Towards Physiologically and Tightly Regulated Vectored Antibody Therapies



Abstract
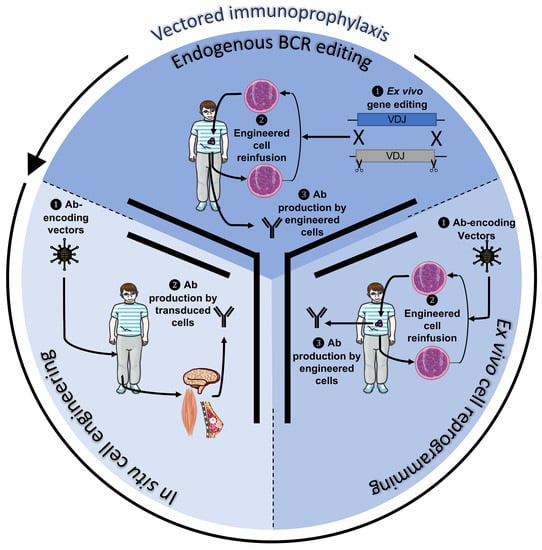
1. Introduction
2. Active Immunotherapy Approaches for Antibody Secretion (Vectored Immunoprophylaxis)
2.1. General Notions of Vector Design
2.2. Applications for Cancer Therapy
2.3. Immune Consequences after Vector Infusion
3. Transgenic Antibody Expression by B Cells
3.1. Constitutive Expression of Transgenic Antibodies by B Cells
3.2. Toward the Physiologically Regulated Expression of Ectopic Antibodies
4. Direct Gene Editing of the Endogenous BCR
4.1. General Principle
4.2. Current Challenges
5. Conclusion and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Antibodies to Restrict Vector Tropism to Tumor Cells in Gene Therapy
Appendix B. New Tools for Genetic Modifications of B Cells
References
- Coley, W.B. Contribution to the knowledge on Sarcoma. Ann. Surg. 1891, 14, 199–220. [Google Scholar] [CrossRef]
- Coley, W.B. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. Clin. Orthop. 1893, 1991, 3–11. [Google Scholar]
- Fehleisen, F. Die Aetiologie des Erysipels; Theodor Fisch: Berlin, Germany, 1883. [Google Scholar]
- Powles, L.M.; Wilson, K.; Xiang, S.D.; Selomulya, C.; Plebanski, M. New strategies in vaccine design for the induction of CD8 T cell responses using biodegradable iron oxide nanoparticles. J. Immunol. 2017, 198, 79.23. [Google Scholar]
- Siegrist, C.-A. Vaccine immunology. In Vaccines; Elsevier: Amsterdam, The Netherlands, 2013; pp. 14–32. ISBN 978-1-4557-0090-5. [Google Scholar]
- Aspeslagh, S.; Postel-Vinay, S.; Rusakiewicz, S.; Soria, J.-C.; Zitvogel, L.; Marabelle, A. Rationale for anti-OX40 cancer immunotherapy. Eur. J. Cancer 2016, 52, 50–66. [Google Scholar] [CrossRef] [PubMed]
- Marhelava, K.; Pilch, Z.; Bajor, M.; Graczyk-Jarzynka, A.; Zagozdzon, R. Targeting Negative and Positive Immune Checkpoints with Monoclonal Antibodies in Therapy of Cancer. Cancers 2019, 11, 1756. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 165. [Google Scholar] [CrossRef]
- Macor, P.; Secco, E.; Mezzaroba, N.; Zorzet, S.; Durigutto, P.; Gaiotto, T.; De Maso, L.; Biffi, S.; Garrovo, C.; Capolla, S.; et al. Bispecific antibodies targeting tumor-associated antigens and neutralizing complement regulators increase the efficacy of antibody-based immunotherapy in mice. Leukemia 2015, 29, 406–414. [Google Scholar] [CrossRef]
- Wu, H.; Yao, L.; Chou, L.; Yang, J.-H.; Zhang, Y.-X.; Li, X.-L.; Shan, B.-E. Construction and functional analysis of an anti-human cervical carcinoma/anti-human CD3 single-chain bispecific antibody. Mol. Med. Rep. 2016, 14, 804–810. [Google Scholar] [CrossRef]
- Wu, L.; Seung, E.; Xu, L.; Rao, E.; Lord, D.M.; Wei, R.R.; Cortez-Retamozo, V.; Ospina, B.; Posternak, V.; Ulinski, G.; et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat. Cancer 2019. [Google Scholar] [CrossRef]
- Kellner, C.; Derer, S.; Klausz, K.; Rosskopf, S.; Wirt, T.; Rösner, T.; Otte, A.; Cappuzzello, E.; Peipp, M. Fc Glyco- and Fc Protein-Engineering: Design of Antibody Variants with Improved ADCC and CDC Activity. In Antibody Engineering; Nevoltris, D., Chames, P., Eds.; Springer: New York, NY, USA, 2018; Volume 1827, pp. 381–397. ISBN 978-1-4939-8647-7. [Google Scholar]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef]
- Saunders, K.O. Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front. Immunol. 2019, 10, 1296. [Google Scholar] [CrossRef] [PubMed]
- Zalevsky, J.; Chamberlain, A.K.; Horton, H.M.; Karki, S.; Leung, I.W.L.; Sproule, T.J.; Lazar, G.A.; Roopenian, D.C.; Desjarlais, J.R. Enhanced antibody half-life improves in vivo activity. Nat. Biotechnol. 2010, 28, 157–159. [Google Scholar] [CrossRef]
