MicroRNAs as Emerging Regulators of Signaling in the Tumor Microenvironment


{kind=link}
{kind=link}
Abstract
1. Introduction
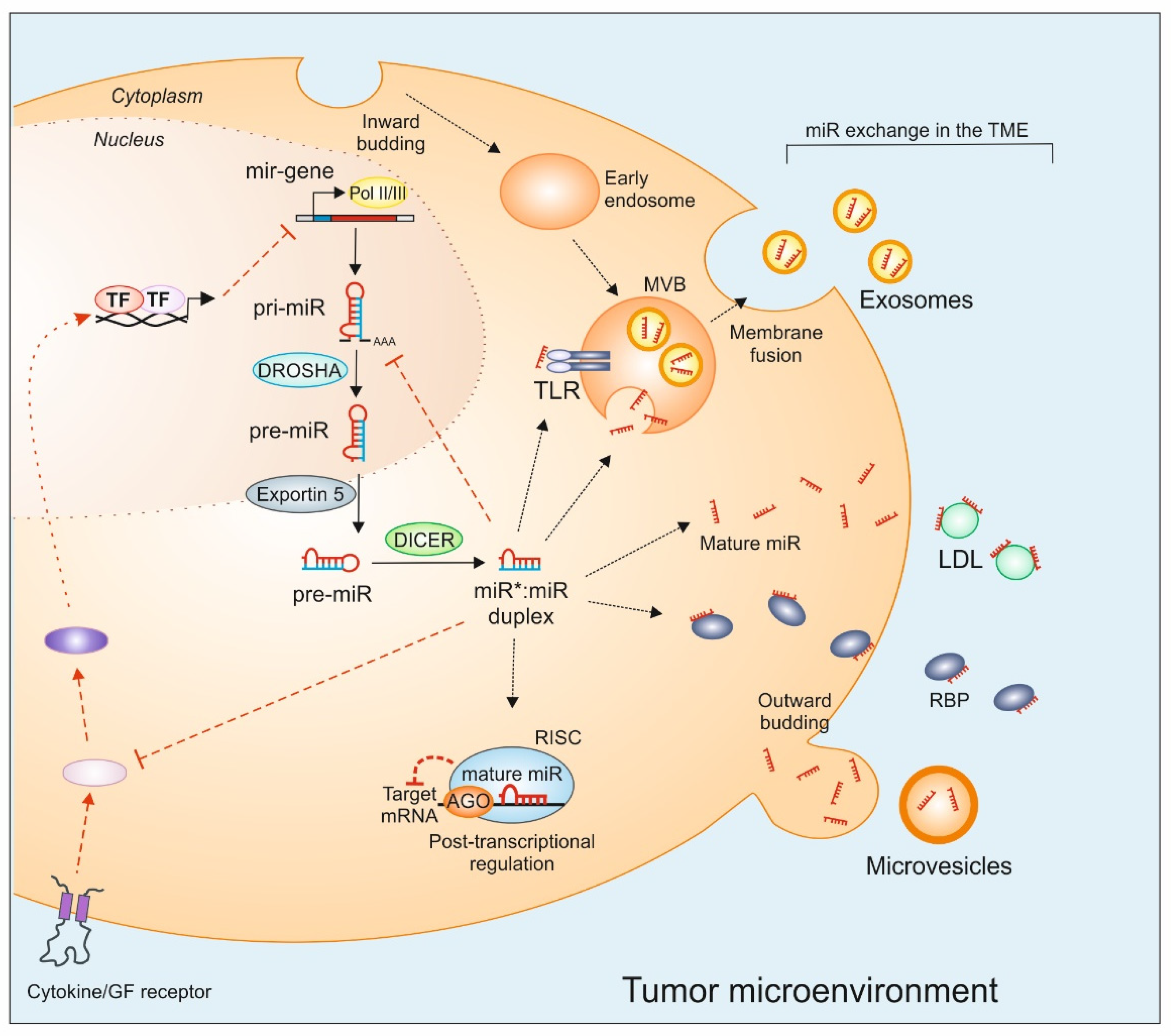
2. MiRs Biogenesis
3. MiRs Mobility in the TME
4. MiRs and TME Signaling
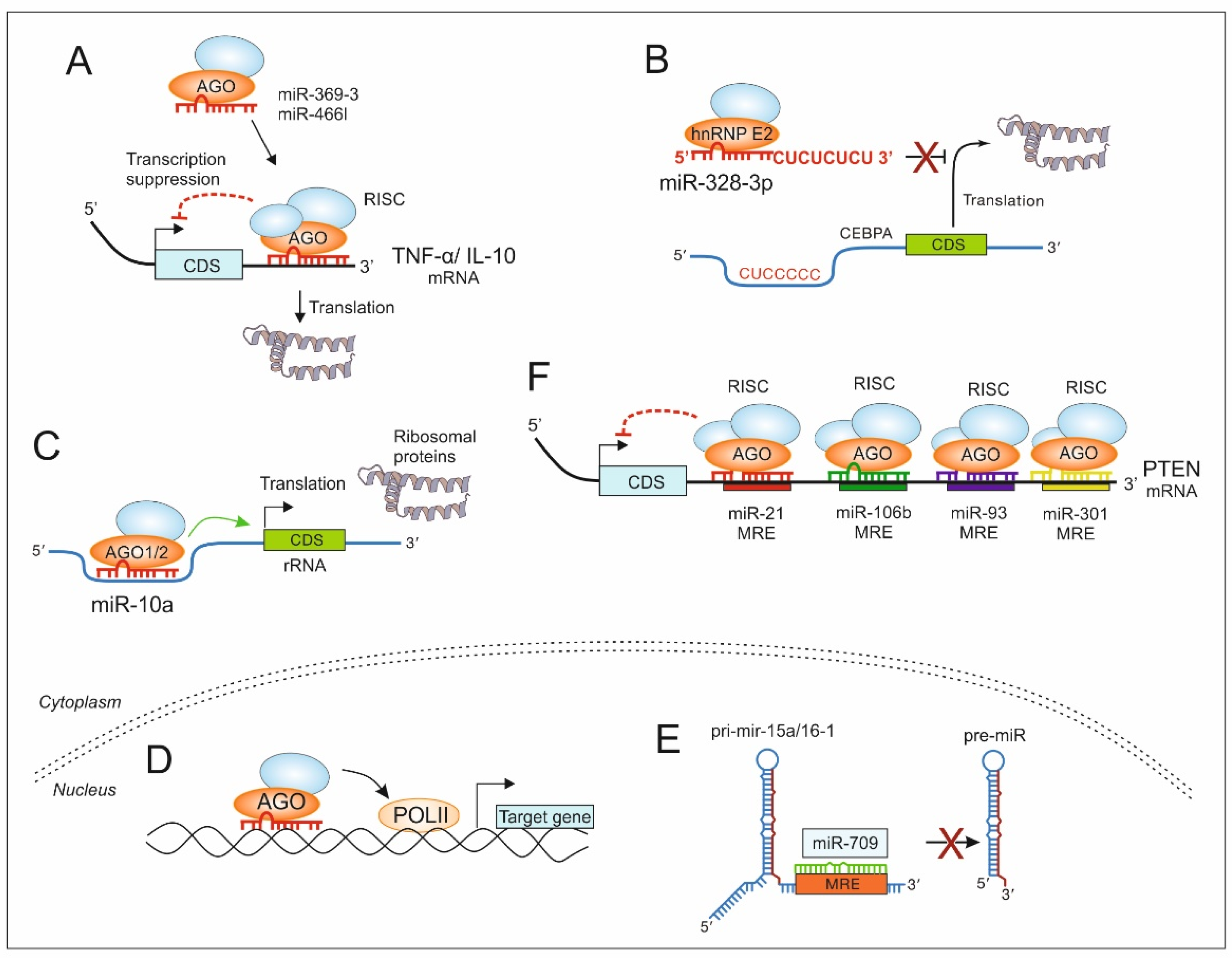
5. Non-Canonical MiRs in the TME
6. Cascading Effects of MiRs in the TME
7. MiRs in the TME—A Road Ahead!
8. Conclusions and Prospective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Odes, E.J.; Randolph-Quinney, P.S.; Steyn, M.; Throckmorton, Z.; Smilg, J.S.; Zipfel, B.; Augustine, T.N.; Beer, F.d.; Hoffman, J.W.; Franklin, R.D.; et al. Earliest hominin cancer: 1.7-million-year-old osteosarcoma from Swartkrans Cave, South Africa. S. Afr. J. Sci. 2016, 112. [Google Scholar] [CrossRef]
- Randolph-Quinney, P.S.; Williams, S.A.; Steyn, M.; Meyer, M.R.; Smilg, J.S.; Churchill, S.E.; Odes, E.J.; Augustine, T.; Tafforeau, P.; Berger, L.R. Osteogenic tumour in Australopithecus sediba: Earliest hominin evidence for neoplastic disease. S. Afr. J. Sci. 2016, 112. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.; Woodward, W.A.; Wang, X.; Reuben, J.M.; Ueno, N.T. Inflammatory breast cancer biology: The tumour microenvironment is key. Nat. Rev. Cancer 2018, 18, 485–499. [Google Scholar] [CrossRef]
- Yang, L.; Li, A.; Lei, Q.; Zhang, Y. Tumor-intrinsic signaling pathways: Key roles in the regulation of the immunosuppressive tumor microenvironment. J. Hematol. Oncol. 2019, 12, 125. [Google Scholar] [CrossRef]
- Najafi, M.; Goradel, N.H.; Farhood, B.; Salehi, E.; Solhjoo, S.; Toolee, H.; Kharazinejad, E.; Mortezaee, K. Tumor microenvironment: Interactions and therapy. J. Cell. Physiol. 2019, 234, 5700–5721. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Moreno-Altamirano, M.M.B.; Prado-Garcia, H.; Sánchez-García, F.J. Lactate Contribution to the Tumor Microenvironment: Mechanisms, Effects on Immune Cells and Therapeutic Relevance. Front. Immunol. 2016, 7, 52. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef]
