BCL-2 Proteins in Pathogenesis and Therapy of B-Cell Non-Hodgkin Lymphomas


Abstract
1. Introduction
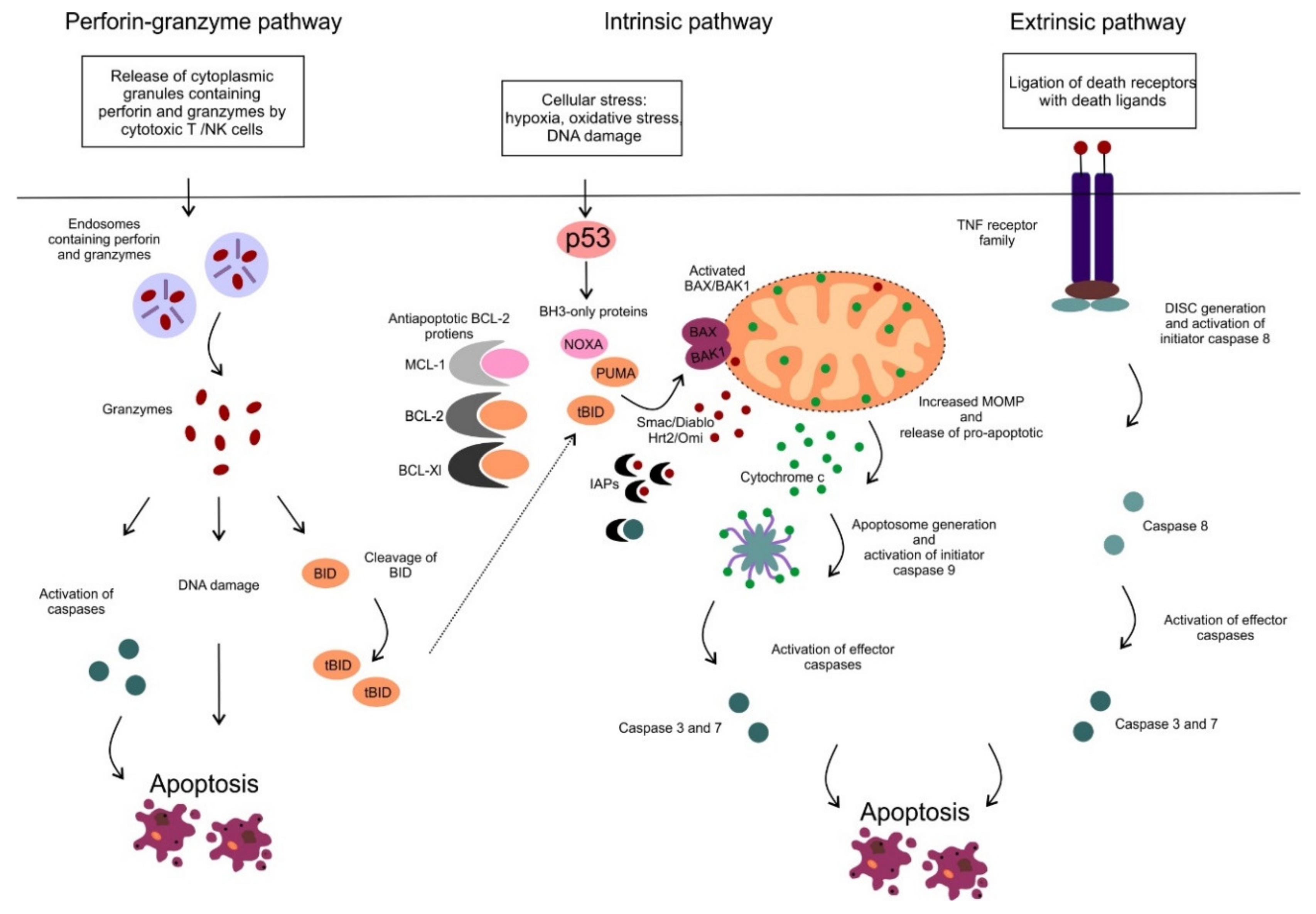
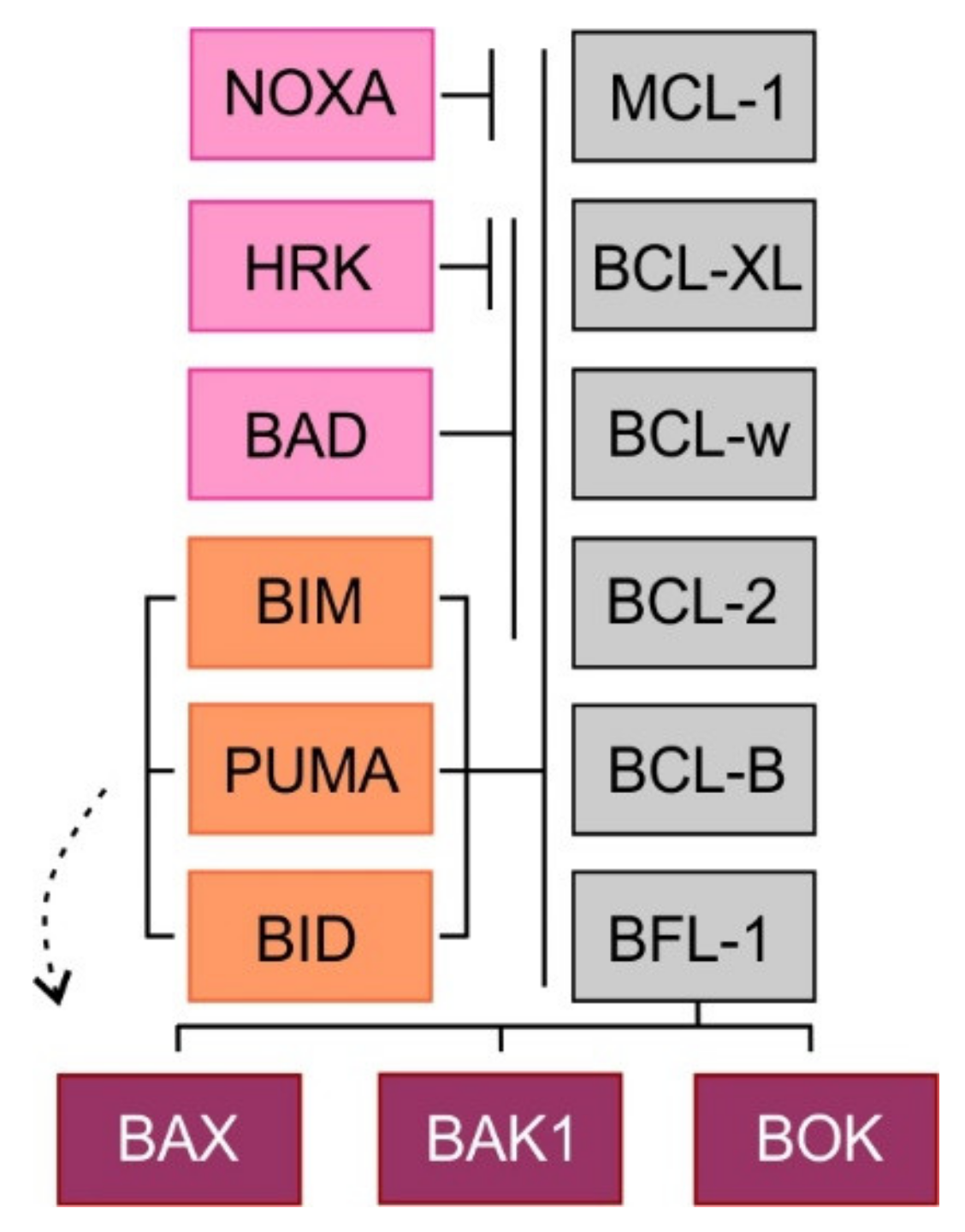
2. Mitochondrial Apoptotic Pathway
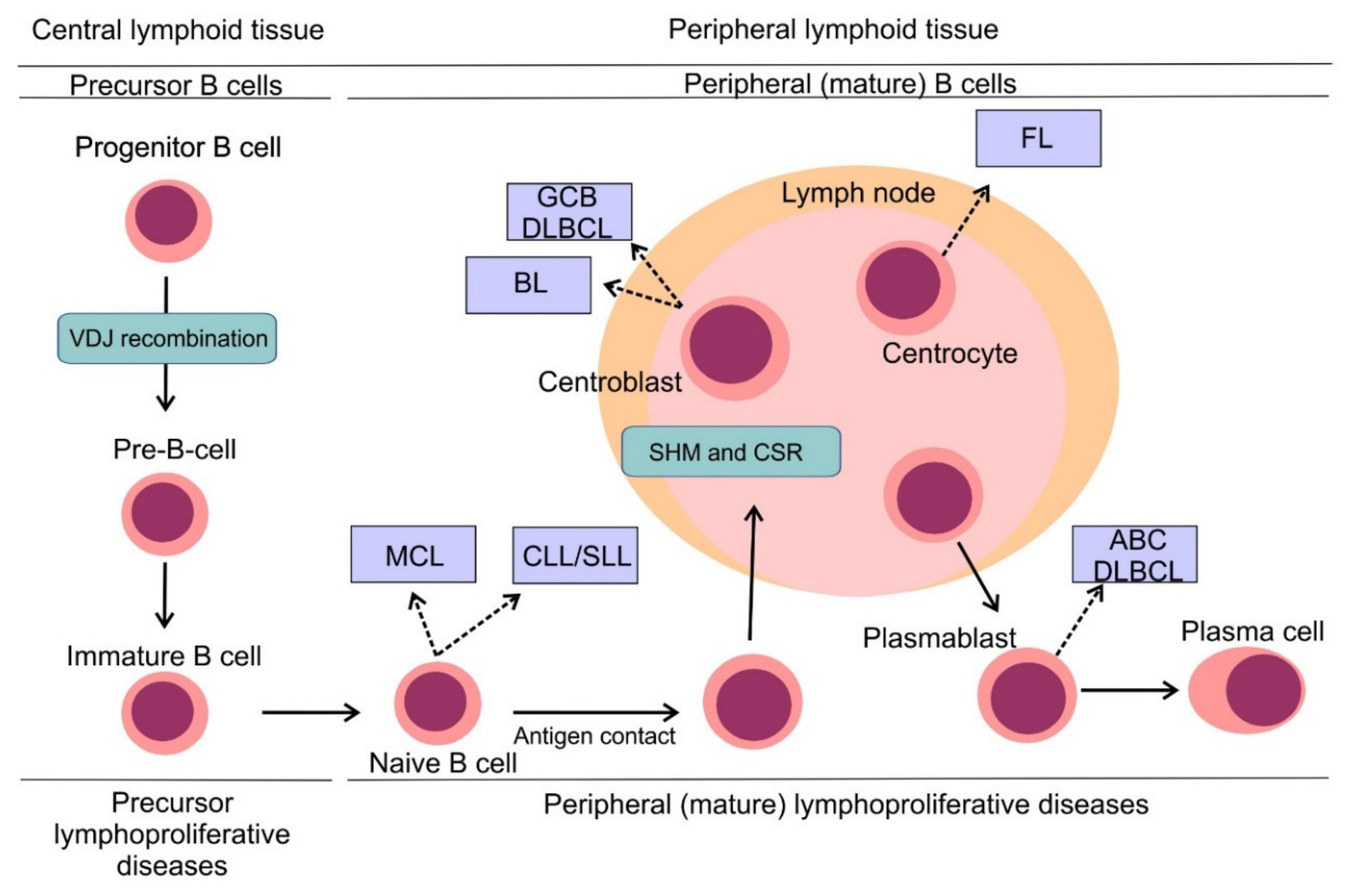
3. B-Cell Non-Hodgkin Lymphomas: Pathogenesis and Classification
4. Deregulation of BCL-2 Proteins in B-Cell Non-Hodgkin Lymphomas
5. Therapeutic Inhibition of Anti-Apoptotic BCL-2 Proteins
6. Venetoclax in Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
7. Venetoclax in B-Cell Non-Hodgkin Lymphomas
8. Selective Inhibitors of MCL-1 and BCL-XL Inhibitors in Chronic Lymphocytic Leukemia and B-Cell Non-Hodgkin Lymphomas
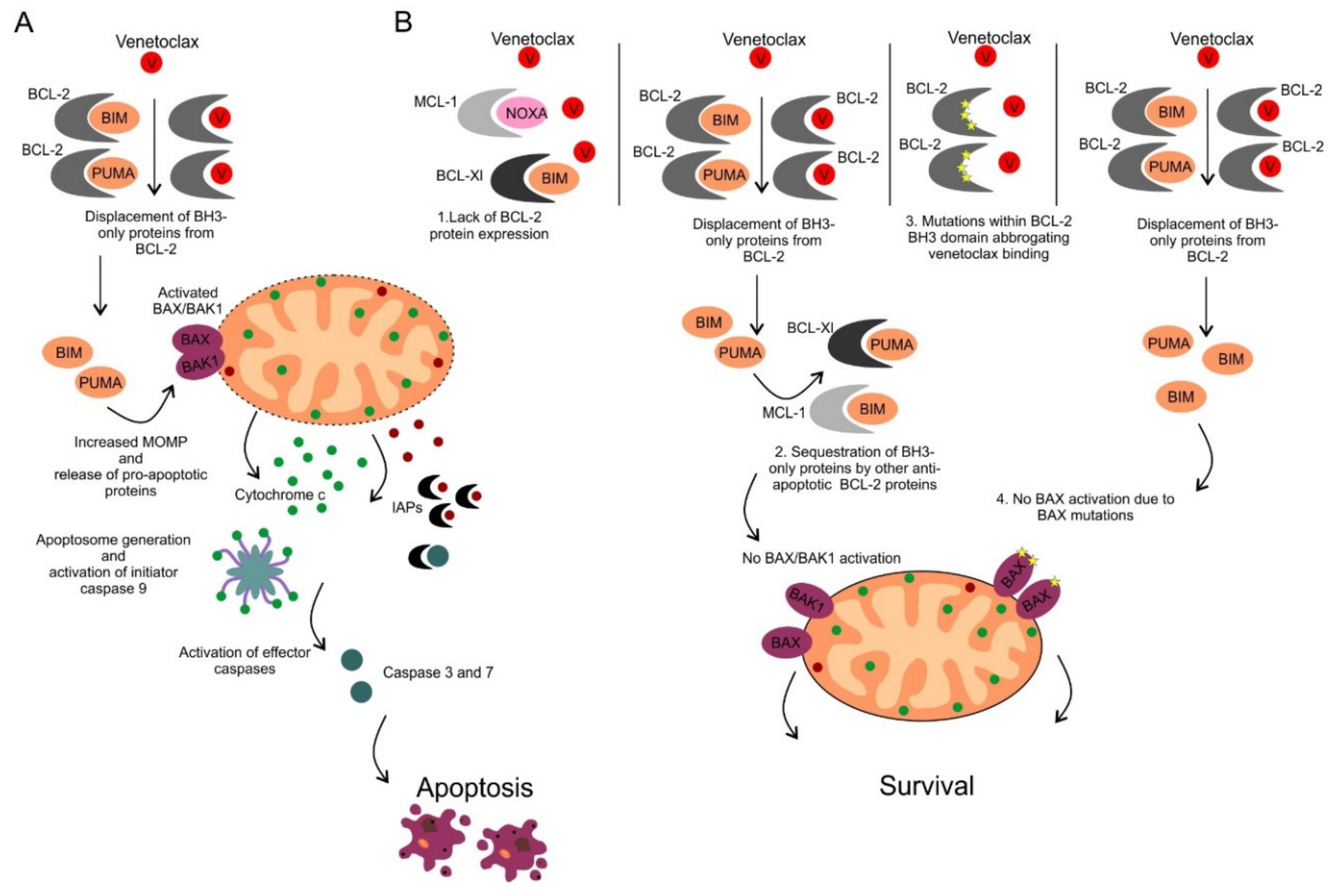
9. Determinants of Sensitivity and Resistance to BCL-2 Inhibition by Venetoclax
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J. Ferroptosis: Bug or feature? Immunol. Rev. 2017, 277, 150–157. [Google Scholar] [CrossRef]
- Ashkenazi, A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat. Rev. Cancer 2002, 2, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Trapani, J.A.; Smyth, M.J. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2002, 2, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Voskoboinik, I.; Whisstock, J.C.; Trapani, J.A. Perforin and granzymes: Function, dysfunction and human pathology. Nat. Rev. Immunol. 2015, 15, 388–400. [Google Scholar] [CrossRef]
- Sutton, V.R.; Davis, J.E.; Cancilla, M.; Johnstone, R.W.; Ruefli, A.A.; Sedelies, K.; Browne, K.A.; Trapani, J.A. Initiation of apoptosis by granzyme B requires direct cleavage of bid, but not direct granzyme B-mediated caspase activation. J. Exp. Med. 2000, 192, 1403–1414. [Google Scholar] [CrossRef]
- Beresford, P.J.; Xia, Z.; Greenberg, A.H.; Lieberman, J. Granzyme A loading induces rapid cytolysis and a novel form of DNA damage independently of caspase activation. Immunity 1999, 10, 585–594. [Google Scholar] [CrossRef]
- Huppertz, B.; Frank, H.G.; Kaufmann, P. The apoptosis cascade--morphological and immunohistochemical methods for its visualization. Anat. Embryol. (Berl) 1999, 200, 1–18. [Google Scholar] [CrossRef]
- Hegde, R.; Srinivasula, S.M.; Zhang, Z.; Wassell, R.; Mukattash, R.; Cilenti, L.; DuBois, G.; Lazebnik, Y.; Zervos, A.S.; Fernandes-Alnemri, T.; et al. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. 2002, 277, 432–438. [Google Scholar] [CrossRef]
- Yang, Q.H.; Church-Hajduk, R.; Ren, J.; Newton, M.L.; Du, C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev. 2003, 17, 1487–1496. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, C.; Olejniczak, E.T.; Meadows, R.P.; Betz, S.F.; Oost, T.; Herrmann, J.; Wu, J.C.; Fesik, S.W. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature 2000, 408, 1004–1008. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Warren, C.F.A.; Wong-Brown, M.W.; Bowden, N.A. BCL-2 family isoforms in apoptosis and cancer. Cell Death Dis. 2019, 10, 177. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Kim, J.H.; Bae, J. MCL-1ES induces MCL-1L-dependent BAX- and BAK-independent mitochondrial apoptosis. PLoS ONE 2013, 8, e79626. [Google Scholar] [CrossRef]
- Nechushtan, A.; Smith, C.L.; Lamensdorf, I.; Yoon, S.H.; Youle, R.J. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J. Cell Biol. 2001, 153, 1265–1276. [Google Scholar] [CrossRef]
- Ke, F.; Bouillet, P.; Kaufmann, T.; Strasser, A.; Kerr, J.; Voss, A.K. Consequences of the combined loss of BOK and BAK or BOK and BAX. Cell Death Dis. 2013, 4, e650. [Google Scholar] [CrossRef]
- Certo, M.; Del Gaizo Moore, V.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef]