- Guan, M.; Zhou, Y.-P.; Sun, J.-L.; Chen, S.-C. Adverse Events of Monoclonal Antibodies Used for Cancer Therapy. BioMed Res. Int. 2015, 2015, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Chaudhary, N.; Garg, M.; Floudas, C.S.; Soni, P.; Chandra, A.B. Current Diagnosis and Management of Immune Related Adverse Events (irAEs) Induced by Immune Checkpoint Inhibitor Therapy. Front. Pharmacol. 2017, 8. [Google Scholar]
- Lewis, A.D.; Chen, R.; Montefiori, D.C.; Johnson, P.R.; Clark, K.R. Generation of Neutralizing Activity against Human Immunodeficiency Virus Type 1 in Serum by Antibody Gene Transfer. J. Virol. 2002, 76, 8769–8775. [Google Scholar] [CrossRef]
- Brady, J.M.; Baltimore, D.; Balazs, A.B. Antibody gene transfer with adeno-associated viral vectors as a method for HIV prevention. Immunol. Rev. 2017, 275, 324–333. [Google Scholar] [CrossRef]
- Fuchs, S.P.; Martinez-Navio, J.M.; Piatak, M.; Lifson, J.D.; Gao, G.; Desrosiers, R.C. AAV-Delivered Antibody Mediates Significant Protective Effects against SIVmac239 Challenge in the Absence of Neutralizing Activity. PLOS Pathog. 2015, 11, e1005090. [Google Scholar] [CrossRef]
- De Jong, Y.P.; Dorner, M.; Mommersteeg, M.C.; Xiao, J.W.; Balazs, A.B.; Robbins, J.B.; Winer, B.Y.; Gerges, S.; Vega, K.; Labitt, R.N.; et al. Broadly neutralizing antibodies abrogate established hepatitis C virus infection. Sci. Transl. Med. 2014, 6, ra129–ra254. [Google Scholar] [CrossRef]
- De Felipe, P. Skipping the co-expression problem: The new 2A “CHYSEL” technology. Genet. Vaccines Ther. 2004, 2, 13. [Google Scholar] [CrossRef]
- De Felipe, P.; Hughes, L.E.; Ryan, M.D.; Brown, J.D. Co-translational, Intraribosomal Cleavage of Polypeptides by the Foot-and-mouth Disease Virus 2A Peptide. J. Biol. Chem. 2003, 278, 11441–11448. [Google Scholar] [CrossRef] [PubMed]
- Szymczak, A.L.; Vignali, D.A. Development of 2A peptide-based strategies in the design of multicistronic vectors. Expert Opin. Biol. Ther. 2005, 5, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Doronina, V.A.; de Felipe, P.; Wu, C.; Sharma, P.; Sachs, M.S.; Ryan, M.D.; Brown, J.D. Dissection of a co-translational nascent chain separation event. Biochem. Soc. Trans. 2008, 36, 712–716. [Google Scholar] [CrossRef]
- Doronina, V.A.; Wu, C.; de Felipe, P.; Sachs, M.S.; Ryan, M.D.; Brown, J.D. Site-Specific Release of Nascent Chains from Ribosomes at a Sense Codon. Mol. Cell. Biol. 2008, 28, 4227–4239. [Google Scholar] [CrossRef]
- Atkins, J.F.; Wills, N.M.; Loughran, G.; Wu, C.-Y.; Parsawar, K.; Ryan, M.D.; Wang, C.-H.; Nelson, C.C. A case for “StopGo”: Reprogramming translation to augment codon meaning of GGN by promoting unconventional termination (Stop) after addition of glycine and then allowing continued translation (Go). RNA 2007, 13, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hicks, M.J.; Kaminsky, S.M.; Moore, M.A.S.; Crystal, R.G.; Rafii, A. AAV-mediated persistent bevacizumab therapy suppresses tumor growth of ovarian cancer. Gynecol. Oncol. 2014, 135, 325–332. [Google Scholar] [CrossRef]
- Zafir-Lavie, I.; Sherbo, S.; Goltsman, H.; Badinter, F.; Yeini, E.; Ofek, P.; Miari, R.; Tal, O.; Liran, A.; Shatil, T.; et al. Successful intracranial delivery of trastuzumab by gene-therapy for treatment of HER2-positive breast cancer brain metastases. J. Controlled Release 2018, 291, 80–89. [Google Scholar] [CrossRef]
- Rothwell, W.T.; Bell, P.; Richman, L.K.; Limberis, M.P.; Tretiakova, A.P.; Li, M.; Wilson, J.M. Intrathecal Viral Vector Delivery of Trastuzumab Prevents or Inhibits Tumor Growth of Human HER2-Positive Xenografts in Mice. Cancer Res. 2018, 78, 6171–6182. [Google Scholar] [CrossRef]
- Ballesteros-Briones, M.C.; Martisova, E.; Casales, E.; Silva-Pilipich, N.; Buñuales, M.; Galindo, J.; Mancheño, U.; Gorraiz, M.; Lasarte, J.J.; Kochan, G.; et al. Short-Term Local Expression of a PD-L1 Blocking Antibody from a Self-Replicating RNA Vector Induces Potent Antitumor Responses. Mol. Ther. 2019, 27, 1892–1905. [Google Scholar] [CrossRef]
- Hamilton, J.R.; Vijayakumar, G.; Palese, P. A Recombinant Antibody-Expressing Influenza Virus Delays Tumor Growth in a Mouse Model. Cell Rep. 2018, 22, 1–7. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Nagasato, M.; Yoshida, T.; Aoki, K. Recent advances in genetic modification of adenovirus vectors for cancer treatment. Cancer Sci. 2017, 108, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Lévy, C.; Verhoeyen, E.; Cosset, F.-L. Surface engineering of lentiviral vectors for gene transfer into gene therapy target cells. Curr. Opin. Pharmacol. 2015, 24, 79–85. [Google Scholar] [CrossRef]
- Basner-Tschakarjan, E.; Mingozzi, F. Cell-Mediated Immunity to AAV Vectors, Evolving Concepts and Potential Solutions. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- György, B.; Fitzpatrick, Z.; Crommentuijn, M.H.W.; Mu, D.; Maguire, C.A. Naturally enveloped AAV vectors for shielding neutralizing antibodies and robust gene delivery in vivo. Biomaterials 2014, 35, 7598–7609. [Google Scholar] [CrossRef]
- Croyle, M.A.; Callahan, S.M.; Brunner, L.J.; Wilson, J.M.; Kobinger, G.P. PEGylation of VSV-G Pseudotyped Lentiviral Vectors Prevents Inactivation by Complement. Mol. Ther. 2002, 5, S291. [Google Scholar]
- Yao, T.; Zhou, X.; Zhang, C.; Yu, X.; Tian, Z.; Zhang, L.; Zhou, D. Site-Specific PEGylated Adeno-Associated Viruses with Increased Serum Stability and Reduced Immunogenicity. Molecules 2017, 22, 1155. [Google Scholar] [CrossRef] [PubMed]
- Muthumani, K.; Marnin, L.; Kudchodkar, S.B.; Perales-Puchalt, A.; Choi, H.; Agarwal, S.; Scott, V.L.; Reuschel, E.L.; Zaidi, F.I.; Duperret, E.K.; et al. Novel prostate cancer immunotherapy with a DNA-encoded anti-prostate-specific membrane antigen monoclonal antibody. Cancer Immunol. Immunother. 2017, 66, 1577–1588. [Google Scholar] [CrossRef]
- Xing, M.; Wang, X.; Chi, Y.; Zhou, D. Gene therapy for colorectal cancer using adenovirus-mediated full-length antibody, cetuximab. Oncotarget 2016, 7, 28262–28272. [Google Scholar] [CrossRef]
- Liu, F.-R.; Bai, S.; Feng, Q.; Pan, X.-Y.; Song, S.-L.; Fang, H.; Cui, J.; Yang, J.-L. Anti-colorectal cancer effects of anti-p21Ras scFv delivered by the recombinant adenovirus KGHV500 and cytokine-induced killer cells. BMC Cancer 2018, 18, 1087. [Google Scholar] [CrossRef]
- Rao, B.; Gao, Y.; Zhou, Q.; Xiao, P.; Xia, S.; Ma, J.; Luo, J.; Xiao, T.; Le, S.; Huang, M.; et al. A recombinant adenovirus vector encoding the light chain of human coagulation factor VII and IgG1 Fc fragment to targeting tissue factor for colorectal cancer immunotherapy in the mouse model. J. Cancer Res. Clin. Oncol. 2013, 139, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Liikanen, I.; Ta htinen, S.; Guse, K.; Gutmann, T.; Savola, P.; Oksanen, M.; Kanerva, A.; Hemminki, A. Oncolytic Adenovirus Expressing Monoclonal Antibody Trastuzumab for Treatment of HER2-Positive. Cancer Mol. Cancer Ther. 2016, 15, 2259–2269. [Google Scholar] [CrossRef] [PubMed]
- Piperno, G.M.; López-Requena, A.; Predonzani, A.; Dorvignit, D.; Labrada, M.; Zentilin, L.; Burrone, O.R.; Cesco-Gaspere, M. Recombinant AAV-mediated in vivo long-term expression and antitumour activity of an anti-ganglioside GM3(Neu5Gc) antibody. Gene Ther. 2015, 22, 960–967. [Google Scholar] [CrossRef]