- Box, C.; Rogers, S.J.; Mendiola, M.; Eccles, S.A. Tumour-microenvironmental interactions: Paths to progression and targets for treatment. Semin. Cancer Biol. 2010, 20, 128–138. [Google Scholar] [CrossRef]
- Frank, A.-C.; Ebersberger, S.; Fink, A.F.; Lampe, S.; Weigert, A.; Schmid, T.; Ebersberger, I.; Syed, S.N.; Brüne, B. Apoptotic tumor cell-derived microRNA-375 uses CD36 to alter the tumor-associated macrophage phenotype. Nat. Commun. 2019, 10, 1135. [Google Scholar] [CrossRef]
- Syed, S.N.; Frank, A.-C.; Raue, R.; Brüne, B. MicroRNA-A Tumor Trojan Horse for Tumor-Associated Macrophages. Cells 2019, 8, 1482. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, A.F.; Lee, E.S. Non-coding RNA: What is functional and what is junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.P.; Glasner, M.E.; Yekta, S.; Burge, C.B.; Bartel, D.P. Vertebrate microRNA genes. Science 2003, 299, 1540. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Lewis, B.P.; Shih, I.-H.; Jones-Rhoades, M.W.; Bartel, D.P.; Burge, C.B. Prediction of mammalian microRNA targets. Cell 2003, 115, 787–798. [Google Scholar] [CrossRef]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of microRNA-target recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Lee, Y.; Kim, M.; Han, J.; Yeom, K.-H.; Lee, S.; Baek, S.H.; Kim, V.N. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004, 23, 4051–4060. [Google Scholar] [CrossRef]
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol. 2006, 13, 1097–1101. [Google Scholar] [CrossRef]