- Nogai, H.; Dorken, B.; Lenz, G. Pathogenesis of non-Hodgkin’s lymphoma. J. Clin. Oncol. 2011, 29, 1803–1811. [Google Scholar] [CrossRef]
- Schatz, D.G.; Oettinger, M.A.; Baltimore, D. The V(D)J recombination activating gene, RAG-1. Cell 1989, 59, 1035–1048. [Google Scholar] [CrossRef]
- LeBien, T.W.; Tedder, T.F. B lymphocytes: How they develop and function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed]
- The Non-Hodgkin’s Lymphoma Classification Project. A Clinical Evaluation of the International Lymphoma Study Group Classification of Non-Hodgkin’s Lymphoma. Blood 1997, 89, 3909–3918. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Vitolo, U.; Trneny, M.; Belada, D.; Burke, J.M.; Carella, A.M.; Chua, N.; Abrisqueta, P.; Demeter, J.; Flinn, I.; Hong, X.; et al. Obinutuzumab or Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone in Previously Untreated Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef]
- Scott, D.W.; Mottok, A.; Ennishi, D.; Wright, G.W.; Farinha, P.; Ben-Neriah, S.; Kridel, R.; Barry, G.S.; Hother, C.; Abrisqueta, P.; et al. Prognostic Significance of Diffuse Large B-Cell Lymphoma Cell of Origin Determined by Digital Gene Expression in Formalin-Fixed Paraffin-Embedded Tissue Biopsies. J. Clin. Oncol. 2015, 33, 2848–2856. [Google Scholar] [CrossRef]
- Hu, S.; Xu-Monette, Z.Y.; Tzankov, A.; Green, T.; Wu, L.; Balasubramanyam, A.; Liu, W.M.; Visco, C.; Li, Y.; Miranda, R.N.; et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: A report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013, 121, 4021–4031. [Google Scholar] [CrossRef] [PubMed]
- Sehn, L.H.; Oestergaard, M.Z.; Trněný, M.; Bosi, A.; Egyed, M.; Illes, A.; Nakamae, H.; Opat, S.; Topp, M.; Zaja, F.; et al. Prognostic impact of bcl2 and myc expression and translocation in untreated dlbcl: Results from the phase iii goya study. Hematol. Oncol. 2017, 35, 131–133. [Google Scholar] [CrossRef]
- Bolen, C.R.; Klanova, M.; Trneny, M.; Sehn, L.H.; He, J.; Tong, J.; Paulson, J.N.; Kim, E.; Vitolo, U.; Di Rocco, A.; et al. Prognostic impact of somatic mutations in diffuse large B-cell lymphoma and relationship to cell-of-origin: Data from the phase III GOYA study. Haematologica 2019. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Neppalli, V.T.; Wright, G.; Dave, B.J.; Horsman, D.E.; Rosenwald, A.; Lynch, J.; Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; et al. BCL2 expression is a prognostic marker for the activated B-cell-like type of diffuse large B-cell lymphoma. J. Clin. Oncol. 2006, 24, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Leich, E.; Salaverria, I.; Bea, S.; Zettl, A.; Wright, G.; Moreno, V.; Gascoyne, R.D.; Chan, W.C.; Braziel, R.M.; Rimsza, L.M.; et al. Follicular lymphomas with and without translocation t(14;18) differ in gene expression profiles and genetic alterations. Blood 2009, 114, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Bende, R.J.; Smit, L.A.; van Noesel, C.J. Molecular pathways in follicular lymphoma. Leukemia 2007, 21, 18–29. [Google Scholar] [CrossRef]
- Guo, Y.; Karube, K.; Kawano, R.; Suzumiya, J.; Takeshita, M.; Kikuchi, M.; Huang, G.-S.; Li, Q.; Ohshima, K. Bcl2-negative follicular lymphomas frequently have Bcl6 translocation and/or Bcl6 or p53 expression. Pathol. Int. 2007, 57, 148–152. [Google Scholar] [CrossRef]
- Davids, M.S.; Roberts, A.W.; Seymour, J.F.; Pagel, J.M.; Kahl, B.S.; Wierda, W.G.; Puvvada, S.; Kipps, T.J.; Anderson, M.A.; Salem, A.H.; et al. Phase I First-in-Human Study of Venetoclax in Patients With Relapsed or Refractory Non-Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 826–833. [Google Scholar] [CrossRef]
- Tagawa, H.; Karnan, S.; Suzuki, R.; Matsuo, K.; Zhang, X.; Ota, A.; Morishima, Y.; Nakamura, S.; Seto, M. Genome-wide array-based CGH for mantle cell lymphoma: Identification of homozygous deletions of the proapoptotic gene BIM. Oncogene 2005, 24, 1348–1358. [Google Scholar] [CrossRef]
- Masque-Soler, N.; Szczepanowski, M.; Kohler, C.W.; Aukema, S.M.; Nagel, I.; Richter, J.; Siebert, R.; Spang, R.; Burkhardt, B.; Klapper, W. Clinical and pathological features of Burkitt lymphoma showing expression of BCL2--an analysis including gene expression in formalin-fixed paraffin-embedded tissue. Br. J. Haematol. 2015, 171, 501–508. [Google Scholar] [CrossRef]
- Majid, A.; Tsoulakis, O.; Walewska, R.; Gesk, S.; Siebert, R.; Kennedy, D.B.; Dyer, M.J. BCL2 expression in chronic lymphocytic leukemia: Lack of association with the BCL2 938A>C promoter single nucleotide polymorphism. Blood 2008, 111, 874–877. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Hanada, M.; Delia, D.; Aiello, A.; Stadtmauer, E.; Reed, J.C. bcl-2 gene hypomethylation and high-level expression in B-cell chronic lymphocytic leukemia. Blood 1993, 82, 1820–1828. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.; Arber, D.A.; Chang, K.L.; Wilson, C.S.; Weiss, L.M. Frequency of bcl-2 expression in non-Hodgkin’s lymphoma: A study of 778 cases with comparison of marginal zone lymphoma and monocytoid B-cell hyperplasia. Mod. Pathol. 1998, 11, 864–869. [Google Scholar] [PubMed]
- Bakhshi, A.; Jensen, J.P.; Goldman, P.; Wright, J.J.; McBride, O.W.; Epstein, A.L.; Korsmeyer, S.J. Cloning the chromosomal breakpoint of t(14;18) human lymphomas: Clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell 1985, 41, 899–906. [Google Scholar] [CrossRef]
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947. [Google Scholar] [CrossRef]
- Schuetz, J.M.; Johnson, N.A.; Morin, R.D.; Scott, D.W.; Tan, K.; Ben-Nierah, S.; Boyle, M.; Slack, G.W.; Marra, M.A.; Connors, J.M.; et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia 2012, 26, 1383–1390. [Google Scholar] [CrossRef]
- Stilgenbauer, S.; Nickolenko, J.; Wilhelm, J.; Wolf, S.; Weitz, S.; Dohner, K.; Boehm, T.; Dohner, H.; Lichter, P. Expressed sequences as candidates for a novel tumor suppressor gene at band 13q14 in B-cell chronic lymphocytic leukemia and mantle cell lymphoma. Oncogene 1998, 16, 1891–1897. [Google Scholar] [CrossRef]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef]
- Robertson, L.E.; Plunkett, W.; McConnell, K.; Keating, M.J.; McDonnell, T.J. Bcl-2 expression in chronic lymphocytic leukemia and its correlation with the induction of apoptosis and clinical outcome. Leukemia 1996, 10, 456–459. [Google Scholar]
- Dohner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Krober, A.; Bullinger, L.; Dohner, K.; Bentz, M.; Lichter, P. Genomic aberrations and survival in chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef]
- Adams, C.M.; Mitra, R.; Gong, J.Z.; Eischen, C.M. Non-Hodgkin and Hodgkin Lymphomas Select for Overexpression of BCLW. Clin. Cancer Res. 2017, 23, 7119–7129. [Google Scholar] [CrossRef]
- Zhou, P.; Levy, N.B.; Xie, H.; Qian, L.; Lee, C.Y.; Gascoyne, R.D.; Craig, R.W. MCL1 transgenic mice exhibit a high incidence of B-cell lymphoma manifested as a spectrum of histologic subtypes. Blood 2001, 97, 3902–3909. [Google Scholar] [CrossRef] [PubMed]
- Cho-Vega, J.H.; Rassidakis, G.Z.; Admirand, J.H.; Oyarzo, M.; Ramalingam, P.; Paraguya, A.; McDonnell, T.J.; Amin, H.M.; Medeiros, L.J. MCL-1 expression in B-cell non-Hodgkin’s lymphomas. Hum. Pathol. 2004, 35, 1095–1100. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, B.; Naresh, K.N. Bcl-2 family of proteins in indolent B-cell non-Hodgkin’s lymphoma: Study of 116 cases. Am. J. Hematol. 2002, 70, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.S.; Grau, M.; Mavis, C.; Hailfinger, S.; Wolf, A.; Madle, H.; Deeb, G.; Dorken, B.; Thome, M.; Lenz, P.; et al. MCL1 is deregulated in subgroups of diffuse large B-cell lymphoma. Leukemia 2013, 27, 1381–1390. [Google Scholar] [CrossRef]
- Katz, S.G.; Labelle, J.L.; Meng, H.; Valeriano, R.P.; Fisher, J.K.; Sun, H.; Rodig, S.J.; Kleinstein, S.H.; Walensky, L.D. Mantle cell lymphoma in cyclin D1 transgenic mice with Bim-deficient B cells. Blood 2014, 123, 884–893. [Google Scholar] [CrossRef]
- Prukova, D.; Andera, L.; Nahacka, Z.; Karolova, J.; Svaton, M.; Klanova, M.; Havranek, O.; Soukup, J.; Svobodova, K.; Zemanova, Z.; et al. Cotargeting of BCL2 with Venetoclax and MCL1 with S63845 Is Synthetically Lethal In Vivo in Relapsed Mantle Cell Lymphoma. Clin. Cancer Res. 2019, 25, 4455–4465. [Google Scholar] [CrossRef]
- Wang, J.D.; Katz, S.G.; Morgan, E.A.; Yang, D.T.; Pan, X.; Xu, M.L. Proapoptotic protein BIM as a novel prognostic marker in mantle cell lymphoma. Hum. Pathol. 2019, 93, 54–64. [Google Scholar] [CrossRef]
- Pfeifer, M.; Grau, M.; Lenze, D.; Wenzel, S.S.; Wolf, A.; Wollert-Wulf, B.; Dietze, K.; Nogai, H.; Storek, B.; Madle, H.; et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 12420–12425. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Davis, R.E.; Brown, K.D.; Siebenlist, U.; Staudt, L.M. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med. 2001, 194, 1861–1874. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.N.; Davis, R.E.; Lamy, L.; Yu, X.; Zhao, H.; Lenz, G.; Lam, L.T.; Dave, S.; Yang, L.; Powell, J.; et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature 2006, 441, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Davis, R.E.; Ngo, V.N.; Lam, L.; George, T.C.; Wright, G.W.; Dave, S.S.; Zhao, H.; Xu, W.; Rosenwald, A.; et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008, 319, 1676–1679. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Willis, T.G.; Jadayel, D.M.; Du, M.Q.; Peng, H.; Perry, A.R.; Abdul-Rauf, M.; Price, H.; Karran, L.; Majekodunmi, O.; Wlodarska, I.; et al. Bcl10 is involved in t(1;14)(p22;q32) of MALT B cell lymphoma and mutated in multiple tumor types. Cell 1999, 96, 35–45. [Google Scholar] [CrossRef]
- Lucas, P.C.; Yonezumi, M.; Inohara, N.; McAllister-Lucas, L.M.; Abazeed, M.E.; Chen, F.F.; Yamaoka, S.; Seto, M.; Nunez, G. Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF-kappa B signaling pathway. J. Biol. Chem. 2001, 276, 19012–19019. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Wayson, S.M.; Dixit, V.M.; Fairbrother, W.J.; Vucic, D. The inhibitor of apoptosis protein fusion c-IAP2.MALT1 stimulates NF-kappaB activation independently of TRAF1 AND TRAF2. J. Biol. Chem. 2006, 281, 29022–29029. [Google Scholar] [CrossRef]
- Luo, J.L.; Kamata, H.; Karin, M. IKK/NF-kappaB signaling: Balancing life and death--a new approach to cancer therapy. J. Clin. Investig. 2005, 115, 2625–2632. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Medeiros, L.J.; Li, Y.; Orlowski, R.Z.; Andreeff, M.; Bueso-Ramos, C.E.; Greiner, T.C.; McDonnell, T.J.; Young, K.H. Dysfunction of the TP53 tumor suppressor gene in lymphoid malignancies. Blood 2012, 119, 3668–3683. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Wu, L.; Visco, C.; Tai, Y.C.; Tzankov, A.; Liu, W.M.; Montes-Moreno, S.; Dybkaer, K.; Chiu, A.; Orazi, A.; et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: Report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2012, 120, 3986–3996. [Google Scholar] [CrossRef] [PubMed]
- Bea, S.; Valdes-Mas, R.; Navarro, A.; Salaverria, I.; Martin-Garcia, D.; Jares, P.; Gine, E.; Pinyol, M.; Royo, C.; Nadeu, F.; et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18250–18255. [Google Scholar] [CrossRef] [PubMed]
- Zenz, T.; Eichhorst, B.; Busch, R.; Denzel, T.; Habe, S.; Winkler, D.; Buhler, A.; Edelmann, J.; Bergmann, M.; Hopfinger, G.; et al. TP53 mutation and survival in chronic lymphocytic leukemia. J. Clin. Oncol. 2010, 28, 4473–4479. [Google Scholar] [CrossRef] [PubMed]
- Sander, C.A.; Yano, T.; Clark, H.M.; Harris, C.; Longo, D.L.; Jaffe, E.S.; Raffeld, M. p53 mutation is associated with progression in follicular lymphomas. Blood 1993, 82, 1994–2004. [Google Scholar] [CrossRef] [PubMed]
- Lo Coco, F.; Gaidano, G.; Louie, D.C.; Offit, K.; Chaganti, R.S.; Dalla-Favera, R. p53 mutations are associated with histologic transformation of follicular lymphoma. Blood 1993, 82, 2289–2295. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Villunger, A.; Michalak, E.M.; Coultas, L.; Mullauer, F.; Bock, G.; Ausserlechner, M.J.; Adams, J.M.; Strasser, A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 2003, 302, 1036–1038. [Google Scholar] [CrossRef]
- Klasa, R.J.; Gillum, A.M.; Klem, R.E.; Frankel, S.R. Oblimersen Bcl-2 antisense: Facilitating apoptosis in anticancer treatment. Antisense Nucleic Acid Drug Dev. 2002, 12, 193–213. [Google Scholar] [CrossRef]
- O’Brien, S.; Moore, J.O.; Boyd, T.E.; Larratt, L.M.; Skotnicki, A.B.; Koziner, B.; Chanan-Khan, A.A.; Seymour, J.F.; Gribben, J.; Itri, L.M.; et al. 5-year survival in patients with relapsed or refractory chronic lymphocytic leukemia in a randomized, phase III trial of fludarabine plus cyclophosphamide with or without oblimersen. J. Clin. Oncol. 2009, 27, 5208–5212. [Google Scholar] [CrossRef]
- Nguyen, M.; Marcellus, R.C.; Roulston, A.; Watson, M.; Serfass, L.; Murthy Madiraju, S.R.; Goulet, D.; Viallet, J.; Belec, L.; Billot, X.; et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 19512–19517. [Google Scholar] [CrossRef]
- Konopleva, M.; Watt, J.; Contractor, R.; Tsao, T.; Harris, D.; Estrov, Z.; Bornmann, W.; Kantarjian, H.; Viallet, J.; Samudio, I.; et al. Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obatoclax). Cancer Res. 2008, 68, 3413–3420. [Google Scholar] [CrossRef]
- Goy, A.; Berger, M.; Ford, P.; Feldman, T.; Mato, A.; Bejot, C.; Fung, H.C. Sequential single-agent obatoclax mesylate (GX15-070MS) followed by combination with rituximab in patients with previously untreated follicular lymphoma. Leuk Lymphoma 2014, 55, 2932–2934. [Google Scholar] [CrossRef]
- Goy, A.; Hernandez-Ilzaliturri, F.J.; Kahl, B.; Ford, P.; Protomastro, E.; Berger, M. A phase I/II study of the pan Bcl-2 inhibitor obatoclax mesylate plus bortezomib for relapsed or refractory mantle cell lymphoma. Leuk Lymphoma 2014, 55, 2761–2768. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, S.M.; Claxton, D.F.; Crump, M.; Faderl, S.; Kipps, T.; Keating, M.J.; Viallet, J.; Cheson, B.D. Phase I study of obatoclax mesylate (GX15-070), a small molecule pan-Bcl-2 family antagonist, in patients with advanced chronic lymphocytic leukemia. Blood 2009, 113, 299–305. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Tesar, B.; Yu, L.; Werner, L.; Takebe, N.; Mikler, E.; Reynolds, H.M.; Thompson, C.; Fisher, D.C.; Neuberg, D.; et al. Obatoclax in combination with fludarabine and rituximab is well-tolerated and shows promising clinical activity in relapsed chronic lymphocytic leukemia. Leuk Lymphoma 2015, 56, 3336–3342. [Google Scholar] [CrossRef] [PubMed]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef]
- Wilson, W.H.; O’Connor, O.A.; Czuczman, M.S.; LaCasce, A.S.; Gerecitano, J.F.; Leonard, J.P.; Tulpule, A.; Dunleavy, K.; Xiong, H.; Chiu, Y.L.; et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: A phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159. [Google Scholar] [CrossRef]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.; Xiong, H.; et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: Results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef]
- Vogler, M.; Hamali, H.A.; Sun, X.M.; Bampton, E.T.; Dinsdale, D.; Snowden, R.T.; Dyer, M.J.; Goodall, A.H.; Cohen, G.M. BCL2/BCL-X(L) inhibition induces apoptosis, disrupts cellular calcium homeostasis, and prevents platelet activation. Blood 2011, 117, 7145–7154. [Google Scholar] [CrossRef]
- Zhang, H.; Nimmer, P.M.; Tahir, S.K.; Chen, J.; Fryer, R.M.; Hahn, K.R.; Iciek, L.A.; Morgan, S.J.; Nasarre, M.C.; Nelson, R.; et al. Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007, 14, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Kipps, T.J.; Eradat, H.; Grosicki, S.; Catalano, J.; Cosolo, W.; Dyagil, I.S.; Yalamanchili, S.; Chai, A.; Sahasranaman, S.; Punnoose, E.; et al. A phase 2 study of the BH3 mimetic BCL2 inhibitor navitoclax (ABT-263) with or without rituximab, in previously untreated B-cell chronic lymphocytic leukemia. Leuk Lymphoma 2015, 56, 2826–2833. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Stilgenbauer, S.; Eichhorst, B.; Schetelig, J.; Coutre, S.; Seymour, J.F.; Munir, T.; Puvvada, S.D.; Wendtner, C.M.; Roberts, A.W.; Jurczak, W.; et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: A multicentre, open-label, phase 2 study. Lancet Oncol. 2016, 17, 768–778. [Google Scholar] [CrossRef]
- Seymour, J.F.; Ma, S.; Brander, D.M.; Choi, M.Y.; Barrientos, J.; Davids, M.S.; Anderson, M.A.; Beaven, A.W.; Rosen, S.T.; Tam, C.S.; et al. Venetoclax plus rituximab in relapsed or refractory chronic lymphocytic leukaemia: A phase 1b study. Lancet Oncol. 2017, 18, 230–240. [Google Scholar] [CrossRef]
- Seymour, J.F.; Kipps, T.J.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Gerecitano, J.; Robak, T.; De la Serna, J.; et al. Venetoclax-Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2018, 378, 1107–1120. [Google Scholar] [CrossRef]
- Flinn, I.W.; Gribben, J.G.; Dyer, M.J.S.; Wierda, W.; Maris, M.B.; Furman, R.R.; Hillmen, P.; Rogers, K.A.; Iyer, S.P.; Quillet-Mary, A.; et al. Phase 1b study of venetoclax-obinutuzumab in previously untreated and relapsed/refractory chronic lymphocytic leukemia. Blood 2019, 133, 2765–2775. [Google Scholar] [CrossRef]
- Fischer, K.; Al-Sawaf, O.; Bahlo, J.; Fink, A.M.; Tandon, M.; Dixon, M.; Robrecht, S.; Warburton, S.; Humphrey, K.; Samoylova, O.; et al. Venetoclax and Obinutuzumab in Patients with CLL and Coexisting Conditions. N. Engl. J. Med. 2019, 380, 2225–2236. [Google Scholar] [CrossRef]
- Zelenetz, A.D.; Salles, G.; Mason, K.D.; Casulo, C.; Le Gouill, S.; Sehn, L.H.; Tilly, H.; Cartron, G.; Chamuleau, M.E.D.; Goy, A.; et al. Venetoclax plus R- or G-CHOP in non-Hodgkin lymphoma: Results from the CAVALLI phase 1b trial. Blood 2019, 133, 1964–1976. [Google Scholar] [CrossRef]
- Morschhauser, F.; Feugier, P.; Flinn, I.W.; Gasiorowski, R.E.; Greil, R.; Illés, Á.; Johnson, N.A.; Larouche, J.-F.; Lugtenburg, P.J.; Patti, C.; et al. Venetoclax Plus Rituximab, Cyclophosphamide, Doxorubicin, Vincristine and Prednisolone (R-CHOP) Improves Outcomes in BCL2-Positive First-Line Diffuse Large B-Cell Lymphoma (DLBCL): First Safety, Efficacy and Biomarker Analyses from the Phase II CAVALLI Study. Blood 2018, 132, 782. [Google Scholar]
- Zinzani, P.L.; Topp, M.S.; Yuen, S.L.; Rusconi, C.; Fleury, I.; Pro, B.; Gritti, G.; Crump, M.; Hsu, W.; Punnoose, E.A.; et al. Phase 2 Study of Venetoclax Plus Rituximab or Randomized Ven Plus Bendamustine+Rituximab (BR) Versus BR in Patients with Relapsed/Refractory Follicular Lymphoma: Interim Data. Blood 2016, 128, 617. [Google Scholar] [CrossRef]
