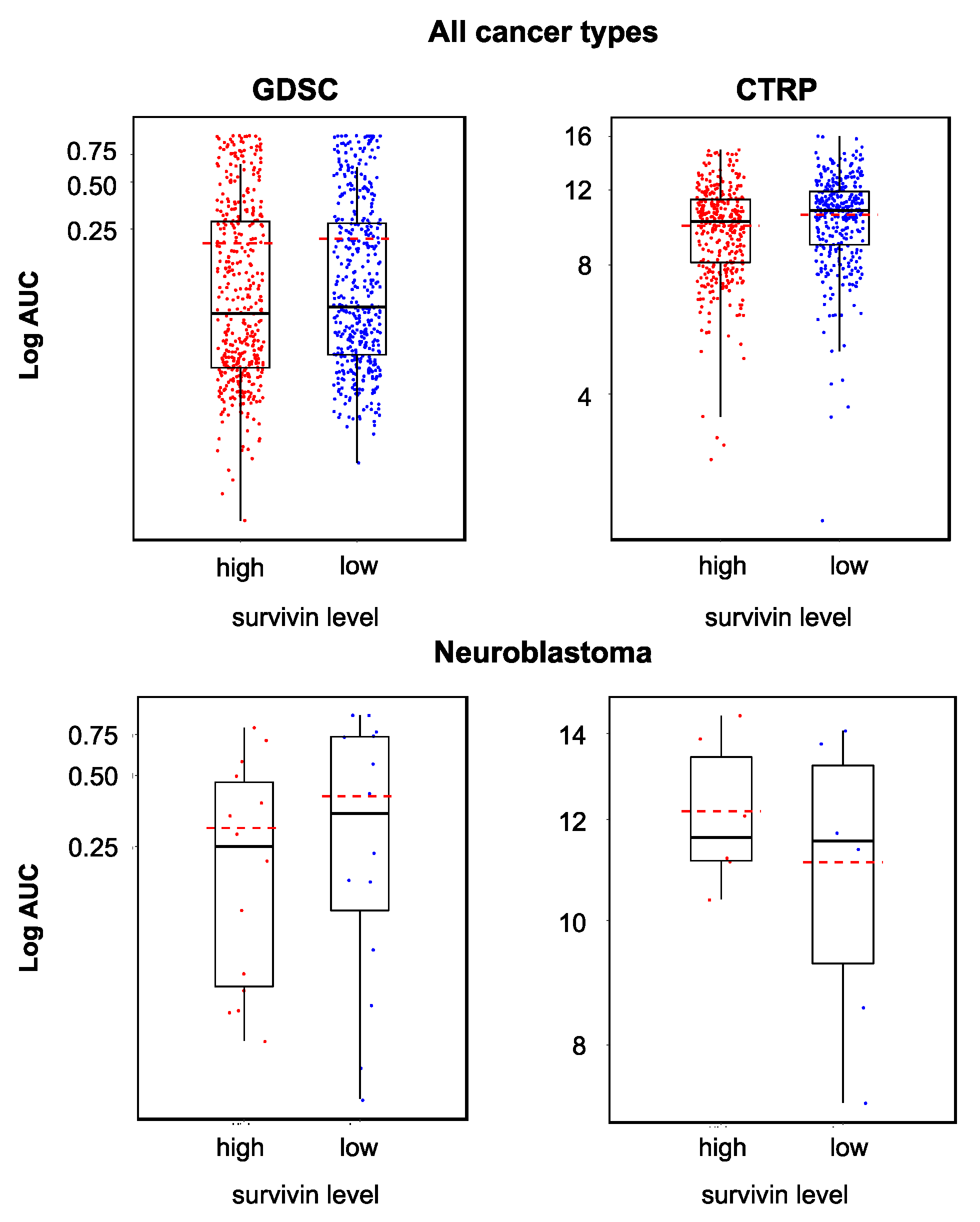
2.3. Cellular Survivin Levels Do Not Reliably Indicate YM155 Response
2.4. Acquired YM155 Resistance is Associated with Decreased Sensitivity to Survivin Depletion
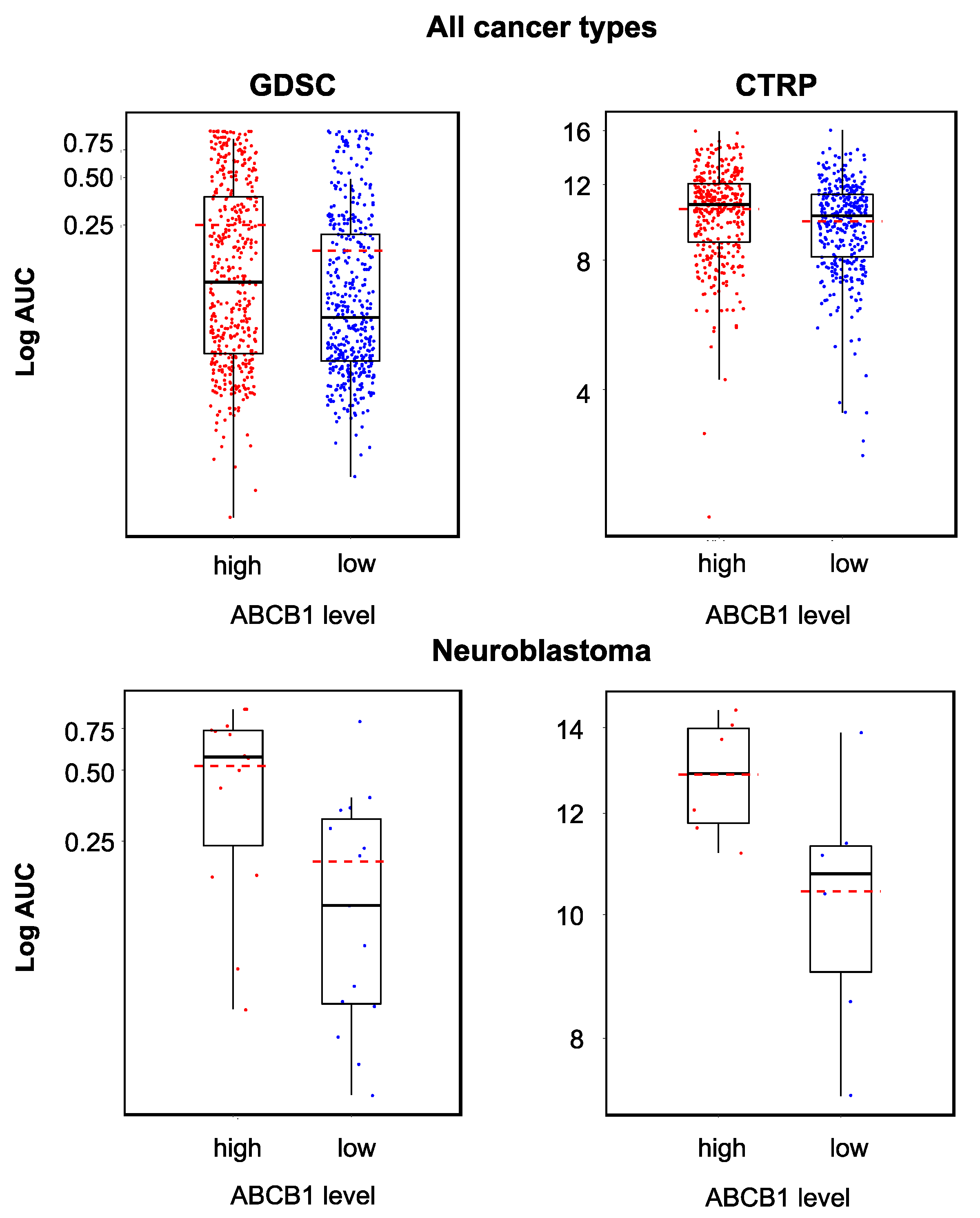
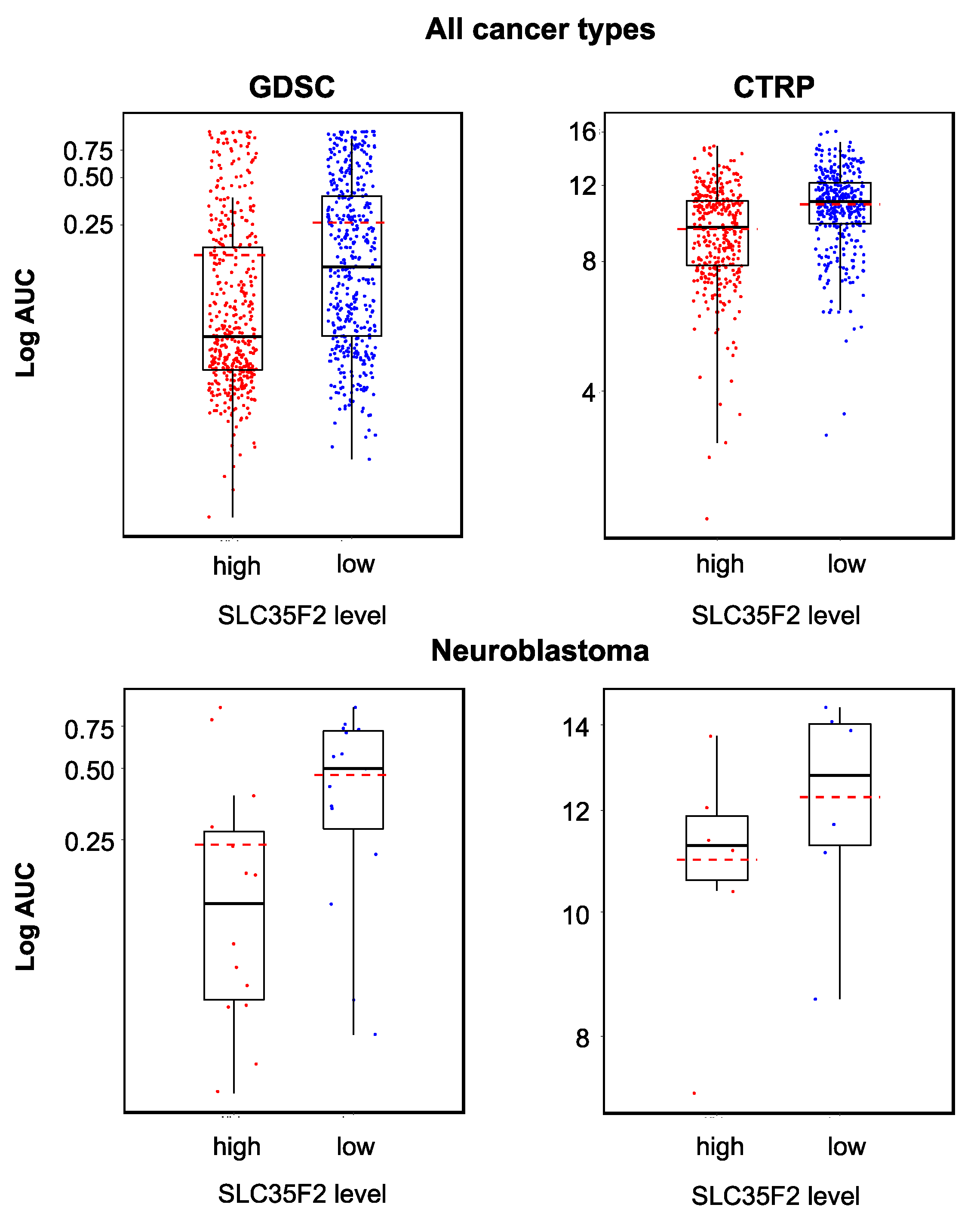
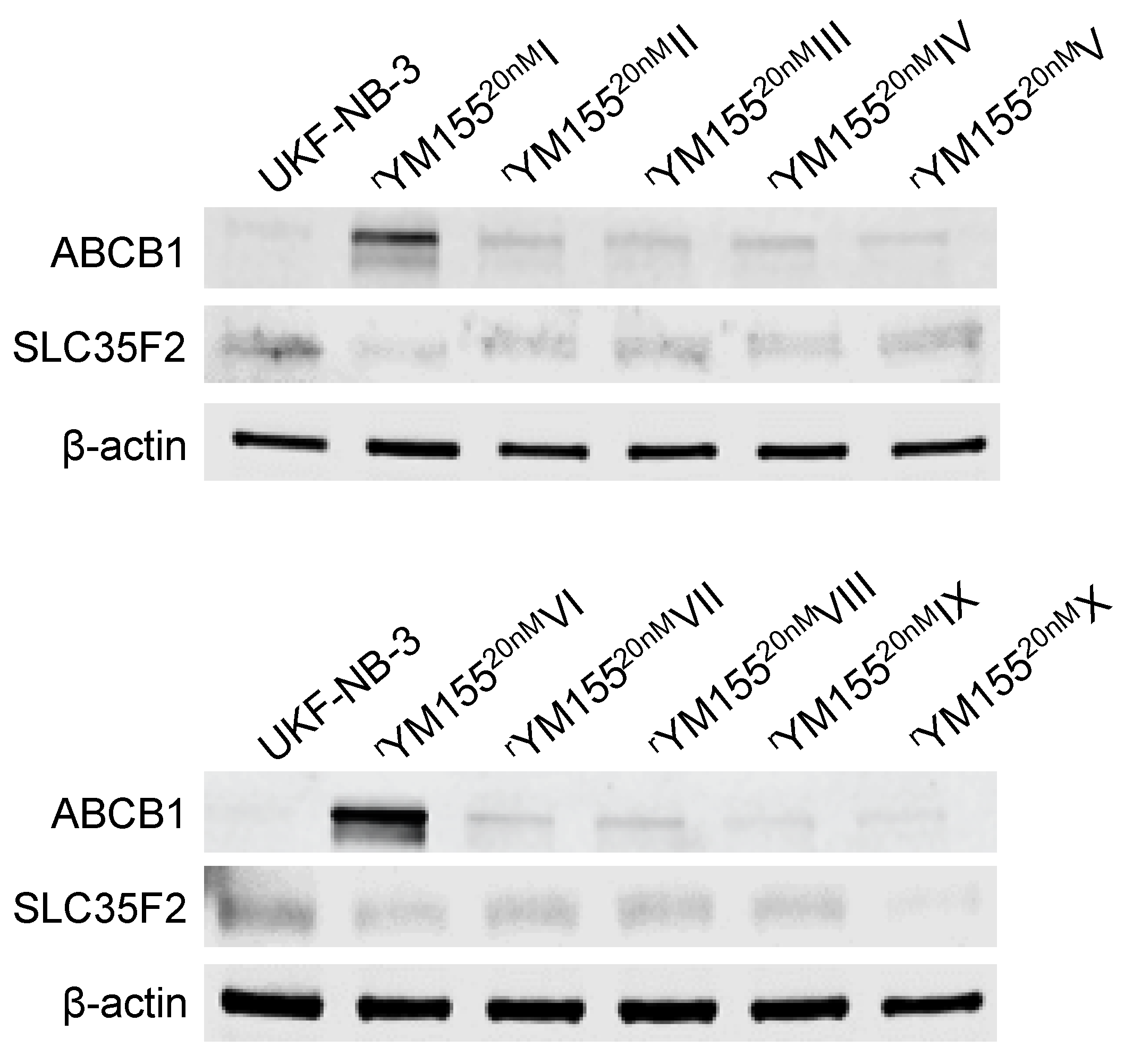
2.5. Relevance of Cellular ABCB1 and SLC35F2 Levels in the Context of YM155 Resistance
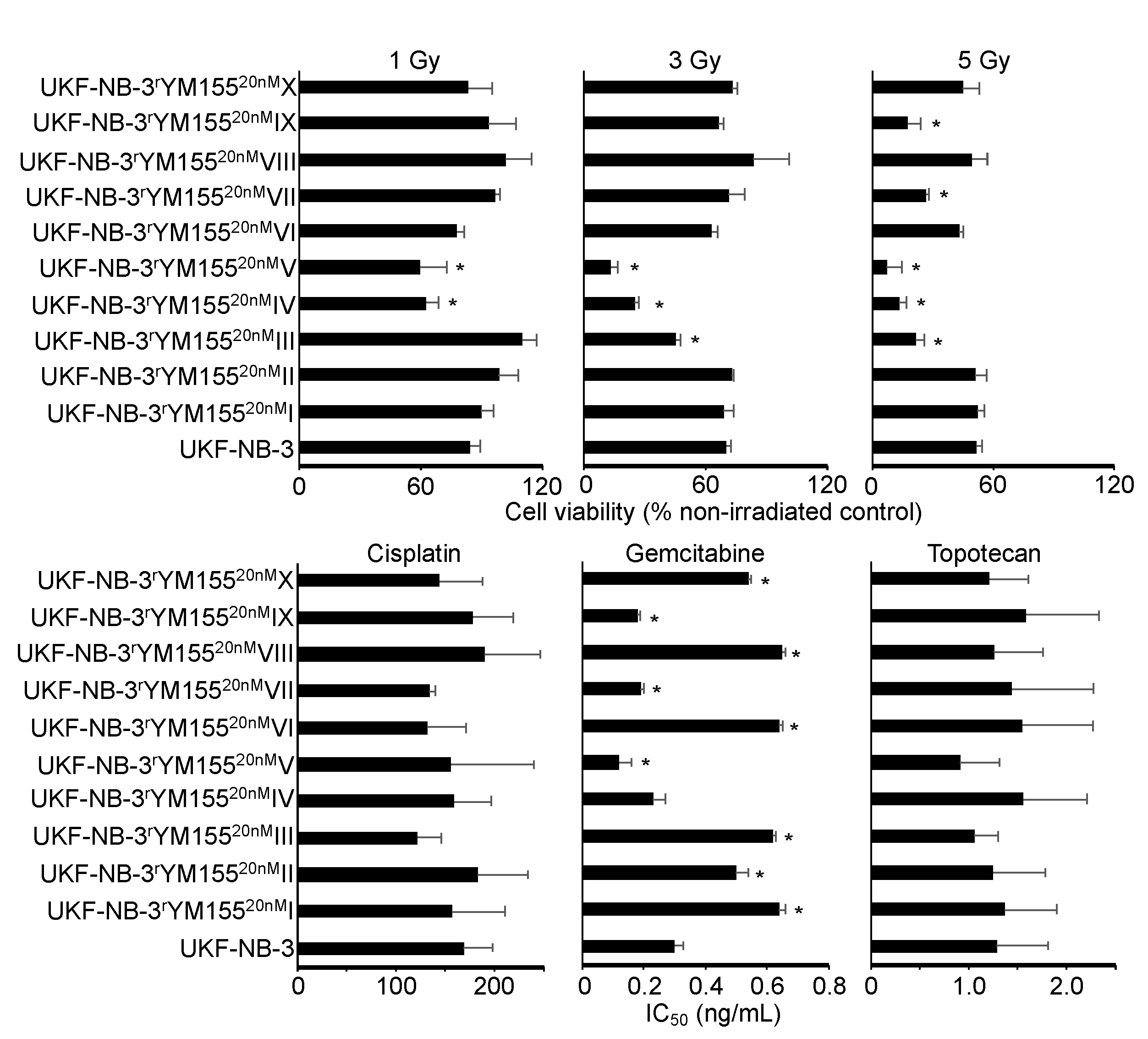
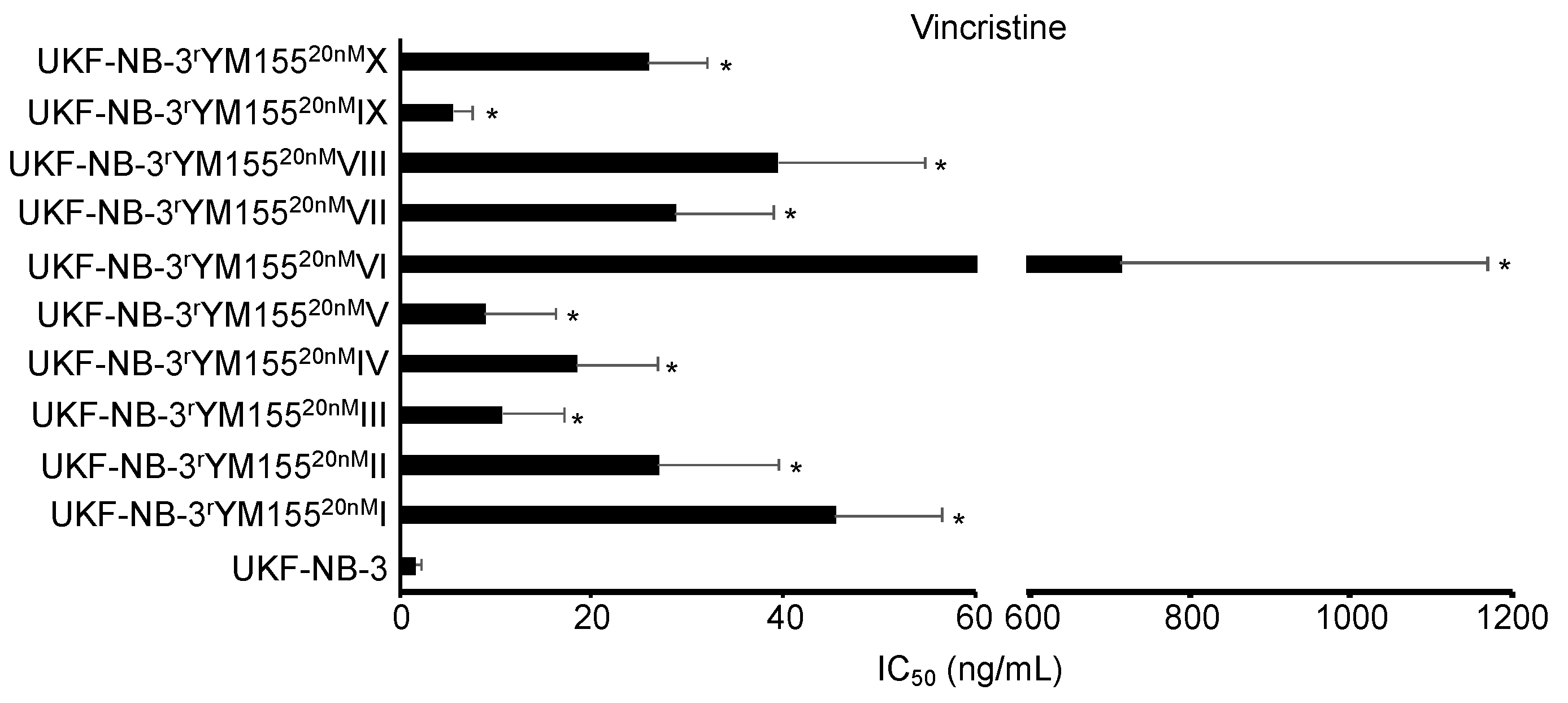
2.6. YM155-Adapted UKF-NB-3 Cells Remain Sensitive to DNA Damage Caused by Irradiation and Cytotoxic Drugs
2.7. Heterogeneity among YM155-Adapted UKF-NB-3 Sublines
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Viability Assay
4.3. TP53 Next-Generation Sequencing
4.4. Western Blot
4.5. RNA Interference Experiments
4.6. Irradiation Procedure
4.7. Analysis of Data Derived from Large Pharmacogenomic Studies
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nakahara, T.; Kita, A.; Yamanaka, K.; Mori, M.; Amino, N.; Takeuchi, M.; Tominaga, F.; Hatakeyama, S.; Kinoyama, I.; Matsuhisa, A.; et al. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007, 67, 8014–8021. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Shao, H.; Yu, C.; Hou, J. Mcl-1 downregulation by YM155 contributes to its synergistic anti-tumor activities with ABT-263. Biochem. Pharmacol. 2011, 82, 1066–1072. [Google Scholar] [CrossRef] [PubMed]
- Glaros, T.G.; Stockwin, L.H.; Mullendore, M.E.; Smith, B.; Morrison, B.L.; Newton, D.L. The “survivin suppressants” NSC 80467 and YM155 induce a DNA damage response. Cancer Chemother. Pharmacol. 2012, 70, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.S.; Yang, S.J.; Kim, S.M.; Jung, K.A.; Moon, J.H.; Shin, J.S.; Yoon, D.H.; Hong, Y.S.; Ryu, M.H.; Lee, J.L.; et al. YM155 induces EGFR suppression in pancreatic cancer cells. PLoS ONE 2012, 7, e38625. [Google Scholar] [CrossRef] [PubMed]
- Rauch, A.; Hennig, D.; Schäfer, C.; Wirth, M.; Marx, C.; Heinzel, T.; Schneider, G.; Krämer, O.H. Survivin and YM155: How faithful is the liaison? Biochim. Biophys. Acta 2014, 1845, 202–220. [Google Scholar] [CrossRef]
- Cheng, S.M.; Chang, Y.C.; Liu, C.Y.; Lee, J.Y.; Chan, H.H.; Kuo, C.W.; Lin, K.Y.; Tsai, S.L.; Chen, S.H.; Li, C.F.; et al. YM155 down-regulates survivin and XIAP, modulates autophagy and induces autophagy-dependent DNA damage in breast cancer cells. Br. J. Pharmacol. 2015, 172, 214–234. [Google Scholar] [CrossRef]
- De Necochea-Campion, R.; Diaz Osterman, C.J.; Hsu, H.W.; Fan, J.; Mirshahidi, S.; Wall, N.R.; Chen, C.S. AML sensitivity to YM155 is modulated through AKT and Mcl-1. Cancer Lett. 2015, 366, 44–51. [Google Scholar] [CrossRef]
- Jung, S.A.; Park, Y.M.; Hong, S.W.; Moon, J.H.; Shin, J.S.; Lee, H.R.; Ha, S.H.; Lee, D.H.; Kim, J.H.; Kim, S.M.; et al. Cellular inhibitor of apoptosis protein 1 (cIAP1) stability contributes to YM155 resistance in human gastric cancer cells. J. Biol. Chem. 2015, 290, 9974–9985. [Google Scholar] [CrossRef]
- Pennati, M.; Sbarra, S.; De Cesare, M.; Lopergolo, A.; Locatelli, S.L.; Campi, E.; Daidone, M.G.; Carlo-Stella, C.; Gianni, A.M.; Zaffaroni, N. YM155 sensitizes triple-negative breast cancer to membrane-bound TRAIL through p38 MAPK- and CHOP-mediated DR5 upregulation. Int. J. Cancer 2015, 136, 299–309. [Google Scholar] [CrossRef]
- Zhao, X.; Puszyk, W.M.; Lu, Z.; Ostrov, D.A.; George, T.J.; Robertson, K.D.; Liu, C. Small molecule inhibitor YM155-mediated activation of death receptor 5 is crucial for chemotherapy-induced apoptosis in pancreatic carcinoma. Mol. Cancer Ther. 2015, 14, 80–89. [Google Scholar] [CrossRef]
- Ho, S.H.; Ali, A.; Chin, T.M.; Go, M.L. Dioxonaphthoimidazoliums AB1 and YM155 disrupt phosphorylation of p50 in the NF-κB pathway. Oncotarget 2016, 7, 11625–11636. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Hayakawa, F.; Morishita, T.; Sugimoto, K.; Minamikawa, Y.; Iwase, M.; Yamamoto, H.; Hirano, D.; Imoto, N.; Shimada, K.; et al. YM155 induces apoptosis through proteasome-dependent degradation of MCL-1 in primary effusion lymphoma. Pharmacol. Res. 2017, 120, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Danielpour, D.; Gao, Z.; Zmina, P.M.; Shankar, E.; Shultes, B.C.; Jobava, R.; Welford, S.M.; Hatzoglou, M. Early Cellular Responses of Prostate Carcinoma Cells to Sepantronium Bromide (YM155) Involve Suppression of mTORC1 by AMPK. Sci. Rep. 2019, 9, 11541. [Google Scholar] [CrossRef] [PubMed]
- Lamers, F.; Schild, L.; Koster, J.; Versteeg, R.; Caron, H.N.; Molenaar, J.J. Targeted BIRC5 silencing using YM155 causes cell death in neuroblastoma cells with low ABCB1 expression. Eur. J. Cancer. 2012, 48, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Zhang, L.; Xu, R.; Ju, X.L. Silencing of survivin using YM155 induces apoptosis and chemosensitization in neuroblastomas cells. Eur. Rev. Med. Pharmacol. Sci. 2013, 17, 2909–2915. [Google Scholar]
- Voges, Y.; Michaelis, M.; Rothweiler, F.; Schaller, T.; Schneider, C.; Politt, K.; Mernberger, M.; Nist, A.; Stiewe, T.; Wass, M.N.; et al. Effects of YM155 on survivin levels and viability in neuroblastoma cells with acquired drug resistance. Cell Death Dis. 2016, 7, e2410. [Google Scholar] [CrossRef]
- Radic-Sarikas, B.; Halasz, M.; Huber, K.V.M.; Winter, G.E.; Tsafou, K.P.; Papamarkou, T.; Brunak, S.; Kolch, W.; Superti-Furga, G. Lapatinib potentiates cytotoxicity of YM155 in neuroblastoma via inhibition of the ABCB1 efflux transporter. Sci. Rep. 2017, 7, 3091. [Google Scholar] [CrossRef]
- Pinto, N.R.; Applebaum, M.A.; Volchenboum, S.L.; Matthay, K.K.; London, W.B.; Ambros, P.F.; Nakagawara, A.; Berthold, F.; Schleiermacher, G.; Park, J.R.; et al. Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J. Clin. Oncol. 2015, 33, 3008–3017. [Google Scholar] [CrossRef]
- Bagatell, R.; Cohn, S.L. Genetic discoveries and treatment advances in neuroblastoma. Curr. Opin. Pediatr. 2016, 28, 19–25. [Google Scholar] [CrossRef]
- Speleman, F.; Park, J.R.; Henderson, T.O. Neuroblastoma: A Tough Nut to Crack. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e548–e557. [Google Scholar] [CrossRef]
- PDQ Pediatric Treatment Editorial Board. Neuroblastoma Treatment (PDQ®): Health Professional Version. In PDQ Cancer Information Summaries [Internet]; National Cancer Institute (US): Bethesda, MD, USA, 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK65747/ (accessed on 13 February 2020).
- Moreno, L.; Caron, H.; Geoerger, B.; Eggert, A.; Schleiermacher, G.; Brock, P.; Valteau-Couanet, D.; Chesler, L.; Schulte, J.H.; De Preter, K.; et al. Accelerating Drug Development for Neuroblastoma—New Drug Development Strategy: An Innovative Therapies for Children with Cancer, European Network for Cancer Research in Children and Adolescents and International Society of Paediatric Oncology Europe Neuroblastoma Project. Expert. Opin. Drug Discov. 2017, 12, 801–811. [Google Scholar] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Perakis, S.; Speicher, M.R. Emerging concepts in liquid biopsies. BMC Med. 2017, 15, 75. [Google Scholar] [CrossRef] [PubMed]
- Miklos, W.; Pelivan, K.; Kowol, C.R.; Pirker, C.; Dornetshuber-Fleiss, R.; Spitzwieser, M.; Englinger, B.; van Schoonhoven, S.; Cichna-Markl, M.; Koellensperger, G.; et al. Triapine-mediated ABCB1 induction via PKC induces widespread therapy unresponsiveness but is not underlying acquired triapine resistance. Cancer Lett. 2015, 361, 112–120. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Carter, L.; Rothwell, D.G.; Mesquita, B.; Smowton, C.; Leong, H.S.; Fernandez-Gutierrez, F.; Li, Y.; Burt, D.J.; Antonello, J.; Morrow, C.J.; et al. Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat. Med. 2017, 23, 114–119. [Google Scholar] [CrossRef]
- Lipinski, K.A.; Barber, L.J.; Davies, M.N.; Ashenden, M.; Sottoriva, A.; Gerlinger, M. Cancer Evolution and the Limits of Predictability in Precision Cancer Medicine. Trends Cancer 2016, 2, 49–63. [Google Scholar] [CrossRef]
- Basu, A.; Bodycombe, N.E.; Cheah, J.H.; Price, E.V.; Liu, K.; Schaefer, G.I.; Ebright, R.Y.; Stewart, M.L.; Ito, D.; Wang, S.; et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 2013, 154, 1151–1161. [Google Scholar] [CrossRef]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef]
- Nakahara, T.; Kita, A.; Yamanaka, K.; Mori, M.; Amino, N.; Takeuchi, M.; Tominaga, F.; Kinoyama, I.; Matsuhisa, A.; Kudou, M.; et al. Broad spectrum and potent antitumor activities of YM155, a novel small-molecule survivin suppressant, in a wide variety of human cancer cell lines and xenograft models. Cancer Sci. 2011, 102, 614–621. [Google Scholar] [CrossRef]
- Ghadimi, M.P.; Young, E.D.; Belousov, R.; Zhang, Y.; Lopez, G.; Lusby, K.; Kivlin, C.; Demicco, E.G.; Creighton, C.J.; Lazar, A.J.; et al. Survivin is a viable target for the treatment of malignant peripheral nerve sheath tumors. Clin. Cancer Res. 2012, 18, 2545–2557. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Chen, J.; Shi, M.; Deivasigamani, A.; Ooi, L.L.; Hui, K.M. The over-expression of survivin enhances the chemotherapeutic efficacy of YM155 in human hepatocellular carcinoma. Oncotarget 2015, 6, 5990–6000. [Google Scholar] [CrossRef]
- Sim, M.Y.; Huynh, H.; Go, M.L.; Yuen, J.S.P. Action of YM155 on clear cell renal cell carcinoma does not depend on survivin expression levels. PLoS ONE 2017, 12, e0178168. [Google Scholar] [CrossRef] [PubMed]
- Winter, G.E.; Radic, B.; Mayor-Ruiz, C.; Blomen, V.A.; Trefzer, C.; Kandasamy, R.K.; Huber, K.V.M.; Gridling, M.; Chen, D.; Klampfl, T.; et al. The solute carrier SLC35F2 enables YM155-mediated DNA damage toxicity. Nat. Chem. Biol. 2014, 10, 768–773. [Google Scholar] [CrossRef]
- Chang, B.H.; Johnson, K.; LaTocha, D.; Rowley, J.S.; Bryant, J.; Burke, R.; Smith, R.L.; Loriaux, M.; Müschen, M.; Mullighan, C.; et al. YM155 potently kills acute lymphoblastic leukemia cells through activation of the DNA damage pathway. J. Hematol. Oncol. 2015, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Kotchetkov, R.; Driever, P.H.; Cinatl, J.; Michaelis, M.; Karaskova, J.; Blaheta, R.; Squire, J.A.; Von Deimling, A.; Moog, J.; Cinatl, J., Jr. Increased malignant behavior in neuroblastoma cells with acquired multi-drug resistance does not depend on P-gp expression. Int. J. Oncol. 2005, 27, 1029–1037. [Google Scholar] [CrossRef]
- Michaelis, M.; Rothweiler, F.; Barth, S.; Cinatl, J.; van Rikxoort, M.; Löschmann, N.; Voges, Y.; Breitling, R.; von Deimling, A.; Rödel, F.; et al. Adaptation of cancer cells from different entities to the MDM2 inhibitor nutlin-3 results in the emergence of p53-mutated multi-drug-resistant cancer cells. Cell Death Dis. 2011, 2, e243. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Mann, H.B.; Whitney, D.R. On a Test of Whether one of Two Random Variables is Stochastically Larger than the Other. Ann. Math. Stat. 1947, 18, 50–60. [Google Scholar] [CrossRef]
- Pearson, K. Notes on regression and inheritance in the case of two parents. Proc. R. Soc. Lond. 1895, 58, 240–242. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GDSC | CTRP | ||||
|---|---|---|---|---|---|
| Gene | Correlation Coefficient | FDR 1 | Gene | Correlation Coefficient | FDR |
| ABCB1 | 0.3792069 | 2.82 × 10−6 | ABCB1 | 0.3624858 | 2.70 × 10−6 |
| FABP1 | 0.2972293 | 5.64 × 10−6 | CST3 | 0.3603422 | 5.39 × 10−6 |
| CDX2 | 0.2927057 | 8.46 × 10−6 | AKR1C3 | 0.3503176 | 8.09 × 10−6 |
| DDC | 0.2922995 | 1.13 × 10−5 | EPS8 | 0.3453892 | 1.08 × 10−5 |
| CDH17 | 0.2652645 | 1.41 × 10−5 | ABHD2 | 0.3309384 | 1.35 × 10−5 |
| ANKS4B | 0.2637047 | 1.97 × 10−5 | S100A6 | 0.3229691 | 1.62 × 10−5 |
| MYO1A | 0.2626518 | 2.26 × 10−5 | ATP1B1 | 0.3111308 | 1.89 × 10−5 |
| PHGR1 | 0.2609317 | 2.54 × 10−5 | CD63 | 0.310712 | 2.16 × 10−5 |
| A1CF | 0.252912 | 2.82 × 10−5 | AKR1C1 | 0.3073306 | 2.43 × 10−5 |
| GUCY2C | 0.2513048 | 3.10 × 10−5 | ACVR1 | 0.30124 | 9.44 × 10−5 |
| GDSC | CTRP | ||||
|---|---|---|---|---|---|
| Gene | Correlation Coefficient | FDR 1 | Gene | Correlation Coefficient | FDR |
| SLC35F2 | −0.2643809 | 1.69 × 10−5 | SLC35F2 | −0.3868041 | 9.17 × 10−5 |
| ALKBH8 | −0.2042557 | 0.000121 | CD19 | −0.349156 | 8.90 × 10−5 |
| CWF19L2 | −0.1926243 | 0.000180 | CD79B | −0.346035 | 8.63 × 10−5 |
| RCSD1 | −0.1855956 | 0.000226 | SPIB | −0.341887 | 8.36 × 10−5 |
| P2RY8 | −0.1834937 | 0.000257 | SNX22 | −0.338893 | 8.09 × 10−5 |
| RGS19 | −0.1831031 | 0.000259 | TCL1A | −0.338393 | 7.82 × 10−5 |
| FLI1 | −0.1825892 | 0.000268 | LOC100130458 | −0.337940 | 7.55 × 10−5 |
| VAV1 | −0.1819569 | 0.000273 | BLK | −0.330471 | 7.28 × 10−5 |
| ATM | −0.1816322 | 0.000276 | CD79A | −0.324465 | 7.01 × 10−5 |
| ARHGAP19 | −0.1813872 | 0.000282 | VPREB3 | −0.322878 | 6.74 × 10−5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michaelis, M.; Wass, M.N.; Reddin, I.; Voges, Y.; Rothweiler, F.; Hehlgans, S.; Cinatl, J.; Mernberger, M.; Nist, A.; Stiewe, T.; et al. YM155-Adapted Cancer Cell Lines Reveal Drug-Induced Heterogeneity and Enable the Identification of Biomarker Candidates for the Acquired Resistance Setting. Cancers 2020, 12, 1080. https://doi.org/10.3390/cancers12051080
Michaelis M, Wass MN, Reddin I, Voges Y, Rothweiler F, Hehlgans S, Cinatl J, Mernberger M, Nist A, Stiewe T, et al. YM155-Adapted Cancer Cell Lines Reveal Drug-Induced Heterogeneity and Enable the Identification of Biomarker Candidates for the Acquired Resistance Setting. Cancers. 2020; 12(5):1080. https://doi.org/10.3390/cancers12051080
Chicago/Turabian StyleMichaelis, Martin, Mark N. Wass, Ian Reddin, Yvonne Voges, Florian Rothweiler, Stephanie Hehlgans, Jaroslav Cinatl, Marco Mernberger, Andrea Nist, Thorsten Stiewe, and et al. 2020. "YM155-Adapted Cancer Cell Lines Reveal Drug-Induced Heterogeneity and Enable the Identification of Biomarker Candidates for the Acquired Resistance Setting" Cancers 12, no. 5: 1080. https://doi.org/10.3390/cancers12051080
APA StyleMichaelis, M., Wass, M. N., Reddin, I., Voges, Y., Rothweiler, F., Hehlgans, S., Cinatl, J., Mernberger, M., Nist, A., Stiewe, T., Rödel, F., & Cinatl, J., Jr. (2020). YM155-Adapted Cancer Cell Lines Reveal Drug-Induced Heterogeneity and Enable the Identification of Biomarker Candidates for the Acquired Resistance Setting. Cancers, 12(5), 1080. https://doi.org/10.3390/cancers12051080