- Chu, C.-Y.; Rana, T.M. Small RNAs: Regulators and guardians of the genome. J. Cell. Physiol. 2007, 213, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Rådmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Lund, E.; Güttinger, S.; Calado, A.; Dahlberg, J.E.; Kutay, U. Nuclear export of microRNA precursors. Science 2004, 303, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Chendrimada, T.P.; Gregory, R.I.; Kumaraswamy, E.; Norman, J.; Cooch, N.; Nishikura, K.; Shiekhattar, R. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature 2005, 436, 740–744. [Google Scholar] [CrossRef]
- Wee, L.M.; Flores-Jasso, C.F.; Salomon, W.E.; Zamore, P.D. Argonaute divides its RNA guide into domains with distinct functions and RNA-binding properties. Cell 2012, 151, 1055–1067. [Google Scholar] [CrossRef]
- Jonas, S.; Izaurralde, E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The biochemical basis of microRNA targeting efficacy. Science 2019, 366. [Google Scholar] [CrossRef]
- Grimson, A.; Farh, K.K.-H.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Molecular Cell 2007, 27, 91–105. [Google Scholar] [CrossRef]
- Schoepp, M.; Ströse, A.J.; Haier, J. Dysregulation of miRNA Expression in Cancer Associated Fibroblasts (CAFs) and Its Consequences on the Tumor Microenvironment. Cancers (Basel) 2017, 9, 54. [Google Scholar] [CrossRef]
- Vojtechova, Z.; Tachezy, R. The Role of miRNAs in Virus-Mediated Oncogenesis. Int. J. Mol. Sci. 2018, 19, 1217. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Tamkovich, S.; Tutanov, O.; Efimenko, A.; Grigor’eva, A.; Ryabchikova, E.; Kirushina, N.; Vlassov, V.; Tkachuk, V.; Laktionov, P. Blood Circulating Exosomes Contain Distinguishable Fractions of Free and Cell-Surface-Associated Vesicles. Curr. Mol. Med. 2019, 19, 273–285. [Google Scholar] [CrossRef]