- Li, M.; Wu, Y.; Qiu, Y.; Yao, Z.; Liu, S.; Liu, Y.; Shi, J.; Zheng, D. 2A Peptide-based, Lentivirus-mediated Anti-death Receptor 5 Chimeric Antibody Expression Prevents Tumor Growth in Nude Mice. Mol. Ther. 2012, 20, 46–53. [Google Scholar] [CrossRef]
- Vigna, E.; Pacchiana, G.; Mazzone, M.; Chiriaco, C.; Fontani, L.; Basilico, C.; Pennacchietti, S.; Comoglio, P.M. “Active” Cancer Immunotherapy by Anti-Met Antibody Gene Transfer. Cancer Res. 2008, 68, 9176–9183. [Google Scholar] [CrossRef]
- Mitchell, L.A.; Yagiz, K.; Hofacre, A.; Viaud, S.; Munday, A.W.; Espinoza, F.L.; Mendoza, D.; Rodriguez-Aguirre, M.E.; Bergqvist, S.; Haghighi, A.; et al. PD-L1 checkpoint blockade delivered by retroviral replicating vector confers anti-tumor efficacy in murine tumor models. Oncotarget 2019, 10, 2252–2269. [Google Scholar] [CrossRef]
- Vijayakumar, G.; McCroskery, S.; Palese, P. Engineering Newcastle disease virus as oncolytic vector for intratumoral delivery of immune checkpoint inhibitors and immunocytokines. J. Virol. 2019. [Google Scholar] [CrossRef]
- Reul, J.; Frisch, J.; Engeland, C.E.; Thalheimer, F.B.; Hartmann, J.; Ungerechts, G.; Buchholz, C.J. Tumor-Specific Delivery of Immune Checkpoint Inhibitors by Engineered AAV Vectors. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef]
- Dias, J.D.; Hemminki, O.; Diaconu, I.; Hirvinen, M.; Bonetti, A.; Guse, K.; Escutenaire, S.; Kanerva, A.; Pesonen, S.; Löskog, A.; et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012, 19, 988–998. [Google Scholar] [CrossRef]
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. CTLA-4 and PD-L1 Checkpoint Blockade Enhances Oncolytic Measles Virus Therapy. Mol. Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Boyer, J.L.; Hackett, N.R.; Qiu, J.; Crystal, R.G. Gene Delivery of the Murine Equivalent of Bevacizumab (Avastin), an Anti-Vascular Endothelial Growth Factor Monoclonal Antibody, to Suppress Growth of Human Tumors in Immunodeficient Mice. Hum. Gene Ther. 2008, 19, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Navio, J.M.; Fuchs, S.P.; Pedreño-López, S.; Rakasz, E.G.; Gao, G.; Desrosiers, R.C. Host Anti-antibody Responses Following Adeno-associated Virus–mediated Delivery of Antibodies Against HIV and SIV in Rhesus Monkeys. Mol. Ther. 2016, 24, 76–86. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.P.; Martinez-Navio, J.M.; Rakasz, E.G.; Gao, G.; Desrosiers, R.C. Liver-Directed but Not Muscle-Directed AAV-Antibody Gene Transfer Limits Humoral Immune Responses in Rhesus Monkeys. Mol. Ther. Methods Clin. Dev. 2020, 16, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Pelegrin, M.; Marin, M.; Noël, D.; Rio, M.D.; Saller, R.; Stange, J.; Mitzner, S.; Günzburg, W.; Piechaczyk, M. Systemic long-term delivery of antibodies in immunocompetent animals using cellulose sulphate capsules containing antibody-producing cells. Gene Ther. 1998, 5, 828–834. [Google Scholar] [CrossRef]
- Noël, D.; Pelegrin, M.; Marin, M.; Biard-Piechaczyk, M.; Ourlin, J.-C.; Mani, J.-C.; Piechaczyk, M. In Vitro and In Vivo Secretion of Cloned Antibodies by Genetically Modified Myogenic Cells. Hum. Gene Ther. 1997, 8, 1219–1229. [Google Scholar] [CrossRef] [PubMed]
- Ochi, A.; Hawley, R.G.; Hawley, T.; Shulman, M.J.; Traunecker, A.; Kohler, G.; Hozumi, N. Functional immunoglobulin M production after transfection of cloned immunoglobulin heavy and light chain genes into lymphoid cells. Proc. Natl. Acad. Sci. USA 1983, 80, 6351–6355. [Google Scholar] [CrossRef]
- Okada, N.; Miyamoto, H.; Yoshioka, T.; Katsume, A.; Saito, H.; Yorozu, K.; Ueda, O.; Itoh, N.; Mizuguchi, H.; Nakagawa, S.; et al. Cytomedical therapy for IgG1 plasmacytosis in human interleukin-6 transgenic mice using hybridoma cells microencapsulated in alginate-poly(l) lysine-alginate membrane. Biochim. Biophys. Acta 1997, 1360, 53–63. [Google Scholar] [CrossRef]
- Frecha, C.; Costa, C.; Lévy, C.; Nègre, D.; Russell, S.J.; Maisner, A.; Salles, G.; Peng, K.-W.; Cosset, F.-L.; Verhoeyen, E. Efficient and stable transduction of resting B lymphocytes and primary chronic lymphocyte leukemia cells using measles virus gp displaying lentiviral vectors. Blood 2009, 114, 3173–3180. [Google Scholar] [CrossRef]
- Girard-Gagnepain, A.; Amirache, F.; Costa, C.; Lévy, C.; Frecha, C.; Fusil, F.; Nègre, D.; Lavillette, D.; Cosset, F.-L.; Verhoeyen, E. Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood 2014, 124, 1221–1231. [Google Scholar] [CrossRef]
- Tittlbach, H.; Schneider, A.; Strobel, J.; Zimmermann, R.; Maas, S.; Gebhardt, B.; Rauser, G.; Mach, M.; Mackensen, A.; Winkler, T.H.; et al. GMP-production of purified human B lymphocytes for the adoptive transfer in patients after allogeneic hematopoietic stem cell transplantation. J. Transl. Med. 2017, 15. [Google Scholar] [CrossRef]
- Luo, X.M.; Maarschalk, E.; O’Connell, R.M.; Wang, P.; Yang, L.; Baltimore, D. Engineering human hematopoietic stem/progenitor cells to produce a broadly neutralizing anti-HIV antibody after in vitro maturation to human B lymphocytes. Blood 2009, 113, 1422–1431. [Google Scholar] [CrossRef]
- Laurie, K.L.; Blundell, M.P.; Baxendale, H.E.; Howe, S.J.; Sinclair, J.; Qasim, W.; Brunsberg, U.; Thrasher, A.J.; Holmdahl, R.; Gustafsson, K. Cell-specific and efficient expression in mouse and human B cells by a novel hybrid immunoglobulin promoter in a lentiviral vector. Gene Ther. 2007, 14, 1623–1631. [Google Scholar] [CrossRef] [PubMed]
- Taher, T.E.; Tulone, C.; Fatah, R.; D’Acquisto, F.; Gould, D.J.; Mageed, R.A. Repopulation of B-lymphocytes with restricted gene expression using haematopoietic stem cells engineered with lentiviral vectors. Gene Ther. 2008, 15, 998–1006. [Google Scholar] [CrossRef][Green Version]
- Freitag, J.; Heink, S.; Roth, E.; Wittmann, J.; Jäck, H.-M.; Kamradt, T. Towards the Generation of B-Cell Receptor Retrogenic Mice. PLoS ONE 2014, 9, e109199. [Google Scholar] [CrossRef]
- Moutai, T.; Yamana, H.; Nojima, T.; Kitamura, D. A Novel and Effective Cancer Immunotherapy Mouse Model Using Antigen-Specific B Cells Selected In Vitro. PLoS ONE 2014, 9, e92732. [Google Scholar] [CrossRef]
- Lin, Q.; Zhao, J.; Song, Y.; Liu, D. Recent updates on CAR T clinical trials for multiple myeloma. Mol. Cancer 2019, 18, 154. [Google Scholar] [CrossRef] [PubMed]
- Suarez, E.R.; Chang, D.-K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor–Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef]
- Peterson, M.L. Immunoglobulin heavy chain gene regulation through polyadenylation and splicing competition: Immunoglobulin heavy chain gene regulation. Wiley Interdiscip. Rev. RNA 2011, 2, 92–105. [Google Scholar] [CrossRef]
- Yu, K.K.; Aguilar, K.; Tsai, J.; Galimidi, R.; Gnanapragasam, P.; Yang, L.; Baltimore, D. Use of Mutated Self-Cleaving 2A Peptides as a Molecular Rheostat to Direct Simultaneous Formation of Membrane and Secreted Anti-HIV Immunoglobulins. PLoS ONE 2012, 7, e50438. [Google Scholar] [CrossRef]
- Ten Dam, E.; Donnelly, M.L.L.; Gani, D.; Luke, G.; Ryan, M.D.; Mendoza, H.; Hughes, L.E. The ‘cleavage’ activities of foot-and-mouth disease virus 2A site-directed mutants and naturally occurring ‘2A-like’ sequences. J. Gen. Virol. 2001, 82, 1027–1041. [Google Scholar]
- Fang, J.; Yi, S.; Simmons, A.; Tu, G.H.; Nguyen, M.; Harding, T.C.; VanRoey, M.; Jooss, K. An antibody delivery system for regulated expression of therapeutic levels of monoclonal antibodies in vivo. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 1153–1159. [Google Scholar] [CrossRef] [PubMed]
- Fusil, F.; Calattini, S.; Amirache, F.; Mancip, J.; Costa, C.; Robbins, J.B.; Douam, F.; Lavillette, D.; Law, M.; Defrance, T.; et al. A Lentiviral Vector Allowing Physiologically Regulated Membrane-anchored and Secreted Antibody Expression Depending on B-cell Maturation Status. Mol. Ther. 2015, 23, 1734–1747. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.L.; Perry, R.P. Regulated production of mu m and mu s mRNA requires linkage of the poly(A) addition sites and is dependent on the length of the mu s-mu m intron. Proc. Natl. Acad. Sci. USA 1986, 83, 8883–8887. [Google Scholar] [CrossRef] [PubMed]
- Galli, G.; Guise, J.W.; McDevitt, M.A.; Tucker, P.W.; Nevins, J.R. Relative position and strengths of poly(A) sites as well as transcription termination are critical to membrane versus secreted mu-chain expression during B-cell development. Genes Amp Dev. 1987, 1, 471–481. [Google Scholar] [CrossRef][Green Version]
- Peterson, M.L.; Bertolino, S.; Davis, F. An RNA Polymerase Pause Site Is Associated with the Immunoglobulin s Poly(A) Site. Mol. Cell. Biol. 2002, 22, 5606–5615. [Google Scholar] [CrossRef][Green Version]
- Yu, K.-R.; Natanson, H.; Dunbar, C.E. Gene Editing of Human Hematopoietic Stem and Progenitor Cells: Promise and Potential Hurdles. Hum. Gene Ther. 2016, 27, 729–740. [Google Scholar] [CrossRef]
- Ott de Bruin, L.M.; Volpi, S.; Musunuru, K. Novel Genome-Editing Tools to Model and Correct Primary Immunodeficiencies. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef]
- Cascalho, M.; Ma, A.; Lee, S.; Masat, L.; Wabl, M. A Quasi-Monoclonal Mouse. Science 1996, 272, 1649–1652. [Google Scholar] [CrossRef]
- Goodnow, C.C.; Crosbie, J.; Adelstein, S.; Lavoie, T.B.; Smith-Gill, S.J.; Brink, R.A.; Pritchard-Briscoe, H.; Wotherspoon, J.S.; Loblay, R.H.; Raphael, K.; et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 1988, 334, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.J.; Laoharawee, K.; Lahr, W.S.; Webber, B.R.; Moriarity, B.S. Engineering of Primary Human B cells with CRISPR/Cas9 Targeted Nuclease. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Moffett, H.F.; Harms, C.K.; Fitzpatrick, K.S.; Tooley, M.R.; Boonyaratanakornkit, J.; Taylor, J.J. B cells engineered to express pathogen-specific antibodies protect against infection. Sci. Immunol. 2019, 4, eaax0644. [Google Scholar] [CrossRef] [PubMed]
- Greiner, V.; Bou Puerto, R.; Liu, S.; Herbel, C.; Carmona, E.M.; Goldberg, M.S. CRISPR-Mediated Editing of the B Cell Receptor in Primary Human B Cells. iScience 2019, 12, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Voss, J.E.; Gonzalez-Martin, A.; Andrabi, R.; Fuller, R.P.; Murrell, B.; McCoy, L.E.; Porter, K.; Huang, D.; Li, W.; Sok, D.; et al. Reprogramming the antigen specificity of B cells using genome-editing technologies. eLife 2019, 8. [Google Scholar] [CrossRef]
- Cheong, T.-C.; Compagno, M.; Chiarle, R. Editing of mouse and human immunoglobulin genes by CRISPR-Cas9 system. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Lin, Y.; Pecetta, S.; Steichen, J.M.; Kratochvil, S.; Melzi, E.; Arnold, J.; Dougan, S.K.; Wu, L.; Kirsch, K.H.; Nair, U.; et al. One-step CRISPR/Cas9 method for the rapid generation of human antibody heavy chain knock-in mice. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Shim, G.; Kim, D.; Park, G.T.; Jin, H.; Suh, S.-K.; Oh, Y.-K. Therapeutic gene editing: Delivery and regulatory perspectives. Acta Pharmacol. Sin. 2017, 38, 738–753. [Google Scholar] [CrossRef]
- Mangeot, P.E.; Risson, V.; Fusil, F.; Marnef, A.; Laurent, E.; Blin, J.; Mournetas, V.; Massouridès, E.; Sohier, T.J.M.; Corbin, A.; et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun. 2019, 10, 45. [Google Scholar] [CrossRef]
- Liu, M.; Rehman, S.; Tang, X.; Gu, K.; Fan, Q.; Chen, D.; Ma, W. Methodologies for Improving HDR Efficiency. Front. Genet. 2019, 9, 691. [Google Scholar] [CrossRef]
- Kuball, J.; Dossett, M.L.; Wolfl, M.; Ho, W.Y.; Voss, R.-H.; Fowler, C.; Greenberg, P.D. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood 2007, 109, 2331–2338. [Google Scholar] [CrossRef]
- Brendel, C.; Rio, P.; Verhoeyen, E. Humanized mice are precious tools for evaluation of hematopoietic gene therapies and preclinical modeling to move towards a clinical trial. Biochem. Pharmacol. 2019, 113711. [Google Scholar] [CrossRef] [PubMed]
- Schiroli, G.; Conti, A.; Ferrari, S.; della Volpe, L.; Jacob, A.; Albano, L.; Beretta, S.; Calabria, A.; Vavassori, V.; Gasparini, P.; et al. Precise Gene Editing Preserves Hematopoietic Stem Cell Function following Transient p53-Mediated DNA Damage Response. Cell Stem Cell 2019, 24, 551–565. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef]
- Dessels, C.; Alessandrini, M.; Pepper, M.S. Factors Influencing the Umbilical Cord Blood Stem Cell Industry: An Evolving Treatment Landscape. Stem Cells Transl. Med. 2018, 7, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.H.; Zarrabi, M.; Abroun, S.; Ahmadipanah, M.; Abbaspanah, B. Umbilical cord blood quality and quantity: Collection up to transplantation. Asian J. Transfus. Sci. 2019, 13, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Pesch, T.; Bonati, L.; Kelton, W.; Parola, C.; Ehling, R.A.; Csepregi, L.; Kitamura, D.; Reddy, S.T. Molecular Design, Optimization, and Genomic Integration of Chimeric B Cell Receptors in Murine B Cells. Front. Immunol. 2019, 10, 2630. [Google Scholar] [CrossRef]
- Leoh, L.S.; Morizono, K.; Kershaw, K.M.; Chen, I.S.Y.; Penichet, M.L.; Daniels-Wells, T.R. Gene delivery in malignant B cells using the combination of lentiviruses conjugated to anti-transferrin receptor antibodies and an immunoglobulin promoter: New dual targeted gene therapies. J. Gene Med. 2014, 16, 11–27. [Google Scholar] [CrossRef]
- Uusi-Kerttula, H.; Davies, J.; Coughlan, L.; Hulin-Curtis, S.; Jones, R.; Hanna, L.; Chester, J.D.; Parker, A.L. Pseudotyped avb6 integrin-targeted adenovirus vectors for ovarian cancer therapies. Oncotarget 2016, 7. [Google Scholar] [CrossRef]
- Ahani, R.; Roohvand, F.; Cohan, R.A.; Etemadzadeh, M.H.; Mohajel, N.; Behdani, M.; Shahosseini, Z.; Madani, N.; Azadmanesh, K. Sindbis Virus-Pseudotyped Lentiviral Vectors Carrying VEGFR2-Specific Nanobody for Potential Transductional Targeting of Tumor Vasculature. Mol. Biotechnol. 2016, 58, 738–747. [Google Scholar] [CrossRef]
- Pelegrin, M.; Marin, M.; Noël, D.; Piechaczyk, M. Genetically Engineered Antibodies in Gene Transfer and Gene Therapy. Hum. Gene Ther. 1998, 9, 2165–2175. [Google Scholar] [CrossRef]
- Kasaraneni, N.; Chamoun-Emanuelli, A.M.; Wright, G.; Chen, Z. Retargeting Lentiviruses via SpyCatcher-SpyTag Chemistry for Gene Delivery into Specific Cell Types. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Verhoeyen, E.; Wiznerowicz, M.; Olivier, D.; Izac, B.; Trono, D.; Dubart-Kupperschmitt, A.; Cosset, F.-L. Novel lentiviral vectors displaying “early-acting cytokines” selectively promote survival and transduction of NOD/SCID repopulating human hematopoietic stem cells. Blood 2005, 106, 3386–3395. [Google Scholar] [CrossRef] [PubMed]
- Hatziioannou, T.; Delahaye, E.; Martin, F.; Russell, S.J.; Cosset, F.-L. Retroviral Display of Functional Binding Domains Fused to the Amino Terminus of Influenza Hemagglutinin. Hum. Gene Ther. 1999, 10, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Maurice, M.; Mazur, S.; Bullough, F.J.; Salvetti, A.; Collins, M.K.; Russell, S.J.; Cosset, F.L. Efficient gene delivery to quiescent interleukin-2 (IL-2)-dependent cells by murine leukemia virus-derived vectors harboring IL-2 chimeric envelope glycoproteins. Blood 1999, 94, 401–410. [Google Scholar]
- Sandrin, V.; Boson, B.; Salmon, P.; Gay, W.; Nègre, D.; Le Grand, R.; Trono, D.; Cosset, F.-L. Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood 2002, 100, 823–832. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Characteristics | Cheong [88] | Lin [89] | Voss [87] | Greiner [86] | Moffett [85] |
|---|---|---|---|---|---|
| Target cell | Human B cells | Mouse ESC | Human B cells | Human B cells | Human B cells |
| Cas9 delivery | Retroviral vector | RNP | Plasmid | RNP | RNP |
| Cutting heavy chain | Yes (1 cut in constant region) | Yes (2 cuts in D and J regions) | Yes (2 cuts in V and after J regions) | Yes (1 cut in V region) | Yes (1 cut after J and before constant region) |
| Cutting light chain | No | No | No | Yes (1 cut in κV region) | No |
| Cutting efficiency | N.D. | 26–54.5% | N.D. | N.D. | 72% |
| Sequences inserted | None | VH | VH | HC and LC | LC fused to VH |
| HDR efficiency | N.D. | 0–50% | 0.21% | 8.5% | 30% |
| Promoter | Endogenous | Exogenous | Endogenous | Endogenous | Exogenous |
| AID/class switching | ? | ? | Yes | ? | ? |
| Target | Endogenous | HIV | HIV | TNF-α | RSV |
| Approach | Infusion of Cells | Target Cell Type | Single Injection | Physiological Regulated | Immune Memory | Other Limitations | Refs |
|---|---|---|---|---|---|---|---|
| Passive immunotherapy (Ab injection) | None | None | No | No | No | Anti-idiotypic Abs Immune escape | [16,17] |
| In situ vectored gene transfer | None | Vector infected cells | Yes | No | No | Anti-idiotypic Abs Immune escape | [28,29,30,31,39,41,42,43,44,46,47,48,49,50,51,52,100,101,102] |
| Molecular rheostat approach | B cells | B cells | Yes | No | No | Random insertions Chimeras Effect on the endogenous response? | [75] |
| FAM2 technology | B cells | B cells | Yes | Yes | Yes | Random insertion Chimeras Effect on the endogenous response? | [72] |
| CRISPR-edited BCR | B cells | B cells | Yes | Yes | Yes | Off-targets HDR efficiency Chimeras Effect on the endogenous response? | [85,86,87,88,89] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Page, A.; Fusil, F.; Cosset, F.-L. Towards Physiologically and Tightly Regulated Vectored Antibody Therapies. Cancers 2020, 12, 962. https://doi.org/10.3390/cancers12040962
Page A, Fusil F, Cosset F-L. Towards Physiologically and Tightly Regulated Vectored Antibody Therapies. Cancers. 2020; 12(4):962. https://doi.org/10.3390/cancers12040962
Chicago/Turabian StylePage, Audrey, Floriane Fusil, and François-Loïc Cosset. 2020. "Towards Physiologically and Tightly Regulated Vectored Antibody Therapies" Cancers 12, no. 4: 962. https://doi.org/10.3390/cancers12040962
APA StylePage, A., Fusil, F., & Cosset, F.-L. (2020). Towards Physiologically and Tightly Regulated Vectored Antibody Therapies. Cancers, 12(4), 962. https://doi.org/10.3390/cancers12040962