Betulinic Acid-Mediated Tuning of PERK/CHOP Signaling by Sp1 Inhibition as a Novel Therapeutic Strategy for Glioblastoma

 , ,
, ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
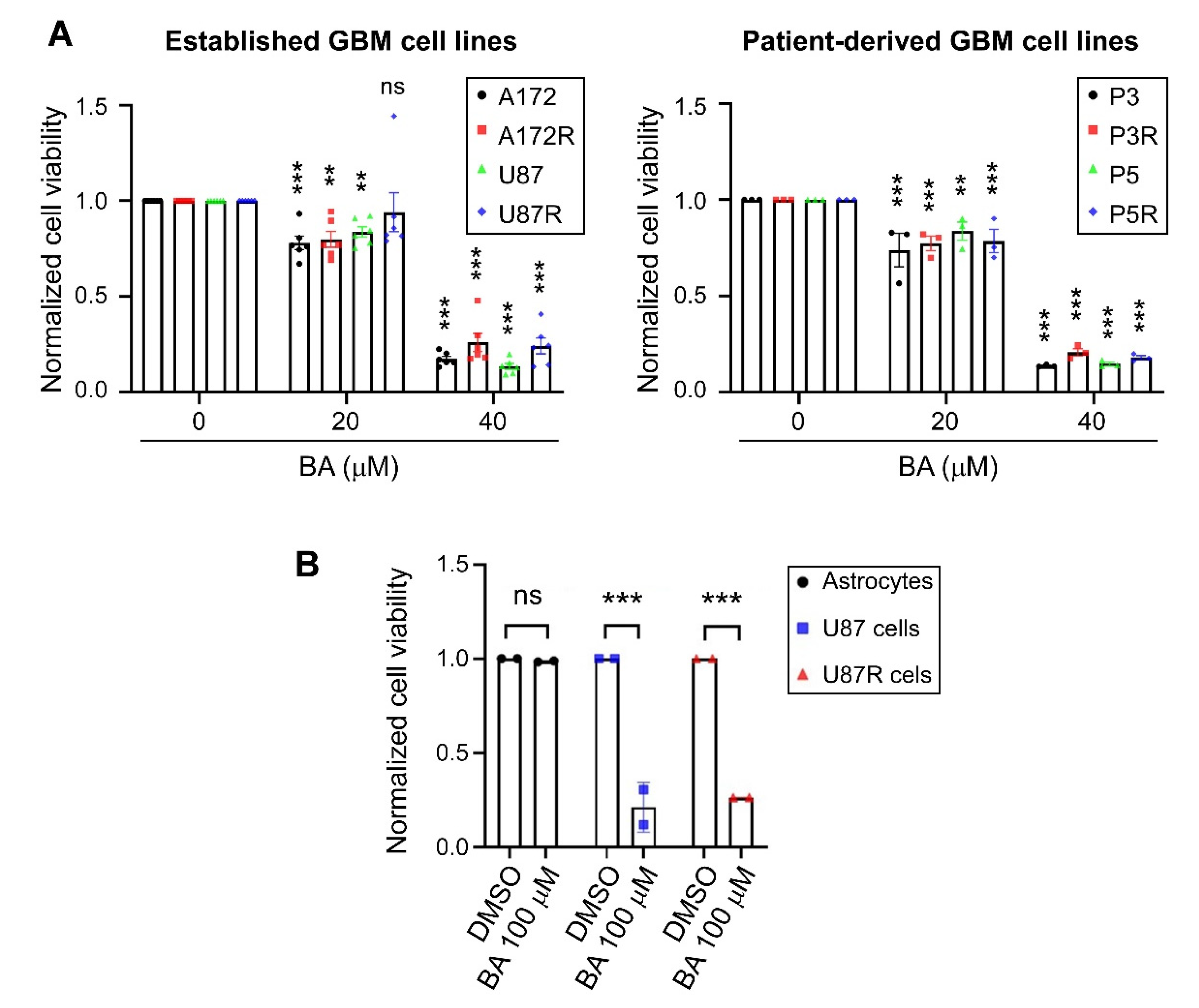
2.1. BA Selectively Targets Brain-Tumor Cells
2.2. BA Sensitizes Resistant GBM Cells to TMZ
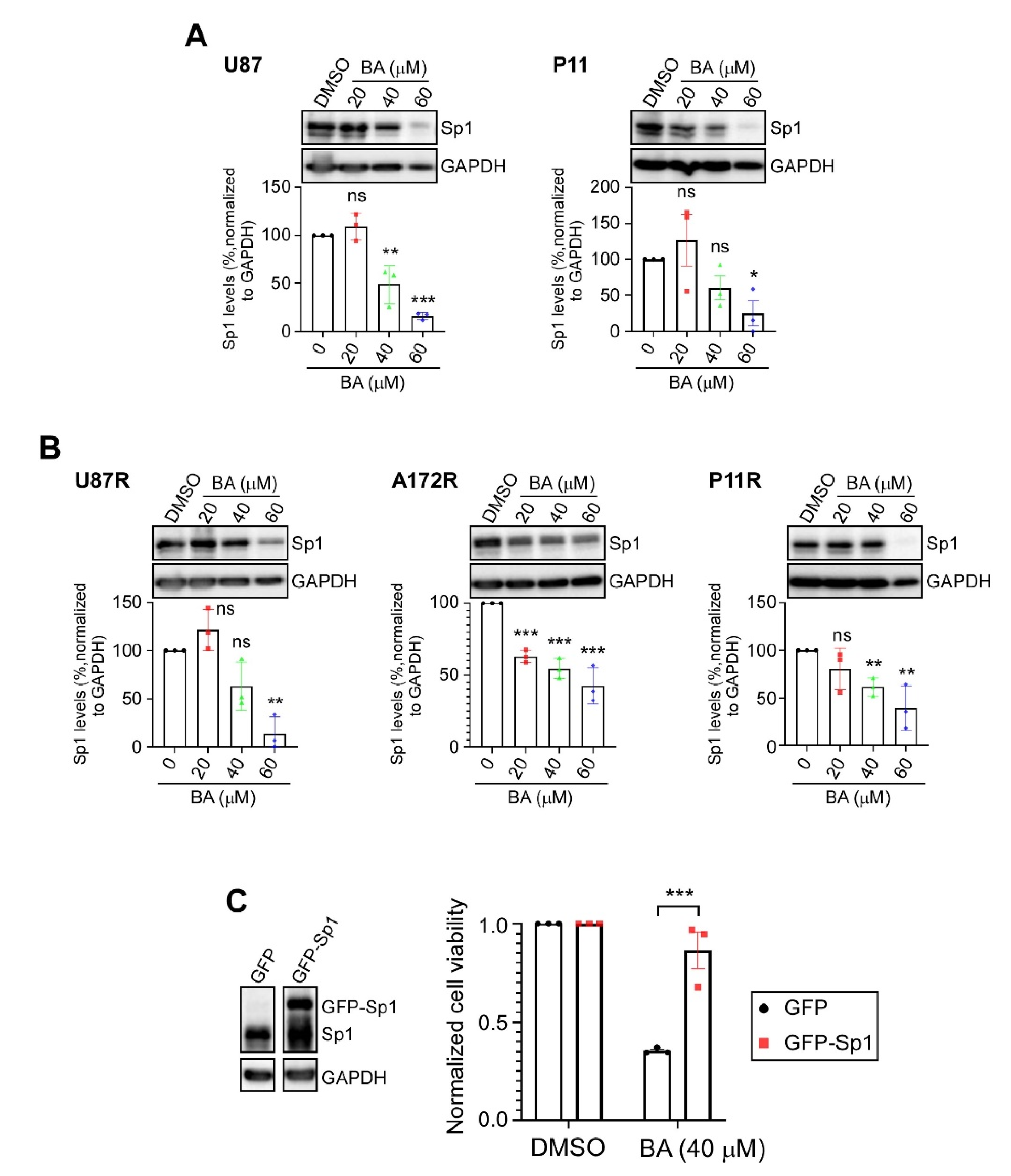
2.3. BA Suppresses GBM Cell Growth via Inhibition of Sp1 Expression
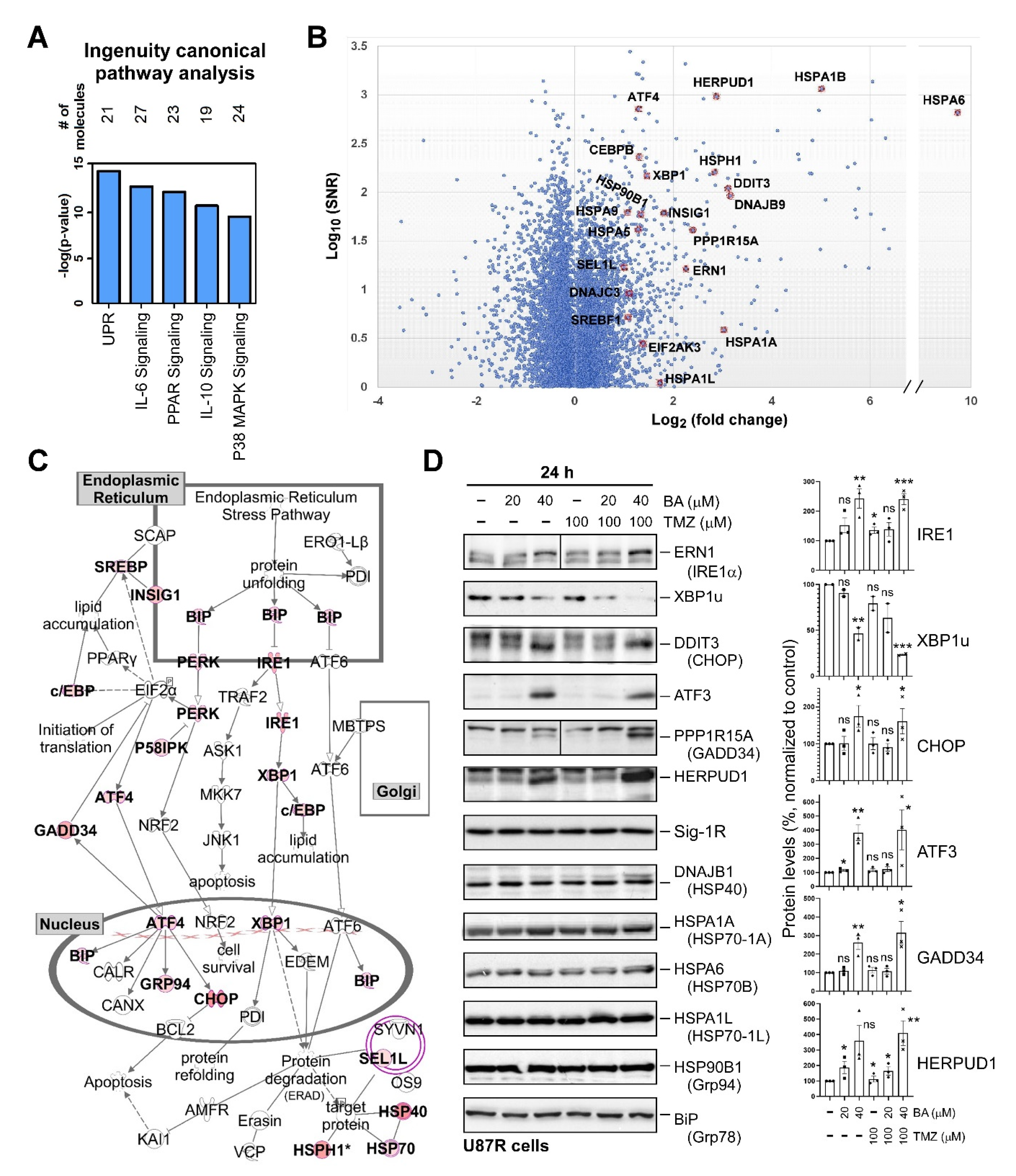
2.4. BA Treatment Alters Expression of ER Stress-Related Genes
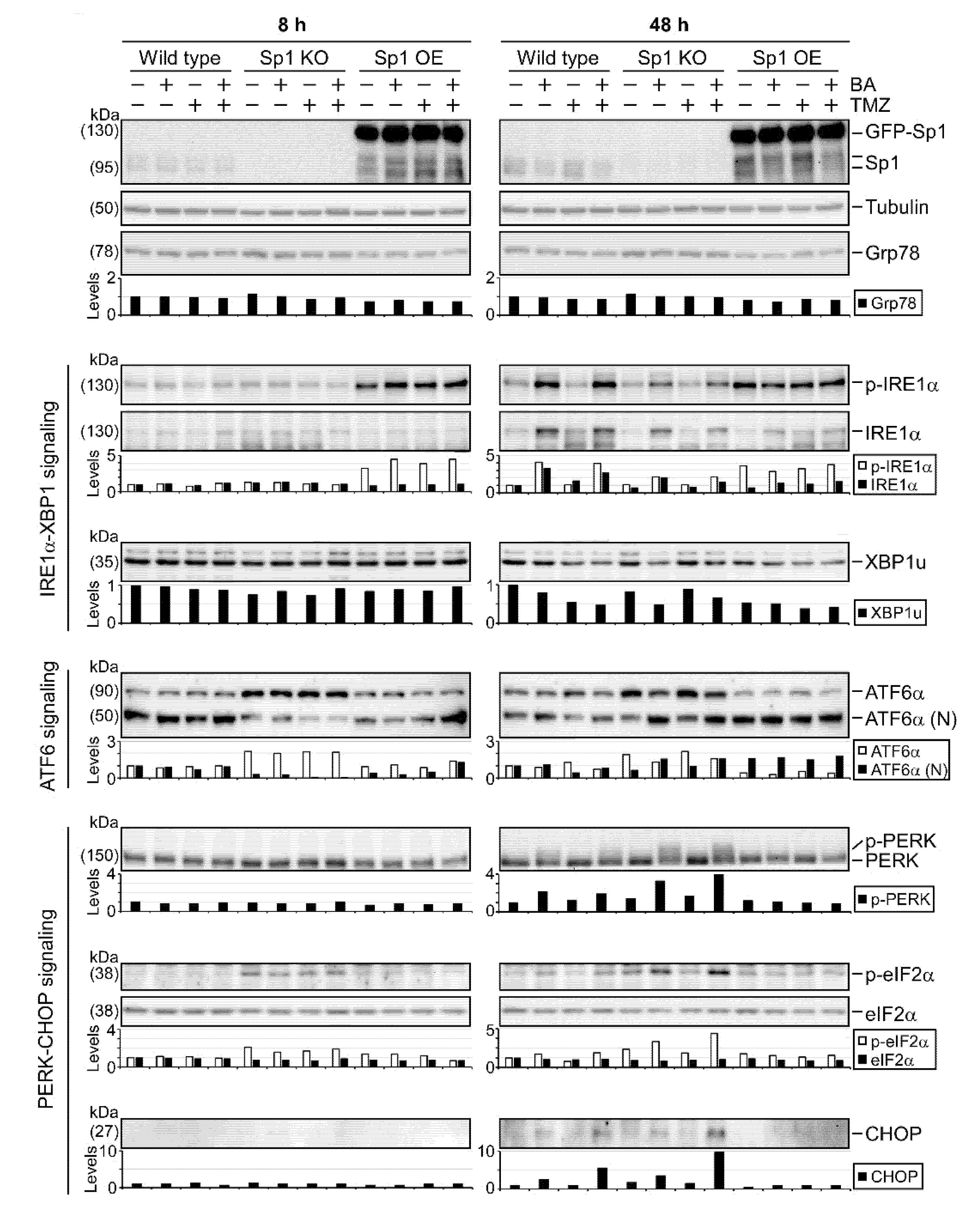
2.5. Sp1 Plays Roles in Regulating UPR Activation
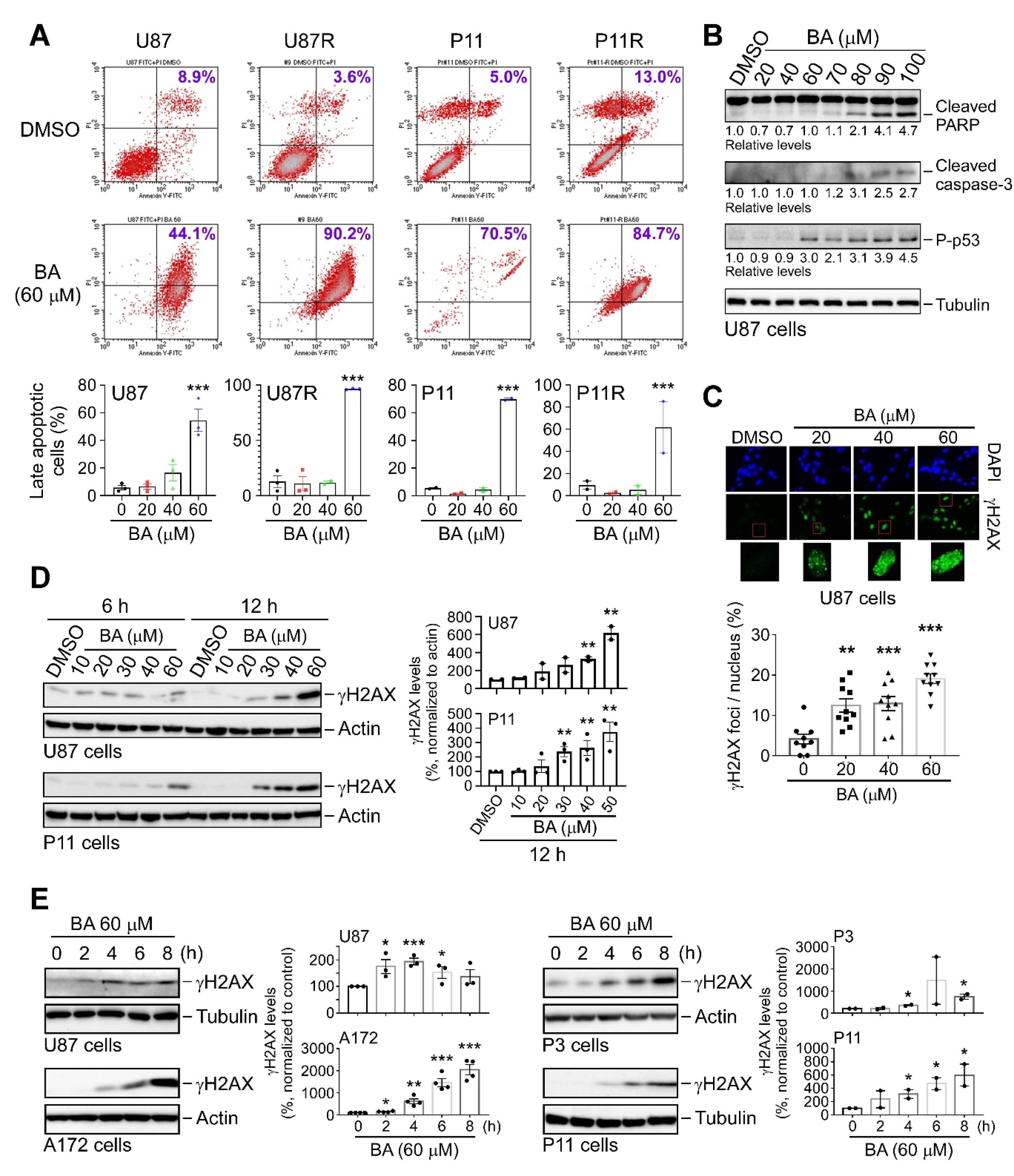
2.6. BA Triggers Apoptosis and DNA Damage in TMZ-Sensitive and -Resistant GBM Cells
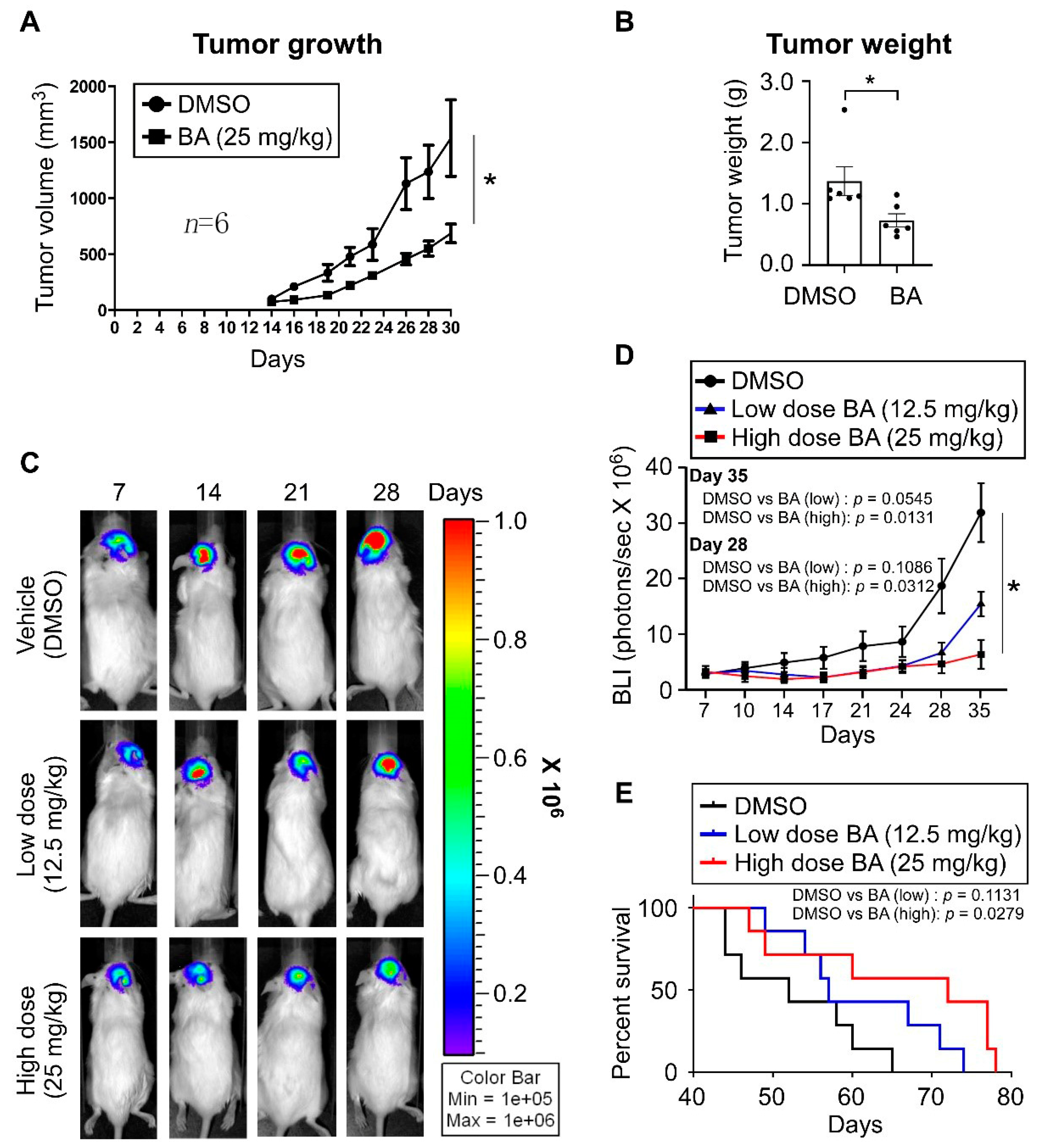
2.7. BA Inhibits GBM Growth and Improves Survival In Vivo
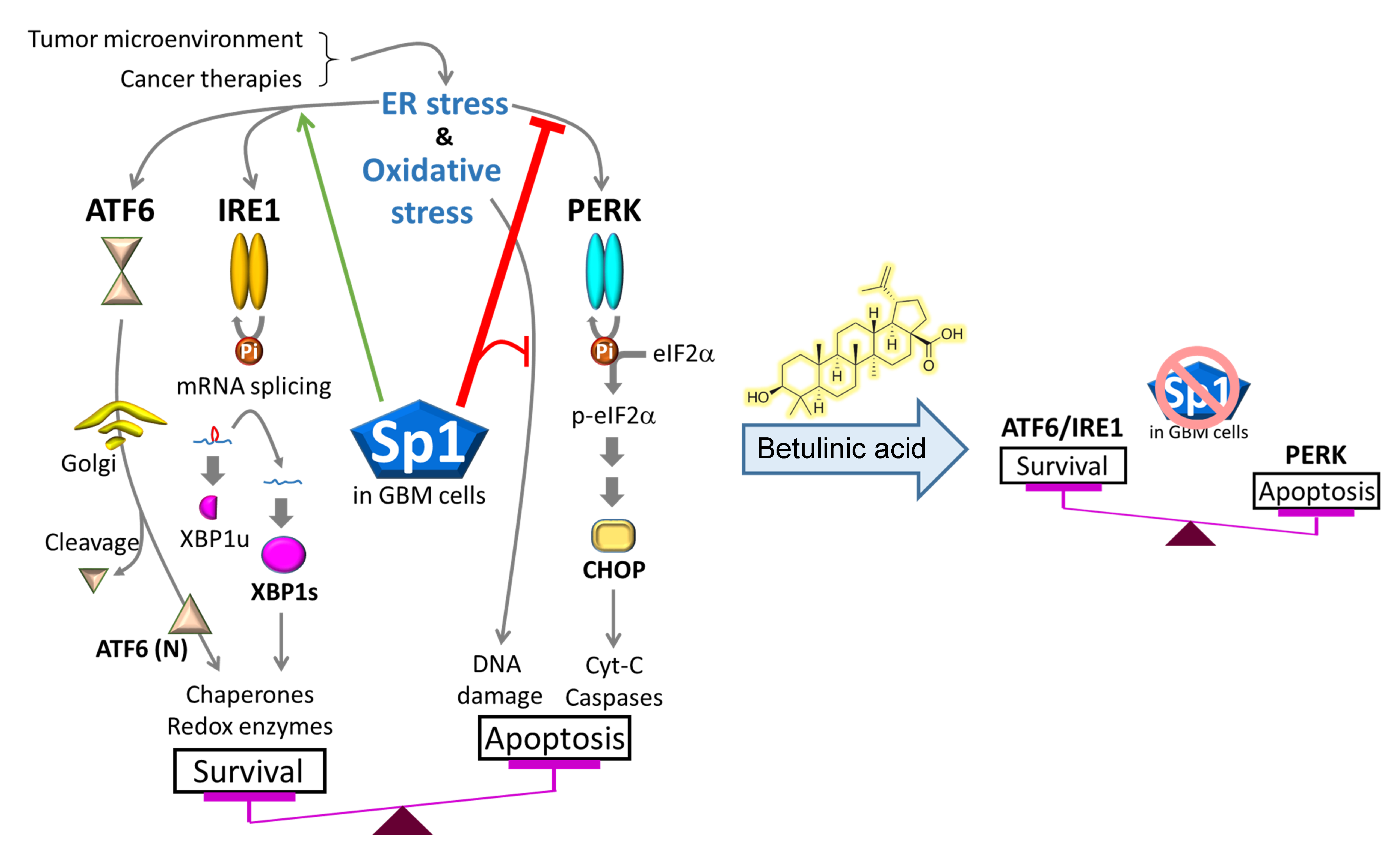
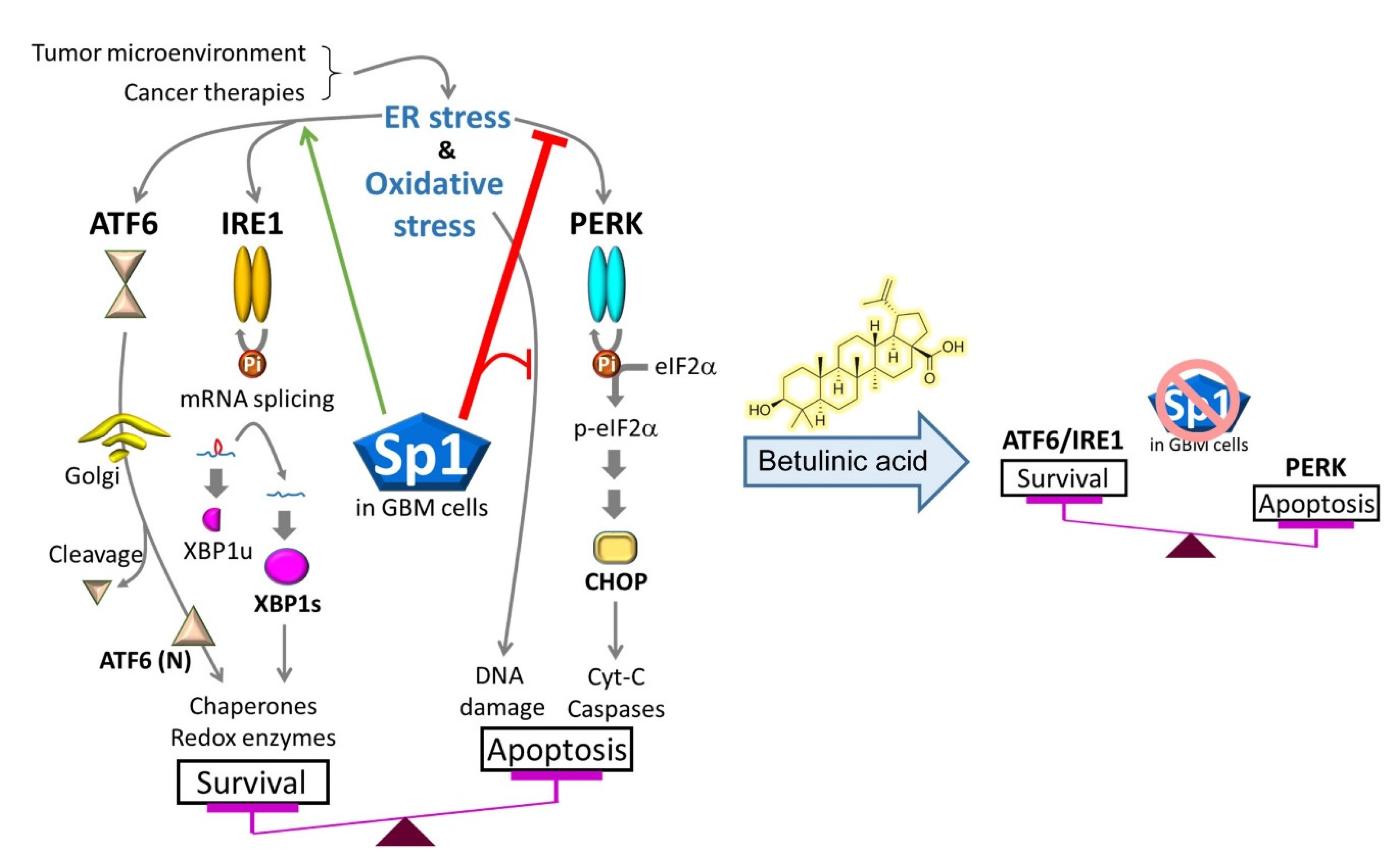
3. Discussion
4. Materials and Methods
4.1. Cell Preparation
4.2. Examination of Cell Viability
4.3. Western Blotting
4.4. Apoptosis Assay
4.5. Microarray Analysis
4.6. In Vivo Animal Model for GBM
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Agarwala, S.S.; Kirkwood, J.M. Temozolomide, a novel alkylating agent with activity in the central nervous system, may improve the treatment of advanced metastatic melanoma. Oncologist 2000, 5, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Stetler, R.A.; Gan, Y.; Zhang, W.; Liou, A.K.; Gao, Y.; Cao, G.; Chen, J. Heat shock proteins: Cellular and molecular mechanisms in the central nervous system. Prog. Neurobiol. 2010, 92, 184–211. [Google Scholar] [CrossRef] [PubMed]
- Biden, T.J.; Boslem, E.; Chu, K.Y.; Sue, N. Lipotoxic endoplasmic reticulum stress, beta cell failure, and type 2 diabetes mellitus. Trends Endocrinol. Metab. 2014, 25, 389–398. [Google Scholar] [CrossRef]
- Oslowski, C.M.; Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 2011, 490, 71–92. [Google Scholar] [CrossRef]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- Pyrko, P.; Schonthal, A.H.; Hofman, F.M.; Chen, T.C.; Lee, A.S. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007, 67, 9809–9816. [Google Scholar] [CrossRef]
- Liu, W.T.; Huang, C.Y.; Lu, I.C.; Gean, P.W. Inhibition of glioma growth by minocycline is mediated through endoplasmic reticulum stress-induced apoptosis and autophagic cell death. Neuro-Oncology 2013, 15, 1127–1141. [Google Scholar] [CrossRef]
- Li, Z.Y.; Zhang, C.; Zhang, Y.; Chen, L.; Chen, B.D.; Li, Q.Z.; Zhang, X.J.; Li, W.P. A novel HDAC6 inhibitor Tubastatin A: Controls HDAC6-p97/VCP-mediated ubiquitination-autophagy turnover and reverses Temozolomide-induced ER stress-tolerance in GBM cells. Cancer Lett. 2017, 391, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Zuco, V.; Supino, R.; Righetti, S.C.; Cleris, L.; Marchesi, E.; Gambacorti-Passerini, C.; Formelli, F. Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett. 2002, 175, 17–25. [Google Scholar] [CrossRef]
- Hsu, T.I.; Wang, M.C.; Chen, S.Y.; Huang, S.T.; Yeh, Y.M.; Su, W.C.; Chang, W.C.; Hung, J.J. Betulinic acid decreases specificity protein 1 (Sp1) level via increasing the sumoylation of sp1 to inhibit lung cancer growth. Mol. Pharmacol. 2012, 82, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Betulinic Acid for cancer treatment and prevention. Int. J. Mol. Sci. 2008, 9, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Hsu, T.I.; Chen, Y.J.; Hung, C.Y.; Wang, Y.C.; Lin, S.J.; Su, W.C.; Lai, M.D.; Kim, S.Y.; Wang, Q.; Qian, K.; et al. A novel derivative of betulinic acid, SYK023, suppresses lung cancer growth and malignancy. Oncotarget 2015, 6, 13671–13687. [Google Scholar] [CrossRef]
- Killinger, B.; Shah, M.; Moszczynska, A. Co-administration of betulinic acid and methamphetamine causes toxicity to dopaminergic and serotonergic nerve terminals in the striatum of late adolescent rats. J. Neurochem. 2014, 128, 764–775. [Google Scholar] [CrossRef]
- Chang, K.Y.; Hsu, T.I.; Hsu, C.C.; Tsai, S.Y.; Liu, J.J.; Chou, S.W.; Liu, M.S.; Liou, J.P.; Ko, C.Y.; Chen, K.Y.; et al. Specificity protein 1-modulated superoxide dismutase 2 enhances temozolomide resistance in glioblastoma, which is independent of O(6)-methylguanine-DNA methyltransferase. Redox Biol. 2017, 13, 655–664. [Google Scholar] [CrossRef]
- Wu, C.C.; Lee, P.T.; Kao, T.J.; Chou, S.Y.; Su, R.Y.; Lee, Y.C.; Yeh, S.H.; Liou, J.P.; Hsu, T.I.; Su, T.P.; et al. Upregulation of Znf179 acetylation by SAHA protects cells against oxidative stress. Redox Biol. 2018, 19, 74–80. [Google Scholar] [CrossRef]
- Hsu, C.C.; Chang, W.C.; Hsu, T.I.; Liu, J.J.; Yeh, S.H.; Wang, J.Y.; Liou, J.P.; Ko, C.Y.; Chang, K.Y.; Chuang, J.Y. Suberoylanilide hydroxamic acid represses glioma stem-like cells. J. Biomed. Sci. 2016, 23, 81. [Google Scholar] [CrossRef]
- Chuang, J.Y.; Lo, W.L.; Ko, C.Y.; Chou, S.Y.; Chen, R.M.; Chang, K.Y.; Hung, J.J.; Su, W.C.; Chang, W.C.; Hsu, T.I. Upregulation of CYP17A1 by Sp1-mediated DNA demethylation confers temozolomide resistance through DHEA-mediated protection in glioma. Oncogenesis 2017, 6, e339. [Google Scholar] [CrossRef]
- Yang, W.B.; Chuang, J.Y.; Ko, C.Y.; Chang, W.C.; Hsu, T.I. Dehydroepiandrosterone Induces Temozolomide Resistance Through Modulating Phosphorylation and Acetylation of Sp1 in Glioblastoma. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, E.; Cheng, Y.; Jin, U.H.; Kim, K.; Safe, S. Specificity protein (Sp) transcription factors Sp1, Sp3 and Sp4 are non-oncogene addiction genes in cancer cells. Oncotarget 2016, 7, 22245–22256. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Rao, X.; Xu, J.F.; Yang, P.; Wang, C.Y. The role of endoplasmic reticulum stress in autoimmune-mediated beta-cell destruction in type 1 diabetes. Exp. Diabetes Res. 2012, 2012, 238980. [Google Scholar] [CrossRef] [PubMed]
- Roy, G.; Zhang, S.; Li, L.; Higham, E.; Wu, H.; Marelli, M.; Bowen, M.A. Development of a fluorescent reporter system for monitoring ER stress in Chinese hamster ovary cells and its application for therapeutic protein production. PLoS ONE 2017, 12, e0183694. [Google Scholar] [CrossRef] [PubMed]
- Bhat, T.A.; Chaudhary, A.K.; Kumar, S.; O’Malley, J.; Inigo, J.R.; Kumar, R.; Yadav, N.; Chandra, D. Endoplasmic reticulum-mediated unfolded protein response and mitochondrial apoptosis in cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1867, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.Y.; Huang, C.T.; Hsu, T.I.; Hsu, C.C.; Liu, J.J.; Chuang, C.K.; Hung, J.J.; Chang, W.C.; Tsai, K.K.; Chuang, J.Y. Stress stimuli induce cancer-stemness gene expression via Sp1 activation leading to therapeutic resistance in glioblastoma. Biochem. Biophys. Res. Commun. 2017, 493, 14–19. [Google Scholar] [CrossRef]
- Zhao, C.; Meng, A. Sp1-like transcription factors are regulators of embryonic development in vertebrates. Dev. Growth Differ. 2005, 47, 201–211. [Google Scholar] [CrossRef]
- Vizcaino, C.; Mansilla, S.; Portugal, J. Sp1 transcription factor: A long-standing target in cancer chemotherapy. Pharmacol. Ther. 2015, 152, 111–124. [Google Scholar] [CrossRef]
- Bedolla, R.G.; Gong, J.; Prihoda, T.J.; Yeh, I.T.; Thompson, I.M.; Ghosh, R.; Kumar, A.P. Predictive value of Sp1/Sp3/FLIP signature for prostate cancer recurrence. PLoS ONE 2012, 7, e44917. [Google Scholar] [CrossRef]
- Dauer, P.; Gupta, V.K.; McGinn, O.; Nomura, A.; Sharma, N.S.; Arora, N.; Giri, B.; Dudeja, V.; Saluja, A.K.; Banerjee, S. Inhibition of Sp1 prevents ER homeostasis and causes cell death by lysosomal membrane permeabilization in pancreatic cancer. Sci. Rep. 2017, 7, 1564. [Google Scholar] [CrossRef]
- Gao, Y.; Jia, Z.; Kong, X.; Li, Q.; Chang, D.Z.; Wei, D.; Le, X.; Suyun, H.; Huang, S.; Wang, L.; et al. Combining betulinic acid and mithramycin a effectively suppresses pancreatic cancer by inhibiting proliferation, invasion, and angiogenesis. Cancer Res. 2011, 71, 5182–5193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.B.; Wang, Z.; Shu, F.; Jin, Y.H.; Liu, H.Y.; Wang, Q.J.; Yang, Y. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J. Biol. Chem. 2010, 285, 40461–40471. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Lee, C.C.; Shih, Y.L.; Lin, C.H.; Wang, S.H.; Chen, T.H.; Shih, C.M. Inhibition of mitochondria- and endoplasmic reticulum stress-mediated autophagy augments temozolomide-induced apoptosis in glioma cells. PLoS ONE 2012, 7, e38706. [Google Scholar] [CrossRef] [PubMed]
- Ryabaya, O.; Prokofieva, A.; Khochenkov, D.; Abramov, I.; Zasedatelev, A.; Stepanova, E. Inhibition of endoplasmic reticulum stress-induced autophagy sensitizes melanoma cells to temozolomide treatment. Oncol. Rep. 2018, 40, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Erasimus, H.; Gobin, M.; Niclou, S.; Van Dyck, E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutat. Res. Rev. Mutat. Res. 2016, 769, 19–35. [Google Scholar] [CrossRef]
- Chien, C.H.; Chuang, J.Y.; Yang, S.T.; Yang, W.B.; Chen, P.Y.; Hsu, T.I.; Huang, C.Y.; Lo, W.L.; Yang, K.Y.; Liu, M.S.; et al. Enrichment of superoxide dismutase 2 in glioblastoma confers to acquisition of temozolomide resistance that is associated with tumor-initiating cell subsets. J. Biomed. Sci. 2019, 26, 77. [Google Scholar] [CrossRef]
- Kitange, G.J.; Mladek, A.C.; Schroeder, M.A.; Pokorny, J.C.; Carlson, B.L.; Zhang, Y.; Nair, A.A.; Lee, J.H.; Yan, H.; Decker, P.A.; et al. Retinoblastoma Binding Protein 4 Modulates Temozolomide Sensitivity in Glioblastoma by Regulating DNA Repair Proteins. Cell. Rep. 2016, 14, 2587–2598. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lo, W.-L.; Hsu, T.-I.; Yang, W.-B.; Kao, T.-J.; Wu, M.-H.; Huang, Y.-N.; Yeh, S.-H.; Chuang, J.-Y. Betulinic Acid-Mediated Tuning of PERK/CHOP Signaling by Sp1 Inhibition as a Novel Therapeutic Strategy for Glioblastoma. Cancers 2020, 12, 981. https://doi.org/10.3390/cancers12040981
Lo W-L, Hsu T-I, Yang W-B, Kao T-J, Wu M-H, Huang Y-N, Yeh S-H, Chuang J-Y. Betulinic Acid-Mediated Tuning of PERK/CHOP Signaling by Sp1 Inhibition as a Novel Therapeutic Strategy for Glioblastoma. Cancers. 2020; 12(4):981. https://doi.org/10.3390/cancers12040981
Chicago/Turabian StyleLo, Wei-Lun, Tsung-I Hsu, Wen-Bin Yang, Tzu-Jen Kao, Ming-Hsiao Wu, Yung-Ning Huang, Shiu-Hwa Yeh, and Jian-Ying Chuang. 2020. "Betulinic Acid-Mediated Tuning of PERK/CHOP Signaling by Sp1 Inhibition as a Novel Therapeutic Strategy for Glioblastoma" Cancers 12, no. 4: 981. https://doi.org/10.3390/cancers12040981
APA StyleLo, W.-L., Hsu, T.-I., Yang, W.-B., Kao, T.-J., Wu, M.-H., Huang, Y.-N., Yeh, S.-H., & Chuang, J.-Y. (2020). Betulinic Acid-Mediated Tuning of PERK/CHOP Signaling by Sp1 Inhibition as a Novel Therapeutic Strategy for Glioblastoma. Cancers, 12(4), 981. https://doi.org/10.3390/cancers12040981