- Xu, L.; Wu, L.-F.; Deng, F.-Y. Exosome: An Emerging Source of Biomarkers for Human Diseases. Curr. Mol. Med. 2019, 19, 387–394. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Greening, D.W.; Gopal, S.K.; Xu, R.; Simpson, R.J.; Chen, W. Exosomes and their roles in immune regulation and cancer. Semin. Cell Dev. Biol. 2015, 40, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, K.; Inoue, K.; Fujiwara, A.; Hatakeyama, K.; Kanto, K.; Watanabe, Y.; Muramatsu, K.; Fukuda, Y.; Ogura, S.-I.; Yamaguchi, K.; et al. Let-7 microRNA family is selectively secreted into the extracellular environment via exosomes in a metastatic gastric cancer cell line. PLoS ONE 2010, 5, e13247. [Google Scholar] [CrossRef]
- Pigati, L.; Yaddanapudi, S.C.S.; Iyengar, R.; Kim, D.-J.; Hearn, S.A.; Danforth, D.; Hastings, M.L.; Duelli, D.M. Selective release of microRNA species from normal and malignant mammary epithelial cells. PLoS ONE 2010, 5, e13515. [Google Scholar] [CrossRef]
- Dai, R.; Ahmed, S.A. MicroRNA, a new paradigm for understanding immunoregulation, inflammation, and autoimmune diseases. Transl. Res. 2011, 157, 163–179. [Google Scholar] [CrossRef]
- Turchinovich, A.; Weiz, L.; Burwinkel, B. Extracellular miRNAs: The mystery of their origin and function. Trends Biochem. Sci. 2012, 37, 460–465. [Google Scholar] [CrossRef]
- Turchinovich, A.; Weiz, L.; Langheinz, A.; Burwinkel, B. Characterization of extracellular circulating microRNA. Nucleic Acids Res. 2011, 39, 7223–7233. [Google Scholar] [CrossRef]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Janas, T.; Janas, M.M.; Sapoń, K.; Janas, T. Mechanisms of RNA loading into exosomes. FEBS Lett. 2015, 589, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Poon, I.K.H.; Lucas, C.D.; Rossi, A.G.; Ravichandran, K.S. Apoptotic cell clearance: Basic biology and therapeutic potential. Nat. Rev. Immunol. 2014, 14, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Atkin-Smith, G.K.; