Analysis of the Circadian Regulation of Cancer Hallmarks by a Cross-Platform Study of Colorectal Cancer Time-Series Data Reveals an Association with Genes Involved in Huntington’s Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
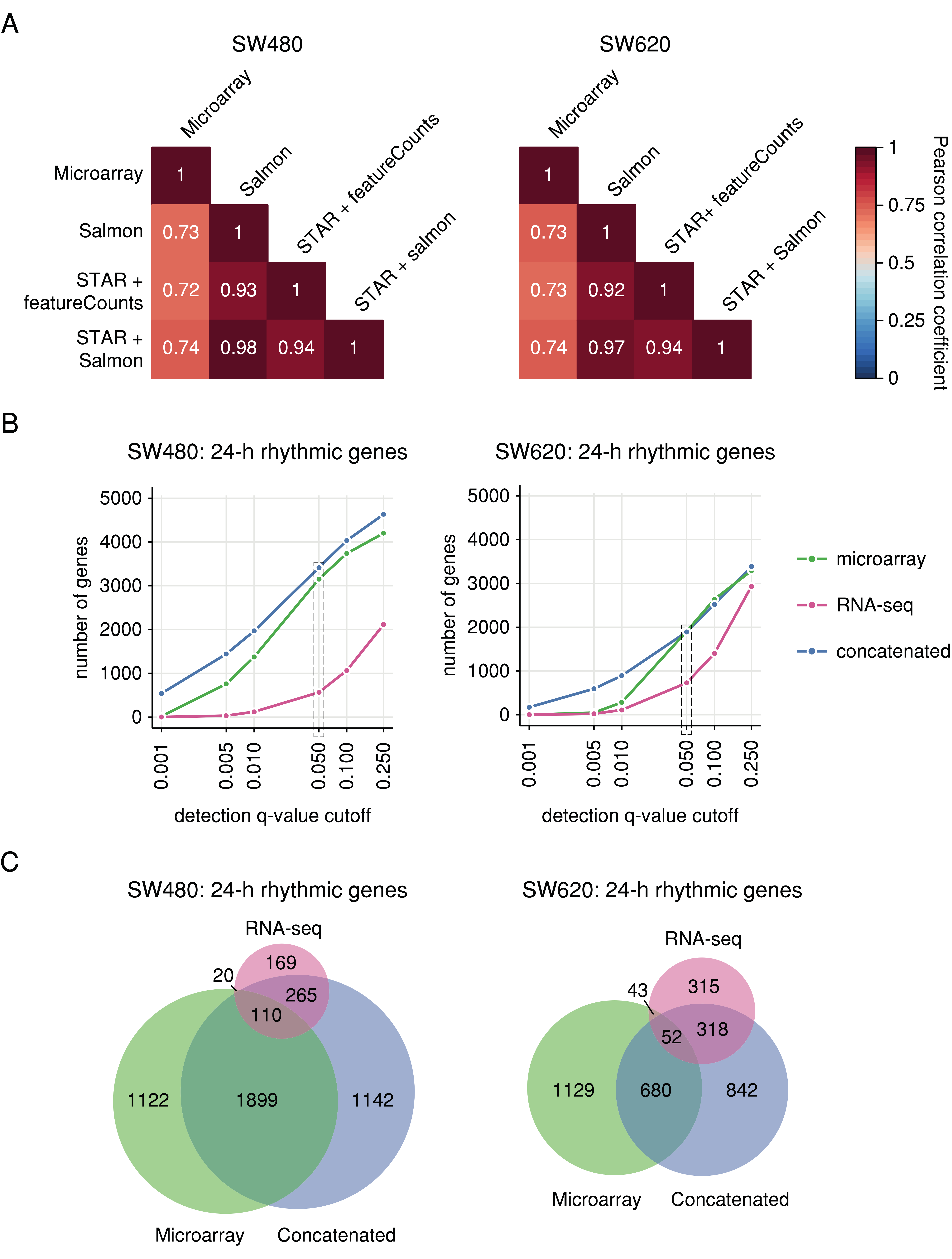
2.1. Correlation of Gene Expression between Circadian Microarray and RNA-seq Data of Human CRC Cell Lines
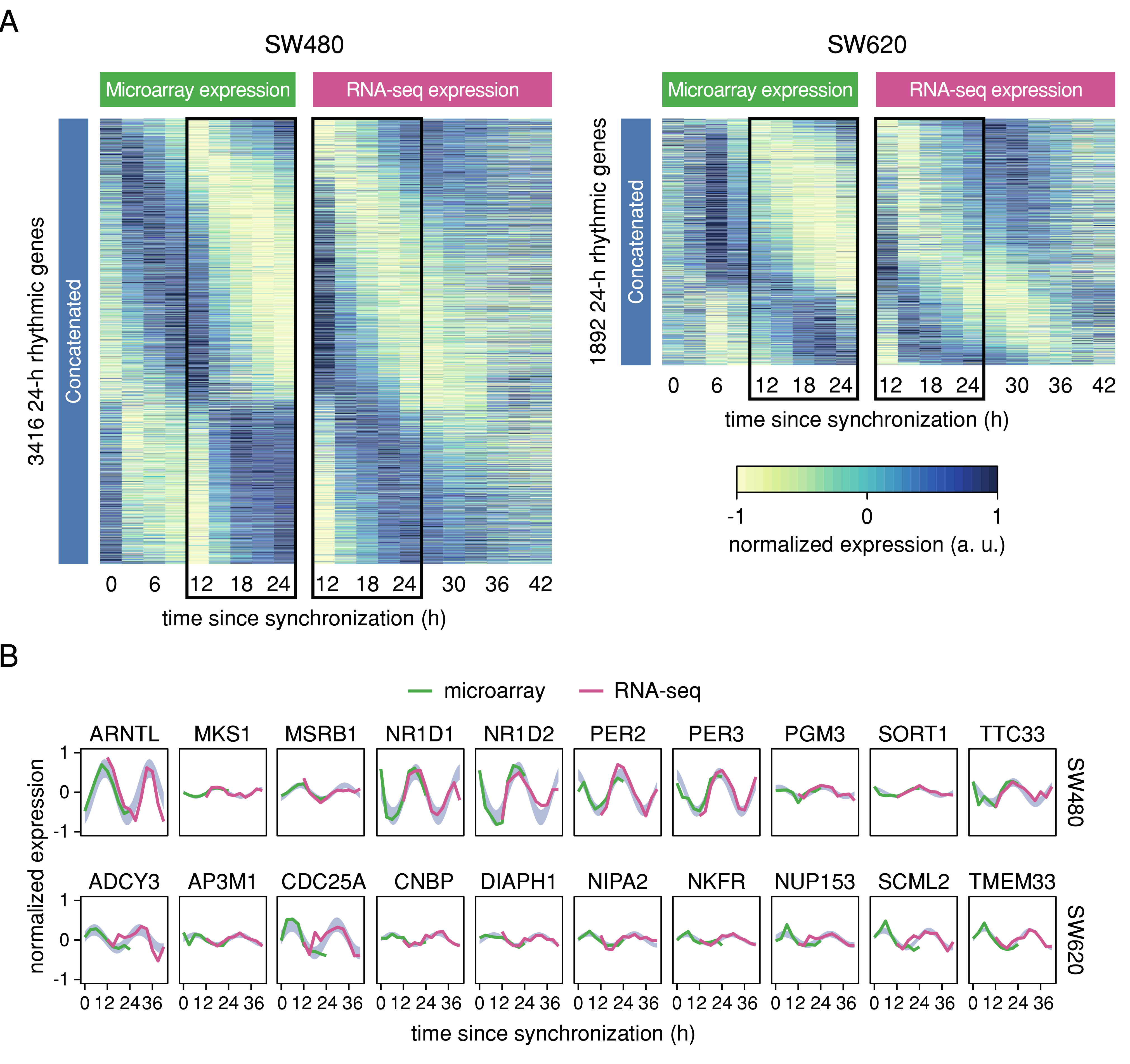
2.2. Cross-Platform Concatenation of Time Series Reveals Robustly Circadian Gene Sets for SW480 and SW620 Cells
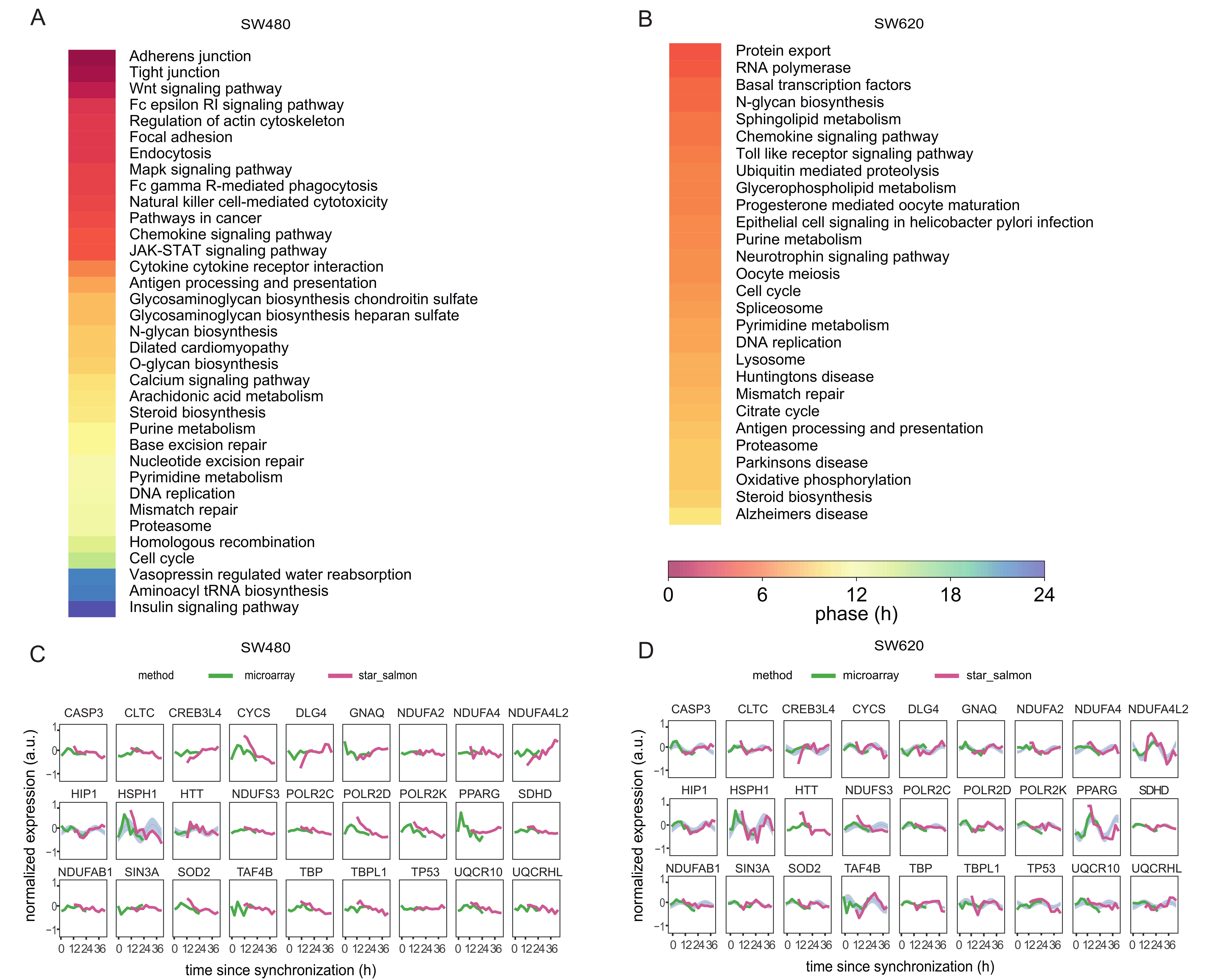
2.3. Oscillatory Expression of Cancer Hallmark-Associated Genes and Drug Target Genes Differs between the CRC Cell Lines
3. Discussion
3.1. Cross-Platform Concatenation of Time Series Reveals Differential Circadian Expression in a CRC Model
3.2. Neurodegeneration-Related Pathways Are Clock-Regulated in the Metastatic CRC Cells
3.3. Circadian Regulation of Cancer Hallmarks and Molecular Drug Targets
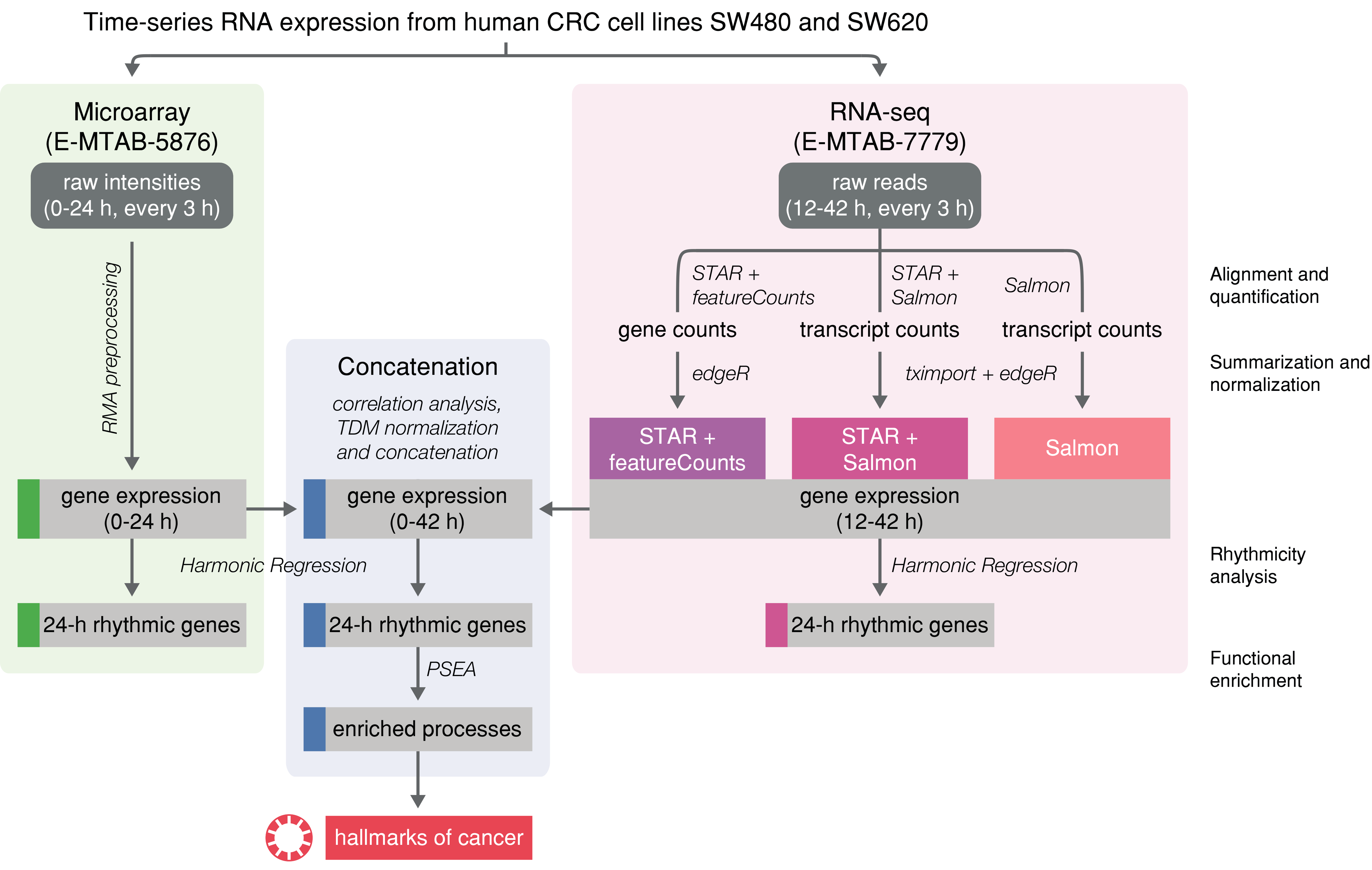
4. Materials and Methods
4.1. Microarray Data Pre-Processing
4.2. RNA-seq Data Pre-Processing
4.3. Cross-Platform Normalization and Concatenation of Time Series
4.4. Rhythmicity Analysis
4.5. Correlation Analysis
4.6. Functional Enrichment
4.7. Analysis of CRC Data From a Cohort of Patients Retrieved from the TCGA Data Base
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, W.K. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; Hickman, J.A. Limits to personalized cancer medicine. N. Engl. J. Med. 2016, 375, 1289–1294. [Google Scholar] [CrossRef] [Green Version]
- Masri, S.; Sassone-Corsi, P. The emerging link between cancer, metabolism, and circadian rhythms. Nat. Med. 2018, 24, 1795–1803. [Google Scholar] [CrossRef]
- Sulli, G.; Lam, M.T.Y.; Panda, S. Interplay between circadian clock and cancer: New frontiers for cancer treatment. Trends Cancer 2019, 5, 475–494. [Google Scholar] [CrossRef]
- Cederroth, C.R.; Albrecht, U.; Bass, J.; Brown, S.A.; Dyhrfjeld-Johnsen, J.; Gachon, F.; Green, C.B.; Hastings, M.H.; Helfrich-Forster, C.; Hogenesch, J.B.; et al. Medicine in the fourth dimension. Cell Metab. 2019, 30, 238–250. [Google Scholar] [CrossRef]
- Pittendrigh, C.S. Circadian Rhythms and the Circadian Organization of Living Systems; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1960. [Google Scholar]
- Antle, M.C.; Silver, R. Neural basis of timing and anticipatory behaviors. Eur. J. Neurosci. 2009, 30, 1643–1649. [Google Scholar] [CrossRef] [Green Version]
- Eckel-Mahan, K.; Sassone-Corsi, P. Metabolism and the circadian clock converge. Physiol. Rev. 2013, 93, 107–135. [Google Scholar] [CrossRef] [PubMed]
- Smarr, B.L.; Jennings, K.J.; Driscoll, J.R.; Kriegsfeld, L.J. A time to remember: The role of circadian clocks in learning and memory. Behav. Neurosci. 2014, 128, 283–303. [Google Scholar] [CrossRef] [Green Version]
- Partch, C.L.; Green, C.B.; Takahashi, J.S. Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 2014, 24, 90–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reppert, S.M.; Weaver, D.R. Coordination of circadian timing in mammals. Nature 2002, 418, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Relogio, A.; Westermark, P.O.; Wallach, T.; Schellenberg, K.; Kramer, A.; Herzel, H. Tuning the mammalian circadian clock: Robust synergy of two loops. PLoS Comput. Biol. 2011, 7, e1002309. [Google Scholar] [CrossRef] [Green Version]
- Ueda, H.R.; Hayashi, S.; Chen, W.; Sano, M.; Machida, M.; Shigeyoshi, Y.; Iino, M.; Hashimoto, S. System-level identification of transcriptional circuits underlying mammalian circadian clocks. Nat. Genet. 2005, 37, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Sancar, A. Regulation of apoptosis by the circadian clock through NF-B signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 12036–12041. [Google Scholar] [CrossRef] [Green Version]
- Sancar, A.; Lindsey-Boltz, L.A.; Kang, T.H.; Reardon, J.T.; Lee, J.H.; Ozturk, N. Circadian clock control of the cellular response to DNA damage. FEBS Lett. 2010, 584, 2618–2625. [Google Scholar] [CrossRef] [Green Version]
- Sancar, A.; Lindsey-Boltz, L.A.; Gaddameedhi, S.; Selby, C.P.; Ye, R.; Chiou, Y.Y.; Kemp, M.G.; Hu, J.; Lee, J.H.; Ozturk, N. Circadian clock, cancer, and chemotherapy. Biochemistry 2015, 54, 110–123. [Google Scholar] [CrossRef]
- Ando, T.; Kawabe, T.; Ohara, H.; Ducommun, B.; Itoh, M.; Okamoto, T. Involvement of the interaction between p21 and proliferating cell nuclear antigen for the maintenance of G2/M arrest after DNA damage. J. Biol. Chem. 2001, 276, 42971–42977. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, T.; Yamaguchi, S.; Mitsui, S.; Emi, A.; Shimoda, F.; Okamura, H. Control mechanism of the circadian clock for timing of cell division In Vivo. Science 2003, 302, 255. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Hsieh, A.L.; Sengupta, A.; Krishnanaiah, S.Y.; Stine, Z.E.; Walton, Z.E.; Gouw, A.M.; Venkataraman, A.; Li, B.; Goraksha-Hicks, P.; et al. MYC disrupts the circadian clock and metabolism in cancer cells. Cell Metab. 2015, 22, 1009–1019. [Google Scholar] [CrossRef] [Green Version]
- Shostak, A.; Diernfellner, A.; Brunner, M. MYC inhibits the clock and supports proliferation. Cell Cycle 2016, 15, 3323–3324. [Google Scholar] [CrossRef] [Green Version]
- Gotoh, T.; Vila-Caballer, M.; Liu, J.; Schiffhauer, S.; Finkielstein, C.V. Association of the circadian factor period 2 to p53 influences p53’s function in DNA-damage signaling. Mol. Biol. Cell 2015, 26, 359–372. [Google Scholar] [CrossRef]
- Gotoh, T.; Vila-Caballer, C.; Santos, C.S.; Liu, J.; Yang, J.; Finkielstein, C.V. The circadian factor period 2 modulates p53 stability and transcriptional activity in unstressed cells. Mol. Biol. Cell 2014, 25, 3081–3093. [Google Scholar] [CrossRef] [Green Version]
- Wood, P.A.; Yang, X.; Taber, A.; Oh, E.Y.; Ansell, C.; Ayers, S.E.; Al-Assaad, Z.; Carnevale, K.; Berger, F.G.; Pena, M.M.; et al. Period 2 mutation accelerates ApcMin/+ tumorigenesis. Mol. Cancer Res. 2008, 6, 1786–1793. [Google Scholar] [CrossRef] [Green Version]
- El-Athman, R.; Genov, N.N.; Mazuch, J.; Zhang, K.; Yu, Y.; Fuhr, L.; Abreu, M.; Li, Y.; Wallach, T.; Kramer, A.; et al. The Ink4a/Arf locus operates as a regulator of the circadian clock modulating RAS activity. PLoS Biol. 2017, 15, 34. [Google Scholar] [CrossRef] [Green Version]
- Kowalskaa, E.; Ripperger, J.A.; Dominik, C.; Brueggera, H.P.; Buchd, T.; Birchlerd, T.; Muellere, A.; Albrecht, U.; Contaldoc, C.; Browna, S.A. NONO couples the circadian clock to the cell cycle. PNAS 2013, 110, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Pelicano, H.; Huang, P.; Liu, J.; Lee, C.C. The circadian gene period2 plays a role in tumor suppression and DNA damage response In Vivo. Cell 2002, 111, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Papagiannakopoulos, T.; Bauer, M.R.; Davidson, S.M.; Heimann, M.; Subbaraj, L.; Bhutkar, A.; Bartlebaugh, J.; Heiden, V.M.G.; Jacks, T. Circadian rhythm disruption promotes lung tumorigenesis. Cell Metab. 2016, 24, 324–331. [Google Scholar] [CrossRef] [Green Version]
- Dallmann, R.; Okyar, A.; Lévi, F. Dosing-time makes the poison: Circadian regulation and pharmacotherapy. Trends Mol. Med. 2016, 22, 430–445. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, R.; Childs, L.; Thomas, P.; Abreu, M.; Fuhr, L.; Herzel, H.; Leser, U.; Relogio, A. Assembly of a comprehensive regulatory network for the mammalian circadian clock: A bioinformatics approach. PLoS ONE 2015, 10, e0126283. [Google Scholar] [CrossRef]
- El-Athman, R.; Fuhr, L.; Relógio, A. A systems-level analysis reveals circadian regulation of splicing in colorectal cancer. EBioMedicine 2018, 33, 68–81. [Google Scholar] [CrossRef]
- El-Athman, R.; Knezevic, D.; Fuhr, L.; Relógio, A. A computational analysis of alternative splicing across mammalian tissues reveals circadian and ultradian rhythms in splicing events. Int. J. Mol. Sci. 2019, 20, 3977. [Google Scholar] [CrossRef] [Green Version]
- Cela, O.; Scrima, R.; Pazienza, V.; Merla, G.; Benegiamo, G.; Augello, B.; Fugetto, S.; Menga, M.; Rubino, R.; Fuhr, L.; et al. Clock genes-dependent acetylation of complex I sets rhythmic activity of mitochondrial OxPhos. Biochim. Biophys. Acta 2016, 1863, 596–606. [Google Scholar] [CrossRef]
- Fuhr, L.; Abreu, M.; Carbone, A.; El-Athman, R.; Bianchi, F.; Laukkanen, M.O.; Mazzoccoli, G.; Relogio, A. The interplay between colon cancer cells and tumour-associated stromal cells impacts the biological clock and enhances malignant phenotypes. Cancers 2019, 11, 988. [Google Scholar] [CrossRef] [Green Version]
- Scrima, R.; Cela, O.; Merla, G.; Augello, B.; Rubino, R.; Quarato, G.; Fugetto, S.; Menga, M.; Fuhr, L.; Relogio, A.; et al. Clock-genes and mitochondrial respiratory activity: Evidence of a reciprocal interplay. Biochim Biophys Acta 2016, 1857, 1344–1351. [Google Scholar] [CrossRef]
- Fuhr, L.; El-Athman, R.; Scrima, R.; Cela, O.; Carbone, A.; Knoop, H.; Li, Y.; Hoffmann, K.; Laukkanen, M.O.; Corcione, F.; et al. The circadian clock regulates metabolic phenotype rewiring via hkdc1 and modulates tumor progression and drug response in colorectal cancer. EBioMedicine 2018, 33, 105–121. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Grant, G.R.; Hogenesch, J.B.; Hughes, M.E. Considerations for RNA-seq analysis of circadian rhythms. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 551, pp. 349–367. [Google Scholar]
- Thompson, J.A.; Tan, J.; Greene, C.S. Cross-platform normalization of microarray and RNA-seq data for machine learning applications. Peer J. 2016, 4, e1621. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Li, J.; Zhou, F.; Huang, Q.; Zhang, J.; Guo, X.; Lyu, Z.; Zhang, H.; Xing, J. NPAS2 promotes cell survival of hepatocellular carcinoma by transactivating CDC25A. Cell Death Dis. 2017, 8, e2704. [Google Scholar] [CrossRef]
- Jang, A.R.; Moravcevic, K.; Saez, L.; Young, M.W.; Sehgal, A. Drosophila TIM binds importin α1, and acts as an adapter to transport PER to the nucleus. PLoS Genet. 2015, 11, e1004974. [Google Scholar] [CrossRef]
- Musiek, E.S. Circadian clock disruption in neurodegenerative diseases: cause and effect? Front Pharm. 2015, 6, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNulty, P.; Pilcher, R.; Ramesh, R.; Necuiniate, R.; Hughes, A.; Farewell, D.; Holmans, P.; Jones, L.; REGISTRY Investigators of the European Huntington’s Disease Network. Reduced cancer incidence in huntington’s disease: Analysis in the registry study. J. Huntingt. Dis. 2018, 7, 209–222. [Google Scholar] [CrossRef]
- Sorensen, S.A.; Fenger, K. Causes of death in patients with Huntington’s disease and in unaffected first degree relatives. J. Med. Genet. 1992, 29, 911–914. [Google Scholar] [CrossRef] [Green Version]
- Kalathur, R.K.; Hernandez-Prieto, M.A.; Futschik, M.E. Huntington’s disease and its therapeutic target genes: A global functional profile based on the HD research crossroads database. BMC Neurol. 2012, 12, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.H.; Yu, Z.X.; Li, C.L.; Nguyen, H.P.; Zhou, Y.X.; Deng, C.; Li, X.J. Lack of huntingtin-associated protein-1 causes neuronal death resembling hypothalamic degeneration in huntington’s disease. J. Neurosci. 2003, 23, 6956–6964. [Google Scholar] [CrossRef] [Green Version]
- Scior, A.; Buntru, A.; Arnsburg, K.; Ast, A.; Iburg, M.; Juenemann, K.; Pigazzini, M.L.; Mlody, B.; Puchkov, D.; Priller, J.; et al. Complete suppression of Htt fibrilization and disaggregation of Htt fibrils by a trimeric chaperone complex. EMBO J. 2018, 37, 282–299. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Cancer Genome Atlas Research Network. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, 359–386. [Google Scholar] [CrossRef]
- Levi, M.F.; Missef, J.-L.; Brienza, S.; Adam, R.; Metzger, G.; Itzakhi, M.; Caussanel, J.-P.; Kunstlinger, F.; Lecouturier, S.; Descorps-Declere, A.; et al. A chronopharmacologic phase ii clinical trial with 5-fluorouracil, folinic acid, and oxaliplatin using an ambulatory multichannel programmable pump. Am. Cancer Soc. Cancer 1992, 69, 893–899. [Google Scholar]
- Filipski, E.; Berland, E.; Ozturk, N.; Guettier, C.; van der Horst, G.T.; Levi, F.; Okyar, A. Optimization of irinotecan chronotherapy with P-glycoprotein inhibition. Toxicol. Appl. Pharm. 2014, 274, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Innominato, P.F.; Focan, C.; Gorlia, T.; Moreau, T.; Garufi, C.; Waterhouse, J.; Giacchetti, S.; Coudert, B.; Iacobelli, S.; Genet, D.; et al. Circadian rhythm in rest and activity: A biological correlate of quality of life and a predictor of survival in patients with metastatic colorectal cancer. Cancer Res. 2009, 69, 4700–4707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, S.; Antoch, M.P.; Miller, B.H.; Schook, A.B.; Schultz, P.G.; Su, A.I.; Straume, M.; Kay, S.A.; Takahashi, J.S.; Hogenesch, J.B. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 2002, 109, 307–320. [Google Scholar] [CrossRef] [Green Version]
- Mermet, J.; Yeung, J.; Naef, F. Systems chronobiology: Global analysis of gene regulation in a 24-hour periodic world. Cold Spring Harb. Perspect. Biol. 2017, 9, a028720. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.E.; Abruzzi, K.C.; Allada, R.; Anafi, R.; Arpat, A.B.; Asher, G.; Baldi, P.; De Bekker, C.; Bell-Pedersen, D.; Blau, J. Guidelines for genome-scale analysis of biological rhythms. J. Biol. Rhythm. 2017, 32, 380–393. [Google Scholar] [CrossRef] [Green Version]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Prim. 2015, 1, 15005. [Google Scholar] [CrossRef]
- Thion, M.S.; Humbert, S. Cancer: From wild-type to mutant huntingtin. J. Huntingt. Dis 2018, 7, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Elias, S.; McGuire, J.R.; Yu, H.; Humbert, S. Huntingtin is required for epithelial polarity through RAB11A-mediated apical trafficking of PAR3-aPKC. PLoS Biol. 2015, 13, e1002142. [Google Scholar] [CrossRef]
- Moreira Sousa, C.; McGuire, J.R.; Thion, M.S.; Gentien, D.; de la Grange, P.; Tezenas du Montcel, S.; Vincent-Salomon, A.; Durr, A.; Humbert, S. The Huntington disease protein accelerates breast tumour development and metastasis through ErbB2/HER2 signalling. EMBO Mol. Med. 2013, 5, 309–325. [Google Scholar] [CrossRef]
- Naia, L.; Rego, A.C. Sirtuins: Double players in huntington’s disease. Biochim. Biophys. Acta 2015, 1852, 2183–2194. [Google Scholar] [CrossRef] [Green Version]
- Genetic Modifiers of Huntington’s Disease Consortium. Identification of genetic factors that modify clinical onset of huntington’s disease. Cell 2015, 162, 516–526. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Shen, Z.; Hu, Y.; Xiao, Q.; Bei, D.; Shen, X.; Ding, K. Association between MutL homolog 1 polymorphisms and the risk of colorectal cancer: A meta-analysis. J. Cancer Res. Clin. Oncol. 2015, 141, 2147–2158. [Google Scholar] [CrossRef]
- Peng, H.X.; Xu, X.; Yang, R.; Chu, Y.M.; Yang, D.M.; Xu, Y.; Zhou, F.L.; Ma, W.Z.; Zhang, X.J.; Guan, M.; et al. Molecular analysis of MLH1 variants in Chinese sporadic colorectal cancer patients. Genet. Mol. Res. 2016, 15, 1–10. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef]
- De Goede, P.; Wefers, J.; Brombacher, E.C.; Schrauwen, P.; Kalsbeek, A. Circadian rhythms in mitochondrial respiration. J. Mol. Endocrinol. 2018, 60, 115–130. [Google Scholar] [CrossRef]
- Suhane, S.; Berel, D.; Ramanujan, V.K. Biomarker signatures of mitochondrial NDUFS3 in invasive breast carcinoma. Biochem. Biophys. Res. Commun. 2011, 412, 590–595. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Ye, Z.; Hensch, T.K. Clock genes control cortical critical period timing. Neuron 2015, 86, 264–275. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Lee, C.C. The circadian clock: Pacemaker and tumour suppressor. Nat. Rev. Cancer 2003, 3, 350–361. [Google Scholar] [CrossRef]
- Gaspar, L.S.; Alvaro, A.R.; Carmo-Silva, S.; Mendes, A.F.; Relogio, A.; Cavadas, C. The importance of determining circadian parameters in pharmacological studies. Br. J. Pharm. 2019, 176, 2827–2847. [Google Scholar] [CrossRef]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT pathway as a target for cancer treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef]
- Slomovitz, B.M.; Coleman, R.L. The PI3K/AKT/mTOR pathway as a therapeutic target in endometrial cancer. Clin. Cancer Res. 2012, 18, 5856–5864. [Google Scholar] [CrossRef] [Green Version]
- Jung-Hynes, B.; Ahmad, N. SIRT1 controls circadian clock circuitry and promotes cell survival: A connection with age-related neoplasms. FASEB J. 2009, 23, 2803–2809. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Begley, M.; Michowski, W.; Inuzuka, H.; Ginzberg, M.; Gao, D.; Tsou, P.; Gan, W.; Papa, A.; Kim, B.M.; et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 2014, 508, 541–545. [Google Scholar] [CrossRef]
- Luciano, A.K.; Zhou, W.; Santana, J.M.; Kyriakides, C.; Velazquez, H.; Sessa, W.C. CLOCK phosphorylation by AKT regulates its nuclear accumulation and circadian gene expression in peripheral tissues. J. Biol. Chem. 2018, 293, 9126–9136. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Lahens, N.F.; Zhang, S.; Bedont, J.; Field, J.M.; Sehgal, A. G1/S cell cycle regulators mediate effects of circadian dysregulation on tumor growth and provide targets for timed anticancer treatment. PLoS Biol. 2019, 17, e3000228. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Xiang, Y.; Ozguc, F.M.; Kim, Y.; Liu, C.J.; Park, P.K.; Hu, Q.; Diao, L.; Lou, Y.; Lin, C.; et al. The Genomic landscape and pharmacogenomic interactions of clock genes in cancer chronotherapy. Cell Syst. 2018, 6, 314–328. [Google Scholar] [CrossRef] [Green Version]
- Walton, Z.E.; Patel, C.H.; Brooks, R.C.; Yu, Y.; Ibrahim-Hashim, A.; Riddle, M.; Porcu, A.; Jiang, T.; Ecker, B.L.; Tameire, F.; et al. Acid suspends the circadian clock in hypoxia through inhibition of mTOR. Cell 2018, 174, 72–87. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in growth, metabolism, and disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, H.; Matsunaga, N.; Fujioka, T.; Okazaki, F.; Akagawa, Y.; Tsurudome, Y.; Ono, M.; Kuwano, M.; Koyanagi, S.; Ohdo, S. Circadian regulation of mTOR by the ubiquitin pathway in renal cell carcinoma. Cancer Res. 2014, 74, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Ramanathan, C.; Kathale, N.D.; Liu, D.; Lee, C.; Freeman, D.A.; Hogenesch, J.B.; Cao, R.; Liu, A.C. mTOR signaling regulates central and peripheral circadian clock function. PLoS Genet. 2018, 14, e1007369. [Google Scholar] [CrossRef]
- Cao, R. mTOR signaling, translational control, and the circadian clock. Front Genet. 2018, 9, 367. [Google Scholar] [CrossRef] [PubMed]
- Easton, J.B.; Houghton, P.J. mTOR and cancer therapy. Oncogene 2006, 25, 6436–6446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Relogio, A.; Thomas, P.; Medina-Perez, P.; Reischl, S.; Bervoets, S.; Gloc, E.; Riemer, P.; Mang-Fatehi, S.; Maier, B.; Schafer, R.; et al. Ras-mediated deregulation of the circadian clock in cancer. PLoS Genet. 2014, 10, e1004338. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-K.; Fu, L.-W. Interaction of tyrosine kinase inhibitors with the MDR-Related ABC transporter proteins. Curr. Drug Metab. 2010, 11, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Anreddy, N.; Gupta, P.; Kathawala, R.J.; Patel, A.; Wurpel, J.N.; Chen, Z.S. Tyrosine kinase inhibitors as reversal agents for ABC transporter mediated drug resistance. Molecules 2014, 19, 13848–13877. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.; An, F.; Xu, X.; Zhao, L.; Liu, L.; Liu, N.; Wang, P.; Liu, J.; Wang, L.; Li, M. Chronopharmacodynamics and mechanisms of antitumor effect induced by erlotinib in xenograft-bearing nude mice. Biochem. Biophys. Res. Commun. 2015, 460, 362–367. [Google Scholar] [CrossRef]
- Liu, J.; Wang, C.Y.; Ji, S.G.; Xu, X.; Wang, P.P.; Zhang, B.; Zhao, L.Y.; Liu, L.; Lin, P.P.; Liu, L.K.; et al. Chronopharmacokinetics of erlotinib and circadian rhythms of related metabolic enzymes in lewis tumor-bearing mice. Eur. J. Drug Metab. Pharm. 2016, 41, 627–635. [Google Scholar] [CrossRef]
- Ortiz-Tudela, E.; Mteyrek, A.; Ballesta, A.; Innominato, P.F.; Lévi, F. Cancer chronotherapeutics: Experimental, theoretical, and clinical aspects. In Circadian Clocks; Kramer, A., Merrow, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 261–288. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31, 15. [Google Scholar] [CrossRef]
- Bolstad, B.M.; Irizarry, R.A.; Åstrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Lück, S.; Thurley, K.; Thaben, P.F.; Westermark, P.O. Rhythmic degradation explains and unifies circadian transcriptome and proteome data. Cell Rep. 2014, 9, 741–751. [Google Scholar] [CrossRef] [Green Version]
- Agostinelli, C.; Lund, U. R Package Circular: Circular Statistics (Version 0.4–93); Department of Environmental Sciences, Informatics and Statistics, Ca’Foscari University: Venice, Italy, 2017. [Google Scholar]
- Zhang, R.; Podtelezhnikov, A.A.; Hogenesch, J.B.; Anafi, R.C. Discovering biology in periodic data through phase set enrichment analysis (PSEA). J. Biol. Rhythm. 2016, 31, 244–257. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic. Acids Res. 2016, 44, 71. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.C.; Colaprico, A.; Olsen, C.; D’Angelo, F.; Bontempi, G.; Ceccarelli, M.; Noushmehr, H. TCGA workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Research 2016, 5, 1542. [Google Scholar] [CrossRef] [PubMed]