- Tam, C.S.; Anderson, M.A.; Pott, C.; Agarwal, R.; Handunnetti, S.; Hicks, R.J.; Burbury, K.; Turner, G.; Di Iulio, J.; Bressel, M.; et al. Ibrutinib plus Venetoclax for the Treatment of Mantle-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.J.; Gustine, J.; Meid, K.; Dubeau, T.; Keezer, A.; Allan, J.N.; Furman, R.R.; Siddiqi, T.; Advani, R.; Lam, J.; et al. Multicenter Prospective Phase II Study of Venetoclax in Patients with Previously Treated Waldenstrom Macroglobulinemia. Blood 2018, 132, 2888. [Google Scholar] [CrossRef]
- Klanova, M.; Andera, L.; Brazina, J.; Svadlenka, J.; Benesova, S.; Soukup, J.; Prukova, D.; Vejmelkova, D.; Jaksa, R.; Helman, K.; et al. Targeting of BCL2 Family Proteins with ABT-199 and Homoharringtonine Reveals BCL2- and MCL1-Dependent Subgroups of Diffuse Large B-Cell Lymphoma. Clin. Cancer Res. 2015. [Google Scholar] [CrossRef]
- Li, L.; Pongtornpipat, P.; Tiutan, T.; Kendrick, S.L.; Park, S.; Persky, D.O.; Rimsza, L.M.; Puvvada, S.D.; Schatz, J.H. Synergistic induction of apoptosis in high-risk DLBCL by BCL2 inhibition with ABT-199 combined with pharmacologic loss of MCL1. Leukemia 2015, 29, 1702–1712. [Google Scholar] [CrossRef]
- Dengler, M.A.; Teh, C.E.; Thijssen, R.; Gangoda, L.; Lan, P.; Herold, M.J.; Gray, D.H.; Kelly, G.L.; Roberts, A.W.; Adams, J.M. Potent efficacy of MCL-1 inhibitor-based therapies in preclinical models of mantle cell lymphoma. Oncogene 2020, 39, 2009–2023. [Google Scholar] [CrossRef]
- Yi, X.; Sarkar, A.; Kismali, G.; Aslan, B.; Ayres, M.; Iles, L.R.; Keating, M.J.; Wierda, W.; Long, J.P.; Bertilaccio, M.T.S.; et al. AMG-176, an Mcl-1 antagonist, shows preclinical efficacy in chronic lymphocytic leukemia. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.H.; Chiarparin, E.; Cidado, J.; Embrey, K.J.; Gangl, E.; Gibbons, F.D.; et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018, 9, 5341. [Google Scholar] [CrossRef]
- Khan, S.; Zhang, X.; Lv, D.; Zhang, Q.; He, Y.; Zhang, P.; Liu, X.; Thummuri, D.; Yuan, Y.; Wiegand, J.S.; et al. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019, 25, 1938–1947. [Google Scholar] [CrossRef]
- Phillips, D.C.; Xiao, Y.; Lam, L.T.; Litvinovich, E.; Roberts-Rapp, L.; Souers, A.J.; Leverson, J.D. Loss in MCL-1 function sensitizes non-Hodgkin’s lymphoma cell lines to the BCL-2-selective inhibitor venetoclax (ABT-199). Blood Cancer J. 2016, 6, e403. [Google Scholar] [CrossRef] [PubMed]
- Del Gaizo Moore, V.; Brown, J.R.; Certo, M.; Love, T.M.; Novina, C.D.; Letai, A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J. Clin. Investig. 2007, 117, 112–121. [Google Scholar] [CrossRef] [PubMed]
- De Jong, M.R.W.; Langendonk, M.; Reitsma, B.; Nijland, M.; van den Berg, A.; Ammatuna, E.; Visser, L.; van Meerten, T. Heterogeneous Pattern of Dependence on Anti-Apoptotic BCL-2 Family Proteins upon CHOP Treatment in Diffuse Large B-Cell Lymphoma. Int. J. Mol. Sci. 2019, 20, 6036. [Google Scholar] [CrossRef] [PubMed]
- Touzeau, C.; Dousset, C.; Bodet, L.; Gomez-Bougie, P.; Bonnaud, S.; Moreau, A.; Moreau, P.; Pellat-Deceunynck, C.; Amiot, M.; Le Gouill, S. ABT-737 induces apoptosis in mantle cell lymphoma cells with a Bcl-2high/Mcl-1low profile and synergizes with other antineoplastic agents. Clin. Cancer Res. 2011, 17, 5973–5981. [Google Scholar] [CrossRef]
- Choudhary, G.S.; Al-Harbi, S.; Mazumder, S.; Hill, B.T.; Smith, M.R.; Bodo, J.; Hsi, E.D.; Almasan, A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015, 6, e1593. [Google Scholar] [CrossRef]
- Liu, Y.; Mondello, P.; Erazo, T.; Tannan, N.B.; Asgari, Z.; de Stanchina, E.; Nanjangud, G.; Seshan, V.E.; Wang, S.; Wendel, H.G.; et al. NOXA genetic amplification or pharmacologic induction primes lymphoma cells to BCL2 inhibitor-induced cell death. Proc. Natl. Acad. Sci. USA 2018, 115, 12034–12039. [Google Scholar] [CrossRef]
- Jin, S.; Cojocari, D.; Purkal, J.J.; Popovic, R.; Talaty, N.N.; Xiao, Y.; Solomon, L.R.; Boghaert, E.R.; Leverson, J.D.; Phillips, D.C. 5-Azacitidine Induces NOXA to Prime AML Cells for Venetoclax-mediated Apoptosis. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Chiron, D.; Bellanger, C.; Papin, A.; Tessoulin, B.; Dousset, C.; Maiga, S.; Moreau, A.; Esbelin, J.; Trichet, V.; Chen-Kiang, S.; et al. Rational targeted therapies to overcome microenvironment-dependent expansion of mantle cell lymphoma. Blood 2016, 128, 2808–2818. [Google Scholar] [CrossRef]
- Guieze, R.; Liu, V.M.; Rosebrock, D.; Jourdain, A.A.; Hernandez-Sanchez, M.; Martinez Zurita, A.; Sun, J.; Ten Hacken, E.; Baranowski, K.; Thompson, P.A.; et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 2019, 36, 369–384. [Google Scholar] [CrossRef]
- Fresquet, V.; Rieger, M.; Carolis, C.; Garcia-Barchino, M.J.; Martinez-Climent, J.A. Acquired mutations in BCL2 family proteins conferring resistance to the BH3 mimetic ABT-199 in lymphoma. Blood 2014, 123, 4111–4119. [Google Scholar] [CrossRef]
- Tausch, E.; Close, W.; Dolnik, A.; Bloehdorn, J.; Chyla, B.; Bullinger, L.; Dohner, H.; Mertens, D.; Stilgenbauer, S. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica 2019, 104, e434–e437. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Peifer, M.; Herling, C.D.; Frenzel, L.P.; Hallek, M. Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemia (Comment to Tausch et al.). Haematologica 2019, 104, e540. [Google Scholar] [CrossRef] [PubMed]
- Blombery, P.; Anderson, M.A.; Gong, J.N.; Thijssen, R.; Birkinshaw, R.W.; Thompson, E.R.; Teh, C.E.; Nguyen, T.; Xu, Z.; Flensburg, C.; et al. Acquisition of the Recurrent Gly101Val Mutation in BCL2 Confers Resistance to Venetoclax in Patients with Progressive Chronic Lymphocytic Leukemia. Cancer Discov. 2019, 9, 342–353. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B-NHL Subtype | BCL-2 Positivity * | Mechanism of BCL-2 Overexpression |
|---|---|---|
| DLBCL | 49%–67% [30,31,32] | BCL2 translocation in GCB DLBCL (30% of cases) [33] BCL2 amplification in ABC DLBCL (20% of cases) [34] |
| FL | > 90% [35,36,37] | BCL2 translocation (90% of cases) [35,36] |
| MCL | BCL-2 positive [38] | 13q14.3 loss (55% of cases) [39] 18q21 gains (20% of cases) [37,39] |
| BL | BCL-2 negative, weak expression in up to 20% [40] | NA |
| CLL/SLL | BCL-2 positive (high expression in majority of the cases) [41] | 13q14.3 loss (68% cases) [42] BCL2 gene hypomethylation [43] BCL2 translocation (rare, < 5%) [41] |
| MZL | >80% [44] | unknown |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klanova, M.; Klener, P. BCL-2 Proteins in Pathogenesis and Therapy of B-Cell Non-Hodgkin Lymphomas. Cancers 2020, 12, 938. https://doi.org/10.3390/cancers12040938
Klanova M, Klener P. BCL-2 Proteins in Pathogenesis and Therapy of B-Cell Non-Hodgkin Lymphomas. Cancers. 2020; 12(4):938. https://doi.org/10.3390/cancers12040938
Chicago/Turabian StyleKlanova, Magdalena, and Pavel Klener. 2020. "BCL-2 Proteins in Pathogenesis and Therapy of B-Cell Non-Hodgkin Lymphomas" Cancers 12, no. 4: 938. https://doi.org/10.3390/cancers12040938
APA StyleKlanova, M., & Klener, P. (2020). BCL-2 Proteins in Pathogenesis and Therapy of B-Cell Non-Hodgkin Lymphomas. Cancers, 12(4), 938. https://doi.org/10.3390/cancers12040938