Dexamethasone Attenuates X-Ray-Induced Activation of the Autotaxin-Lysophosphatidate-Inflammatory Cycle in Breast Tissue and Subsequent Breast Fibrosis




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
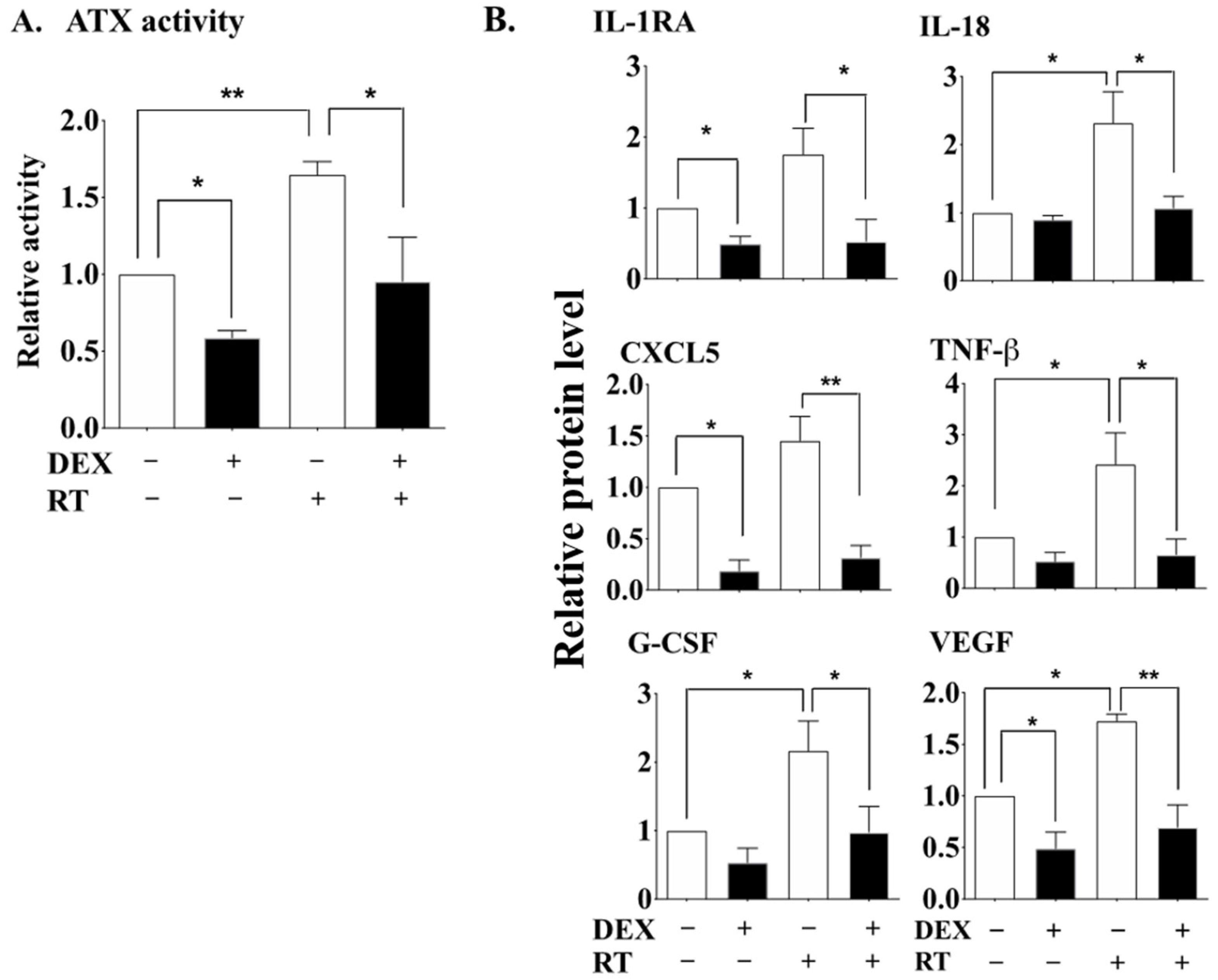
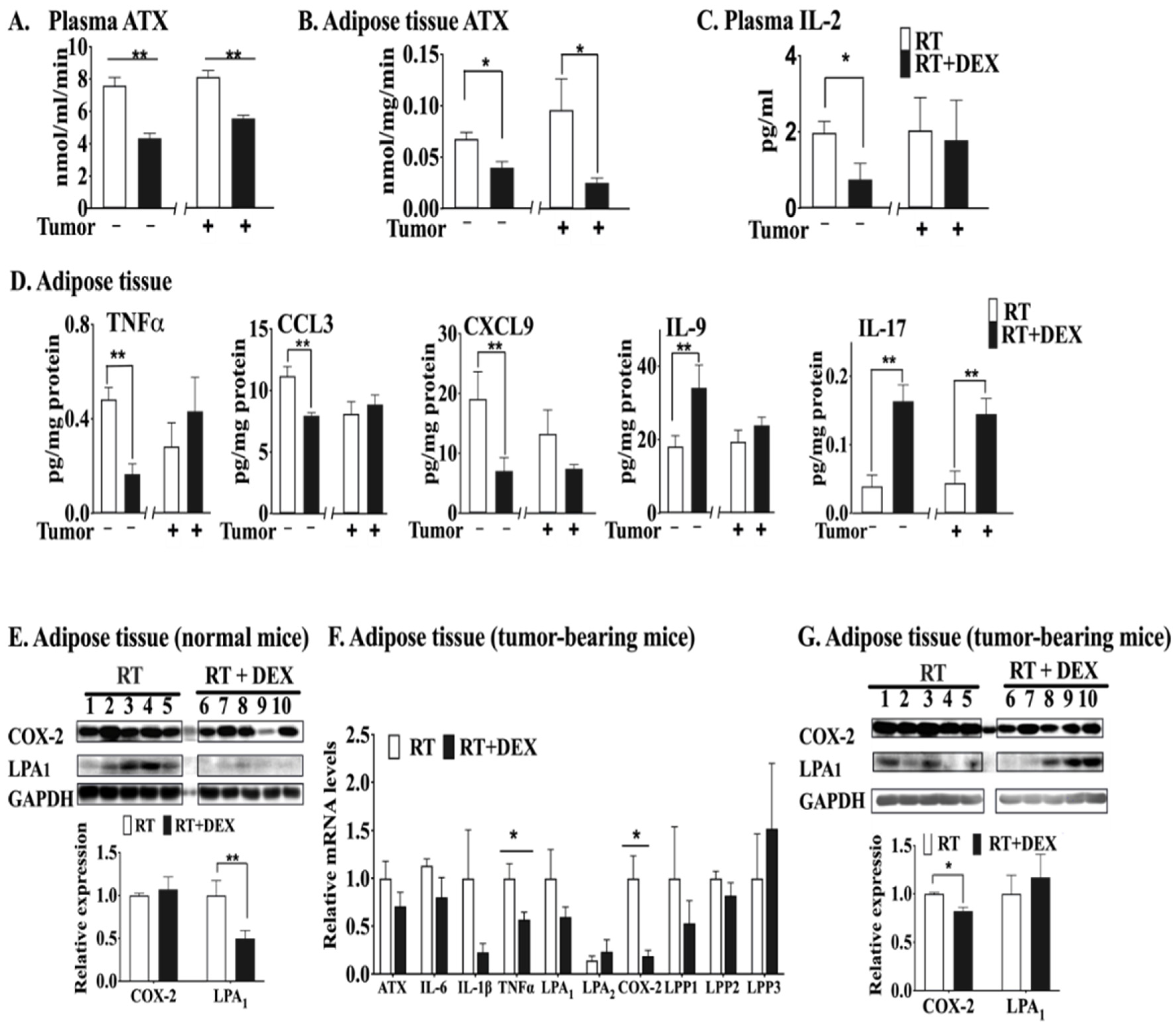
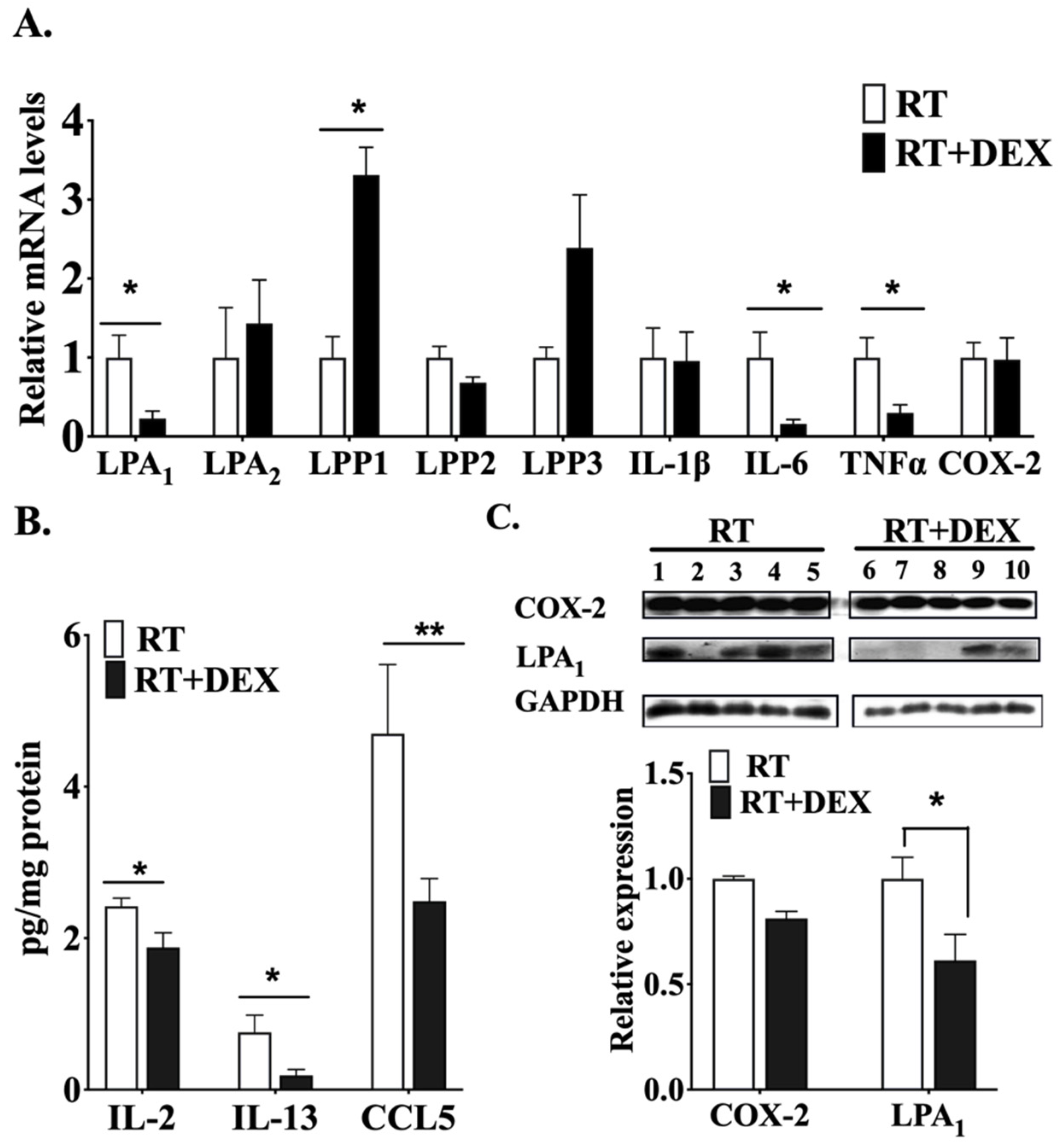
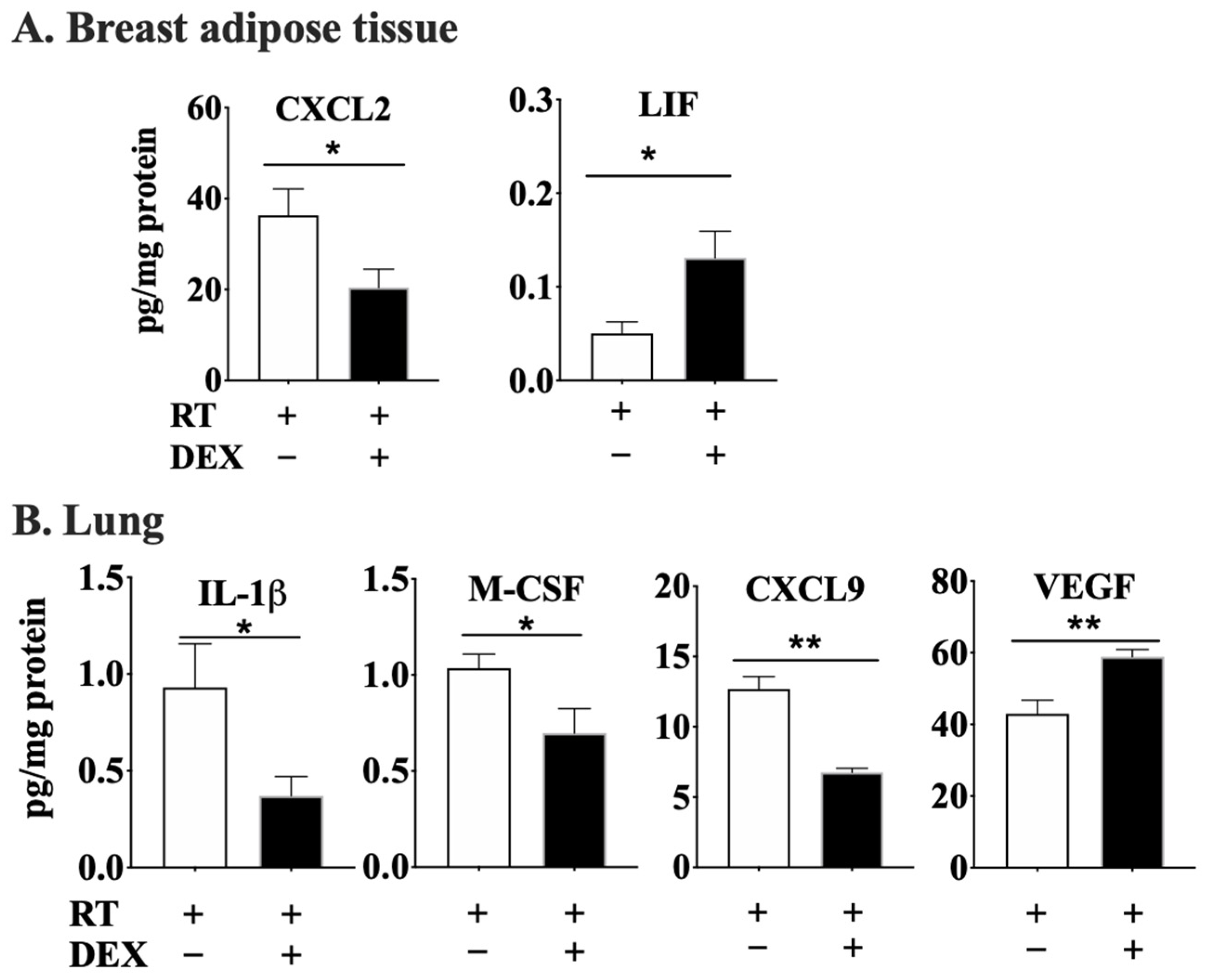
2.1. DEX Attenuates the Activation of the ATX-LPA-Inflammatory Axis in Breast Adipose Tissue and Tumor during RT
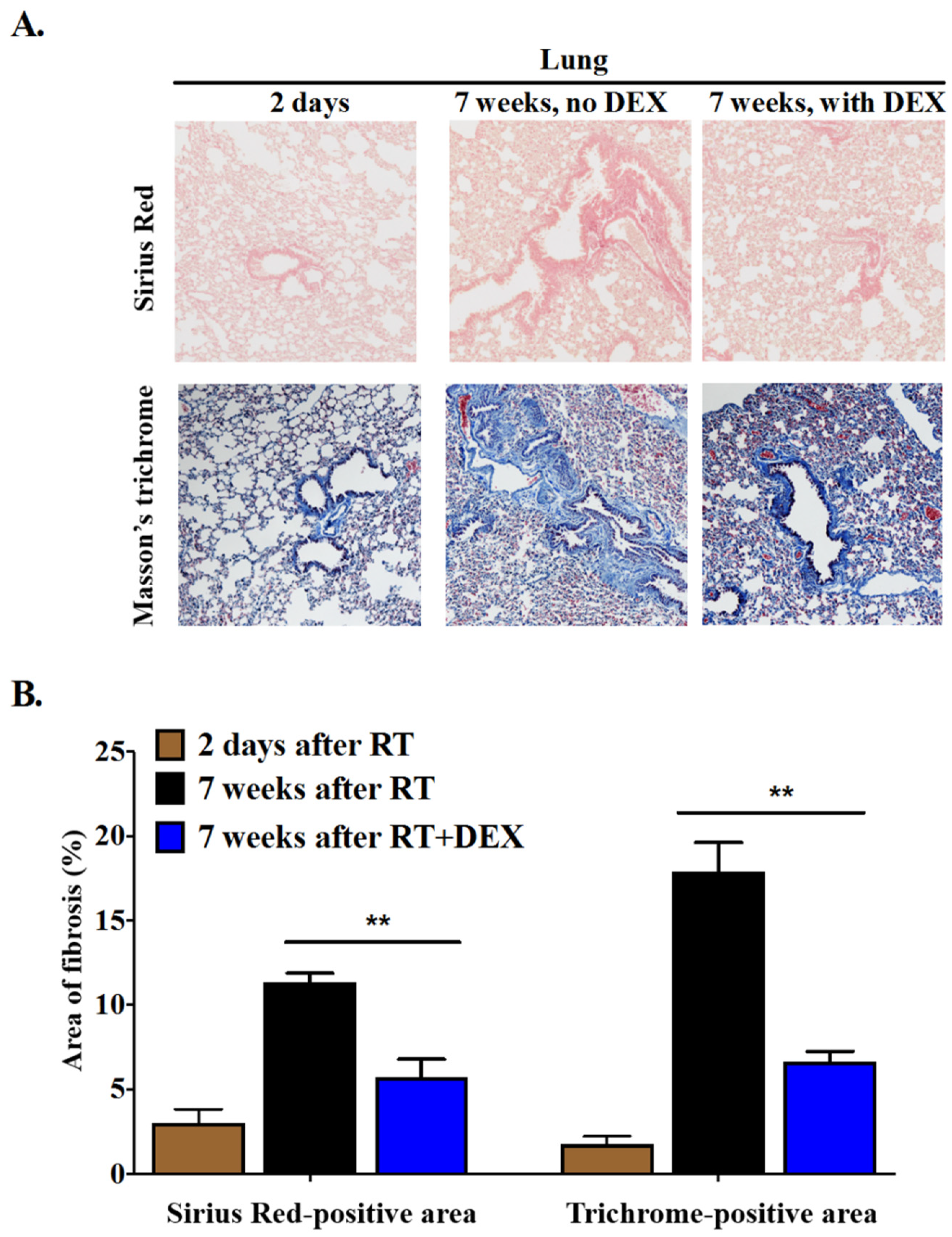
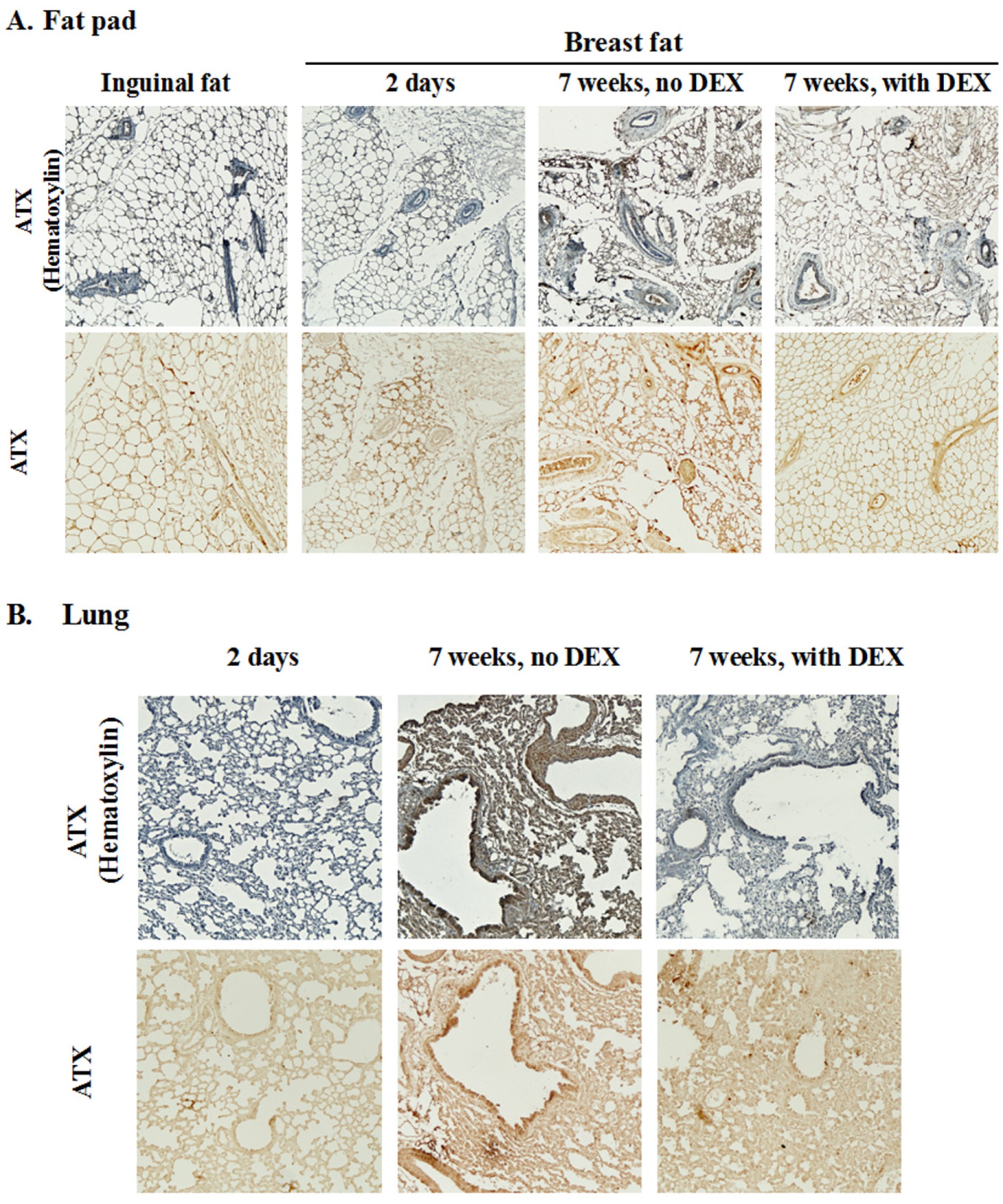
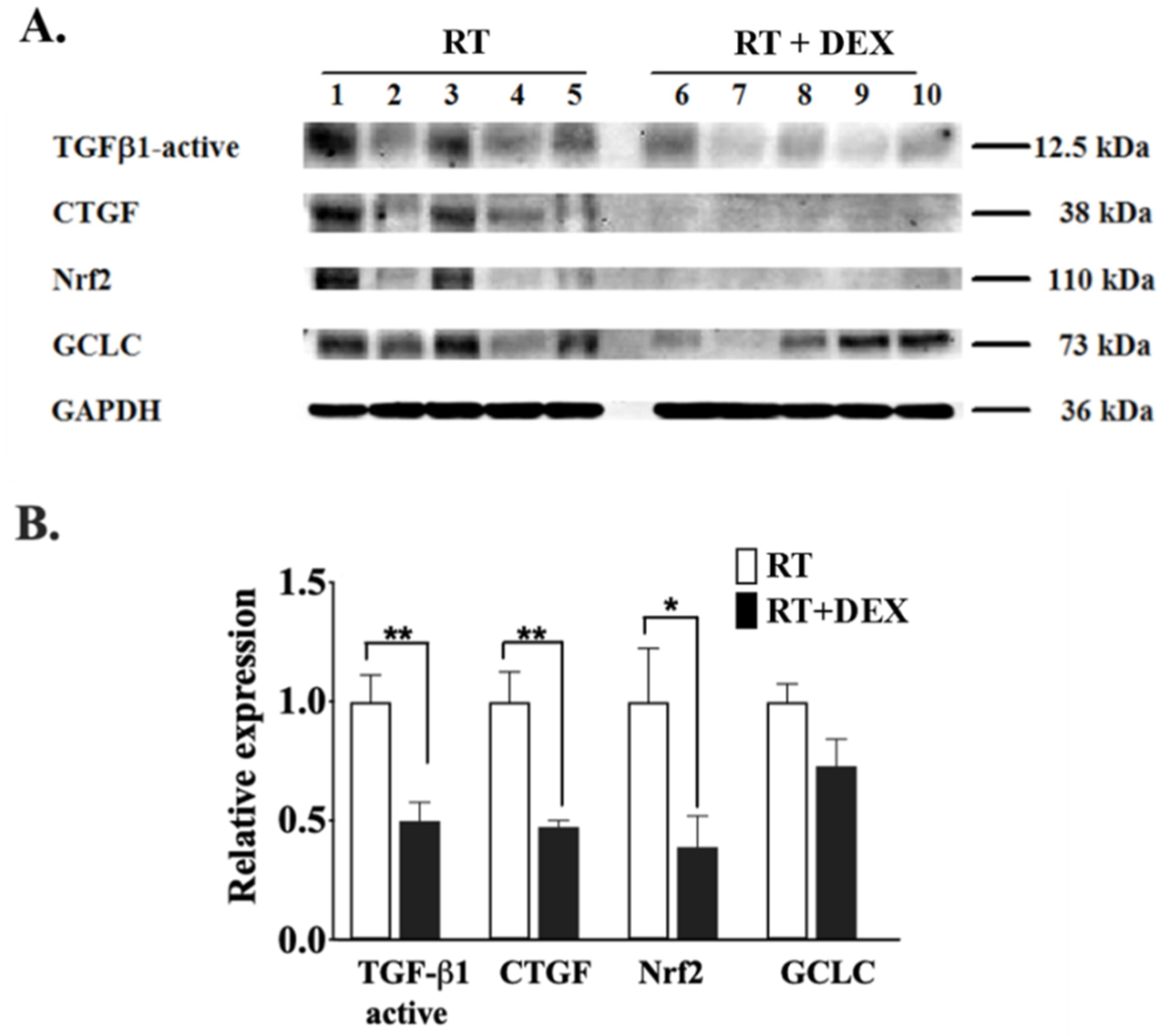
2.2. Dexamethasone Attenuates RT-Induced Fibrosis at 7 Weeks in the Mammary Fat Pad and in the Underlying Lung
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Culture of Human Adipose Tissue and Radiation Exposure
4.3. Measurements of the Effects of DEX on the RT-Induced Inflammatory Responses of Tumor-Bearing and Normal (i.e., Non-Tumor-Bearing) Mice
4.4. Measurements of the Effects of DEX on the RT-Induced Fibrosis in Normal Mice
4.5. Multiplex Analysis of Cytokines and Hormones
4.6. Measurement of ATX Activity
4.7. Quantitative Real-Time PCR (qRT-PCR)
4.8. Immunohistochemistry and Western Blots
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATX | autotaxin |
| BLM | bleomycin |
| COX-2 | cyclooxygenase-2 |
| DEX | dexamethasone |
| CCL | C-C motif chemokine ligand |
| CXCL | C-X-C motif chemokine ligand |
| G-CSF | granulocyte-colony stimulating factor |
| GCLC | glutamate-cysteine ligase catalytic subunit |
| IL | interleukin |
| LPA | lysophosphatidic acid, which at physiological pH is in a salt form, lysophosphatidate |
| LPAR1,2 | LPA receptors types 1− and 2 |
| LPP | lipid phosphate phosphatase |
| Nrf2 | nuclear erythroid 2-like 2 |
| RT | radiation therapy |
| TGF-β | transforming growth factor β |
| TNFα | tumor necrosis factor-α |
| VEGF | vascular endothelial growth factor |
References
- Smith, B.D.; Bellon, J.R.; Blitzblau, R.; Freedman, G.; Haffty, B.; Hahn, C.; Halberg, F.; Hoffman, K.; Horst, K.; Moran, J.; et al. Radiation therapy for the whole breast: Executive summary of an American Society for Radiation Oncology (ASTRO) evidence-based guideline. Pract. Radiat. Oncol. 2018, 8, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Delanian, S.; Lefaix, J.L. The radiation-induced fibroatrophic process: Therapeutic perspective via the antioxidant pathway. Radiother. Oncol. 2004, 73, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Straub, J.M.; New, J.; Hamilton, C.D.; Lominska, C.; Shnayder, Y.; Thomas, S.M. Radiation-induced fibrosis: Mechanisms and implications for therapy. J. Cancer Res. Clin. Oncol. 2015, 141, 1985–1994. [Google Scholar] [CrossRef]
- Olivotto, I.A.; Whelan, T.J.; Parpia, S.; Kim, D.H.; Berrang, T.; Truong, P.T.; Kong, I.; Cochrane, B.; Nichol, A.; Roy, I.; et al. Interim cosmetic and toxicity results from RAPID: A randomized trial of accelerated partial breast irradiation using three-dimensional conformal external beam radiation therapy. J. Clin. Oncol. 2013, 31, 4038–4045. [Google Scholar] [CrossRef] [PubMed]
- Lyngholm, C.D.; Christiansen, P.M.; Damsgaard, T.E.; Overgaard, J. Long-term follow-up of late morbidity, cosmetic outcome and body image after breast conserving therapy. A study from the Danish Breast Cancer Cooperative Group (DBCG). Acta Oncol. 2013, 52, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.R.; Williams, S.; Kanapathy, M.; Naderi, N.; Vavourakis, V.; Mosahebi, A. Radiation-induced fibrosis in breast cancer: A protocol for an observational cross-sectional pilot study for personalised risk estimation and objective assessment. Int. J. Surg. Protoc. 2019, 14, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Erven, K.; Weltens, C.; Nackaerts, K.; Fieuws, S.; Decramer, M.; Lievens, Y. Changes in pulmonary function up to 10 years after locoregional breast irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 701–707. [Google Scholar] [CrossRef]
- Weiss, E. Clinical Manifestations, Prevention, and Treatment of Radiation-Induced Fibrosis. Uptodate.com. 2017. Available online: https://www.uptodate.com/contents/clinical-manifestations-prevention-and-treatment-of-radiation-induced-fibrosis (accessed on 16 April 2020).
- Cao, P.; Aoki, Y.; Badri, L.; Walker, N.M.; Manning, C.M.; Lagstein, A.; Fearon, E.R.; Lama, V.N. Autocrine lysophosphatidic acid signaling activates beta-catenin and promotes lung allograft fibrosis. J. Clin. Investig. 2017, 127, 1517–1530. [Google Scholar] [CrossRef]
- Erstad, D.J.; Tager, A.M.; Hoshida, Y.; Fuchs, B.C. The autotaxin-lysophosphatidic acid pathway emerges as a therapeutic target to prevent liver cancer. Mol. Cell. Oncol. 2017, 4, e1311827. [Google Scholar] [CrossRef]
- Farquhar, M.J.; Humphreys, I.S.; Rudge, S.A.; Wilson, G.K.; Bhattacharya, B.; Ciaccia, M.; Hu, K.; Zhang, Q.; Mailly, L.; Reynolds, G.M.; et al. Autotaxin-lysophosphatidic acid receptor signalling regulates hepatitis C virus replication. J. Hepatol. 2017, 66, 919–929. [Google Scholar] [CrossRef]
- Gan, L.; Xue, J.X.; Li, X.; Liu, D.S.; Ge, Y.; Ni, P.Y.; Deng, L.; Lu, Y.; Jiang, W. Blockade of lysophosphatidic acid receptors LPAR1/3 ameliorates lung fibrosis induced by irradiation. Biochem. Biophys. Res. Commun. 2011, 409, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Kaffe, E.; Katsifa, A.; Xylourgidis, N.; Ninou, I.; Zannikou, M.; Harokopos, V.; Foka, P.; Dimitriadis, A.; Evangelou, K.; Moulas, A.N.; et al. Hepatocyte autotaxin expression promotes liver fibrosis and cancer. Hepatology 2017, 65, 1369–1383. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, N.; Mouratis, M.A.; Tzouvelekis, A.; Kaffe, E.; Valavanis, C.; Vilaras, G.; Karameris, A.; Prestwich, G.D.; Bouros, D.; Aidinis, V. Pulmonary autotaxin expression contributes to the pathogenesis of pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Okudaira, S.; Yukiura, H.; Aoki, J. Biological roles of lysophosphatidic acid signaling through its production by autotaxin. Biochimie 2010, 92, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Pradere, J.P.; Klein, J.; Gres, S.; Guigne, C.; Neau, E.; Valet, P.; Calise, D.; Chun, J.; Bascands, J.L.; Saulnier-Blache, J.S.; et al. LPA1 receptor activation promotes renal interstitial fibrosis. J. Am. Soc. Nephrol. 2007, 18, 3110–3118. [Google Scholar] [CrossRef] [PubMed]
- Rancoule, C.; Pradere, J.P.; Gonzalez, J.; Klein, J.; Valet, P.; Bascands, J.L.; Schanstra, J.P.; Saulnier-Blache, J.S. Lysophosphatidic acid-1-receptor targeting agents for fibrosis. Expert Opin. Investig. Drugs 2011, 20, 657–667. [Google Scholar] [CrossRef]
- Sevastou, I.; Kaffe, E.; Mouratis, M.A.; Aidinis, V. Lysoglycerophospholipids in chronic inflammatory disorders: The PLA(2)/LPC and ATX/LPA axes. Biochim. Biophys. Acta 2013, 1831, 42–60. [Google Scholar] [CrossRef]
- Swaney, J.S.; Chapman, C.; Correa, L.D.; Stebbins, K.J.; Bundey, R.A.; Prodanovich, P.C.; Fagan, P.; Baccei, C.S.; Santini, A.M.; Hutchinson, J.H.; et al. A novel, orally active LPA(1) receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br. J. Pharmacol. 2010, 160, 1699–1713. [Google Scholar] [CrossRef]
- Zhao, Y.; Natarajan, V. Lysophosphatidic acid (LPA) and its receptors: Role in airway inflammation and remodeling. Biochim. Biophys. Acta 2013, 1831, 86–92. [Google Scholar] [CrossRef]
- Meng, G.; Tang, X.; Yang, Z.; Benesch, M.G.K.; Marshall, A.; Murray, D.; Hemmings, D.G.; Wuest, F.; McMullen, T.P.W.; Brindley, D.N. Implications for breast cancer treatment from increased autotaxin production in adipose tissue after radiotherapy. FASEB J. 2017, 31, 4064–4077. [Google Scholar] [CrossRef]
- Tager, A.M.; LaCamera, P.; Shea, B.S.; Campanella, G.S.; Selman, M.; Zhao, Z.; Polosukhin, V.; Wain, J.; Karimi-Shah, B.A.; Kim, N.D.; et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, A.; Ninou, I.; Kokotou, M.G.; Kaffe, E.; Afantitis, A.; Aidinis, V.; Kokotos, G. Hydroxamic acids constitute a novel class of autotaxin inhibitors that exhibit in vivo efficacy in a pulmonary fibrosis model. J. Med. Chem. 2018, 61, 3697–3711. [Google Scholar] [CrossRef] [PubMed]
- Povirk, L.F. DNA damage and mutagenesis by radiomimetic DNA-cleaving agents: Bleomycin, neocarzinostatin and other enediynes. Mutat. Res. 1996, 355, 71–89. [Google Scholar] [CrossRef]
- GLPG1690. Available online: http://www.glpg.com/docs/view/598b6182b414d-en (accessed on 16 April 2020).
- BMS986020. Available online: https://pulmonaryfibrosisnews.com/bms-986020-for-pulmonary-fibrosis/ (accessed on 16 April 2020).
- Brindley, D.N.; Benesch, M.G.K.; Murph, M. (Eds.) Autotaxin: An Enzymatic Augmenter of Malignant Progression Linked to Inflammation; Chapter 12; INTECH: Rijeka, Croatia, 2015. [Google Scholar]
- Benesch, M.G.; Ko, Y.M.; Tang, X.; Dewald, J.; Lopez-Campistrous, A.; Zhao, Y.Y.; Lai, R.; Curtis, J.M.; Brindley, D.N.; McMullen, T.P. Autotaxin is an inflammatory mediator and therapeutic target in thyroid cancer. Endocr. Relat. Cancer 2015, 22, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.; Tang, X.; Maeda, T.; Ohhata, A.; Zhao, Y.Y.; Kok, B.P.; Dewald, J.; Hitt, M.; Curtis, J.M.; McMullen, T.P.; et al. Inhibition of autotaxin delays breast tumor growth and lung metastasis in mice. FASEB J. 2014, 28, 2655–2666. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.; Zhao, Y.Y.; Curtis, J.M.; McMullen, T.P.; Brindley, D.N. Regulation of autotaxin expression and secretion by lysophosphatidate and sphingosine 1-phosphate. J. Lipid. Res. 2015, 56, 1134–1144. [Google Scholar] [CrossRef]
- Tang, X.; Benesch, M.G.; Brindley, D.N. Lipid phosphate phosphatases and their roles in mammalian physiology and pathology. J. Lipid. Res. 2015, 56, 2048–2060. [Google Scholar] [CrossRef]
- Benesch, M.G.K.; Yang, Z.; Tang, X.; Meng, G.; Brindley, D.N. Lysophosphatidate signaling: The tumor microenvironments new nemesis. Trends Cancer 2017, 3, 748–752. [Google Scholar] [CrossRef]
- Benesch, M.G.K.; Tang, X.; Brindley, D.N. Autotaxin and breast cancer: Towards overcoming treatment barriers and sequelae. Cancers 2020, 12, 374. [Google Scholar] [CrossRef]
- Volden, P.A.; Skor, M.N.; Johnson, M.B.; Singh, P.; Patel, F.N.; McClintock, M.K.; Brady, M.J.; Conzen, S.D. Mammary adipose tissue-derived lysophospholipids promote estrogen receptor-negative mammary epithelial cell proliferation. Cancer Prev. Res. 2016, 9, 367–378. [Google Scholar] [CrossRef]
- Schmid, R.; Wolf, K.; Robering, J.W.; Strauss, S.; Strissel, P.L.; Strick, R.; Rubner, M.; Fasching, P.A.; Horch, R.E.; Kremer, A.E.; et al. ADSCs and adipocytes are the main producers in the autotaxin-lysophosphatidic acid axis of breast cancer and healthy mammary tissue in vitro. BMC Cancer 2018, 18, 1273. [Google Scholar] [CrossRef] [PubMed]
- Dusaulcy, R.; Rancoule, C.; Gres, S.; Wanecq, E.; Colom, A.; Guigne, C.; van Meeteren, L.A.; Moolenaar, W.H.; Valet, P.; Saulnier-Blache, J.S. Adipose-specific disruption of autotaxin enhances nutritional fattening and reduces plasma lysophosphatidic acid. J. Lipid. Res. 2011, 52, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Rancoule, C.; Dusaulcy, R.; Treguer, K.; Gres, S.; Guigne, C.; Quilliot, D.; Valet, P.; Saulnier-Blache, J.S. Depot-specific regulation of autotaxin with obesity in human adipose tissue. J. Physiol. Biochem. 2012, 68, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.K.; MacIntyre, I.T.K.; McMullen, T.P.W.; Brindley, D.N. Coming of age for autotaxin and lysophosphatidate signaling: Clinical applications for preventing, detecting and targeting tumor-promoting inflammation. Cancers 2018, 3, 73. [Google Scholar] [CrossRef]
- Meng, G.; Wuest, M.; Tang, X.; Dufour, J.; Zhao, Y.; Curtis, J.M.; McMullen, T.P.W.; Murray, D.; Wuest, F.; Brindley, D.N. Repeated fractions of X-radiation to the breast fat pads of mice augment activation of the autotaxin-lysophosphatidate-inflammatory cycle. Cancers 2019, 11, 1816. [Google Scholar] [CrossRef]
- Benesch, M.G.; Tang, X.; Dewald, J.; Dong, W.F.; Mackey, J.R.; Hemmings, D.G.; McMullen, T.P.; Brindley, D.N. Tumor-induced inflammation in mammary adipose tissue stimulates a vicious cycle of autotaxin expression and breast cancer progression. FASEB J. 2015, 29, 3990–4000. [Google Scholar] [CrossRef]
- Meng, G.; Tang, X.; Yang, Z.; Zhao, Y.; Curtis, J.M.; McMullen, T.P.W.; Brindley, D.N. Dexamethasone decreases the autotaxin-lysophosphatidate-inflammatory axis in adipose tissue: Implications for the metabolic syndrome and breast cancer. FASEB J. 2019, 33, 1899–1910. [Google Scholar] [CrossRef]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids--new mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef]
- Hong, J.H.; Chiang, C.S.; Tsao, C.Y.; Lin, P.Y.; McBride, W.H.; Wu, C.J. Rapid induction of cytokine gene expression in the lung after single and fractionated doses of radiation. Int. J. Radiat. Biol. 1999, 75, 1421–1427. [Google Scholar] [CrossRef]
- Hong, J.H.; Chiang, C.S.; Tsao, C.Y.; Lin, P.Y.; Wu, C.J.; McBride, W.H. Can short-term administration of dexamethasone abrogate radiation-induced acute cytokine gene response in lung and modify subsequent molecular responses? Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 296–303. [Google Scholar] [CrossRef]
- Chen, B.; Na, F.; Yang, H.; Li, R.; Li, M.; Sun, X.; Hu, B.; Huang, G.; Lan, J.; Xu, H.; et al. Ethyl pyruvate alleviates radiation-induced lung injury in mice. Biomed. Pharmacother. 2017, 92, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.P.; Wang, Y.W.; Wang, B.Z.; Sun, G.M.; Wang, X.Y.; Xu, J.L. Expression of interleukin-17A in lung tissues of irradiated mice and the influence of dexamethasone. Sci. World J. 2014, 2014, 251067. [Google Scholar] [CrossRef] [PubMed]
- Levine, S.J.; Benfield, T.; Shelhamer, J.H. Corticosteroids induce intracellular interleukin-1 receptor antagonist type I expression by a human airway epithelial cell line. Am. J. Respir. Cell Mol. Biol. 1996, 15, 245–251. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, W.; Kamanaka, M.; Booth, C.J.; Town, T.; Nakae, S.; Iwakura, Y.; Kolls, J.K.; Flavell, R.A. A protective function for interleukin 17A in T cell-mediated intestinal inflammation. Nat. Immunol. 2009, 10, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Cuneo, A.A.; Autieri, M.V. Expression and function of anti-inflammatory interleukins: The other side of the vascular response to injury. Curr. Vasc. Pharmacol. 2009, 7, 267–276. [Google Scholar] [CrossRef]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Fact. 2011, 29, 196–202. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-beta on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef]
- Ninou, I.; Magkrioti, C.; Aidinis, V. Autotaxin in pathophysiology and pulmonary fibrosis. Front. Med. 2018, 5, 180. [Google Scholar] [CrossRef]
- Ejaz, A.; Greenberger, J.S.; Rubin, P.J. Understanding the mechanism of radiation induced fibrosis and therapy options. Pharmacol. Ther. 2019, 107399. [Google Scholar] [CrossRef]
- Hanania, A.N.; Mainwaring, W.; Ghebre, Y.T.; Hanania, N.A.; Ludwig, M. Radiation-induced lung injury: Assessment and management. Chest 2019, 156, 150–162. [Google Scholar] [CrossRef]
- Sekhar, K.R.; Freeman, M.L. Nrf2 promotes survival following exposure to ionizing radiation. Free Radic. Biol. Med. 2015, 88, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Blom Goldman, U.; Svane, G.; Anderson, M.; Wennberg, B.; Lind, P. Long-term functional and radiological pulmonary changes after radiation therapy for breast cancer. Acta Oncol. 2014, 53, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Tager, A.M. Autotaxin emerges as a therapeutic target for idiopathic pulmonary fibrosis: Limiting fibrosis by limiting lysophosphatidic acid synthesis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Keune, W.J.; Hausmann, J.; Bolier, R.; Tolenaars, D.; Kremer, A.; Heidebrecht, T.; Joosten, R.P.; Sunkara, M.; Morris, A.J.; Matas-Rico, E.; et al. Steroid binding to autotaxin links bile salts and lysophosphatidic acid signalling. Nat. Commun. 2016, 7, 11248. [Google Scholar] [CrossRef] [PubMed]
- Strieter, R.M.; Gomperts, B.N.; Keane, M.P. The role of CXC chemokines in pulmonary fibrosis. J. Clin. Invest. 2007, 117, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.A.; Habiel, D.M.; Hohmann, M.; Camelo, A.; Shang, H.; Zhou, Y.; Coelho, A.L.; Peng, X.; Gulati, M.; Crestani, B.; et al. Antifibrotic role of vascular endothelial growth factor in pulmonary fibrosis. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, Y.; Niu, Y.; Fu, L.; Chin, Y.E.; Yu, C. Leukemia inhibitory factor attenuates renal fibrosis through Stat3-miR-29c. Am. J. Physiol. Renal. Physiol. 2015, 309, F595–F603. [Google Scholar] [CrossRef]
- Fulkerson, Z.; Wu, T.; Sunkara, M.; Kooi, C.V.; Morris, A.J.; Smyth, S.S. Binding of autotaxin to integrins localizes lysophosphatidic acid production to platelets and mammalian cells. J. Biol. Chem. 2011, 286, 34654–34663. [Google Scholar] [CrossRef]
- Hausmann, J.; Kamtekar, S.; Christodoulou, E.; Day, J.E.; Wu, T.; Fulkerson, Z.; Albers, H.M.; van Meeteren, L.A.; Houben, A.J.; van Zeijl, L.; et al. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 2011, 18, 198–204. [Google Scholar] [CrossRef]
- Leblanc, R.; Sahay, D.; Houssin, A.; Machuca-Gayet, I.; Peyruchaud, O. Autotaxin-beta interaction with the cell surface via syndecan-4 impacts on cancer cell proliferation and metastasis. Oncotarget 2018, 9, 33170–33185. [Google Scholar] [CrossRef]
- Van Meeteren, L.A.; Moolenaar, W.H. Regulation and biological activities of the autotaxin-LPA axis. Progress Lipid. Res. 2007, 46, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Shea, B.S.; Tager, A.M. Role of the lysophospholipid mediators lysophosphatidic acid and sphingosine 1-phosphate in lung fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Miyabe, C.; Miyabe, Y.; Nagai, J.; Miura, N.N.; Ohno, N.; Chun, J.; Tsuboi, R.; Ueda, H.; Miyasaka, M.; Miyasaka, N.; et al. Abrogation of lysophosphatidic acid receptor 1 ameliorates murine vasculitis. Arthritis Res. Ther. 2019, 21, 191. [Google Scholar] [CrossRef] [PubMed]
- Funke, M.; Zhao, Z.; Xu, Y.; Chun, J.; Tager, A.M. The lysophosphatidic acid receptor LPA1 promotes epithelial cell apoptosis after lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 46, 355–364. [Google Scholar] [CrossRef]
- Nincheri, P.; Bernacchioni, C.; Cencetti, F.; Donati, C.; Bruni, P. Sphingosine kinase-1/S1P1 signalling axis negatively regulates mitogenic response elicited by PDGF in mouse myoblasts. Cell. Signal. 2010, 22, 1688–1699. [Google Scholar] [CrossRef]
- Evans, M.L.; Graham, M.M.; Mahler, P.A.; Rasey, J.S. Use of steroids to suppress vascular response to radiation. Int. J. Radiat. Oncol. Biol. Phys. 1987, 13, 563–567. [Google Scholar] [CrossRef]
- Graham, M.M.; Evans, M.L.; Dahlen, D.D.; Mahler, P.A.; Rasey, J.S. Pharmacological alteration of the lung vascular response to radiation. Int. J. Radiat. Oncol. Biol. Phys. 1990, 19, 329–339. [Google Scholar] [CrossRef]
- Peterson, L.M.; Evans, M.L.; Graham, M.M.; Eary, J.F.; Dahlen, D.D. Vascular response to radiation injury in the rat lung. Radiat. Res. 1992, 129, 139–148. [Google Scholar] [CrossRef]
- Desroy, N.; Housseman, C.; Bock, X.; Joncour, A.; Bienvenu, N.; Cherel, L.; Labeguere, V.; Rondet, E.; Peixoto, C.; Grassot, J.M.; et al. Discovery of 2-[[2-Ethyl-6-[4-[2-(3-hydroxyazetidin-1-yl)-2-oxoethyl]piperazin-1-yl]-8-methyli midazo[1,2-a]pyridin-3-yl]methylamino]-4-(4-fluorophenyl)thiazole-5-carbonitrile (GLPG1690), a first-in-Class autotaxin inhibitor undergoing clinical evaluation for the treatment of idiopathic pulmonary fibrosis. J. Med. Chem. 2017, 60, 3580–3590. [Google Scholar] [CrossRef]
- Kato, K.; Ikeda, H.; Miyakawa, S.; Futakawa, S.; Nonaka, Y.; Fujiwara, M.; Okudaira, S.; Kano, K.; Aoki, J.; Morita, J.; et al. Structural basis for specific inhibition of autotaxin by a DNA aptamer. Nat. Struct. Mol. Biol. 2016, 23, 395–401. [Google Scholar] [CrossRef]
- Martin, M.; Lefaix, J.; Delanian, S. TGF-beta1 and radiation fibrosis: A master switch and a specific therapeutic target? Int. J. Radiat. Oncol. Biol. Phys. 2000, 47, 277–290. [Google Scholar] [CrossRef]
- Shieh, J.M.; Tsai, Y.J.; Chi, J.C.; Wu, W.B. TGFβ mediates collagen production in human CRSsNP nasal mucosa-derived fibroblasts through Smad2/3-dependent pathway and CTGF induction and secretion. J. Cell. Physiol. 2019, 234, 10489–10499. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef]
- Xu, M.Y.; Porte, J.; Knox, A.J.; Weinreb, P.H.; Maher, T.M.; Violette, S.M.; McAnulty, R.J.; Sheppard, D.; Jenkins, G. Lysophosphatidic acid induces alphavbeta6 integrin-mediated TGF-beta activation via the LPA2 receptor and the small G protein G alpha(q). Am. J. Pathol. 2009, 174, 1264–1279. [Google Scholar] [CrossRef]
- Pyne, N.J.; Dubois, G.; Pyne, S. Role of sphingosine 1-phosphate and lysophosphatidic acid in fibrosis. Biochim. Biophys. Acta 2013, 1831, 228–238. [Google Scholar] [CrossRef]
- Zhao, J.; He, D.; Berdyshev, E.; Zhong, M.; Salgia, R.; Morris, A.J.; Smyth, S.S.; Natarajan, V.; Zhao, Y. Autotaxin induces lung epithelial cell migration through lysoPLD activity-dependent and -independent pathways. Biochem. J. 2011, 439, 45–55. [Google Scholar] [CrossRef]
- Yang, Y.; Mou, L.; Liu, N.; Tsao, M.S. Autotaxin expression in non-small-cell lung cancer. Am. J. Respir. Cell Mol. Biol. 1999, 21, 216–222. [Google Scholar] [CrossRef]
- Ward, H.E.; Kemsley, L.; Davies, L.; Holecek, M.; Berend, N. The effect of steroids on radiation-induced lung disease in the rat. Radiat. Res. 1993, 136, 22–28. [Google Scholar] [CrossRef]
- Liu, Z.H.; Fan, W.; Chen, R.C. 3,4-dihydroxyphenylethanol suppresses irradiation-induced pulmonary fibrosis in adult rats. Int. J. Clin. Exp. Pathol. 2015, 8, 3441–3450. [Google Scholar]
- Ramos-Vara, J.A. Technical aspects of immunohistochemistry. Vet. Pathol. 2005, 42, 405–426. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, G.; Wuest, M.; Tang, X.; Dufour, J.; McMullen, T.P.W.; Wuest, F.; Murray, D.; Brindley, D.N. Dexamethasone Attenuates X-Ray-Induced Activation of the Autotaxin-Lysophosphatidate-Inflammatory Cycle in Breast Tissue and Subsequent Breast Fibrosis. Cancers 2020, 12, 999. https://doi.org/10.3390/cancers12040999
Meng G, Wuest M, Tang X, Dufour J, McMullen TPW, Wuest F, Murray D, Brindley DN. Dexamethasone Attenuates X-Ray-Induced Activation of the Autotaxin-Lysophosphatidate-Inflammatory Cycle in Breast Tissue and Subsequent Breast Fibrosis. Cancers. 2020; 12(4):999. https://doi.org/10.3390/cancers12040999
Chicago/Turabian StyleMeng, Guanmin, Melinda Wuest, Xiaoyun Tang, Jennifer Dufour, Todd P.W. McMullen, Frank Wuest, David Murray, and David N. Brindley. 2020. "Dexamethasone Attenuates X-Ray-Induced Activation of the Autotaxin-Lysophosphatidate-Inflammatory Cycle in Breast Tissue and Subsequent Breast Fibrosis" Cancers 12, no. 4: 999. https://doi.org/10.3390/cancers12040999
APA StyleMeng, G., Wuest, M., Tang, X., Dufour, J., McMullen, T. P. W., Wuest, F., Murray, D., & Brindley, D. N. (2020). Dexamethasone Attenuates X-Ray-Induced Activation of the Autotaxin-Lysophosphatidate-Inflammatory Cycle in Breast Tissue and Subsequent Breast Fibrosis. Cancers, 12(4), 999. https://doi.org/10.3390/cancers12040999