- Anaya, J. OncoLnc: Linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput. Sci. 2016, 2, 67. [Google Scholar] [CrossRef] [Green Version]
- Sahar, S.; Sassone-Corsi, P. Metabolism and cancer: The circadian clock connection. Nat. Rev. Cancer 2009, 9, 886–896. [Google Scholar] [CrossRef]
- El-Athman, R.; Relogio, A. Escaping circadian regulation: An emerging hallmark of cancer? Cell Syst. 2018, 6, 266–267. [Google Scholar] [CrossRef] [Green Version]
- Giacchetti, S.; Perpoint, B.; Zidani, R.; Le Bail, N.; Faggiuolo, R.; Focan, C.; Chollet, P.; Llory, J.F.; Letourneau, Y.; Coudert, B.; et al. Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil–leucovorin as first-line treatment of metastatic colorectal cancer. J. Clin. Oncol. 2016, 18, 136. [Google Scholar] [CrossRef]
- Gholam, D.; Giacchetti, S.; Brezault-Bonnet, C.; Bouchahda, M.; Hauteville, D.; Adam, R.; Ducot, B.; Ghemard, O.; Kustlinger, F.; Jasmin, C.; et al. Chronomodulated irinotecan, oxaliplatin, and leucovorin-modulated 5-Fluorouracil as ambulatory salvage therapy in patients with irinotecan- and oxaliplatin-resistant metastatic colorectal cancer. Oncologist 2006, 11, 1072–1080. [Google Scholar] [CrossRef] [Green Version]
- Lévi, F.; Zidani, R.; Misset, J.-L. Randomised multicentre trial of chronotherapy with oxaliplatin, fluorouracil, and folinic acid in metastatic colorectal cancer. Lancet 1997, 350, 681–686. [Google Scholar] [CrossRef]
- Giacchetti, S.; Dugue, P.A.; Innominato, P.F.; Bjarnason, G.A.; Focan, C.; Garufi, C.; Tumolo, S.; Coudert, B.; Iacobelli, S.; Smaaland, R.; et al. Sex moderates circadian chemotherapy effects on survival of patients with metastatic colorectal cancer: A meta-analysis. Ann. Oncol. 2012, 23, 3110–3116. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yalçin, M.; El-Athman, R.; Ouk, K.; Priller, J.; Relógio, A. Analysis of the Circadian Regulation of Cancer Hallmarks by a Cross-Platform Study of Colorectal Cancer Time-Series Data Reveals an Association with Genes Involved in Huntington’s Disease. Cancers 2020, 12, 963. https://doi.org/10.3390/cancers12040963
Yalçin M, El-Athman R, Ouk K, Priller J, Relógio A. Analysis of the Circadian Regulation of Cancer Hallmarks by a Cross-Platform Study of Colorectal Cancer Time-Series Data Reveals an Association with Genes Involved in Huntington’s Disease. Cancers. 2020; 12(4):963. https://doi.org/10.3390/cancers12040963
Chicago/Turabian StyleYalçin, Müge, Rukeia El-Athman, Koliane Ouk, Josef Priller, and Angela Relógio. 2020. "Analysis of the Circadian Regulation of Cancer Hallmarks by a Cross-Platform Study of Colorectal Cancer Time-Series Data Reveals an Association with Genes Involved in Huntington’s Disease" Cancers 12, no. 4: 963. https://doi.org/10.3390/cancers12040963
APA StyleYalçin, M., El-Athman, R., Ouk, K., Priller, J., & Relógio, A. (2020). Analysis of the Circadian Regulation of Cancer Hallmarks by a Cross-Platform Study of Colorectal Cancer Time-Series Data Reveals an Association with Genes Involved in Huntington’s Disease. Cancers, 12(4), 963. https://doi.org/10.3390/cancers12040963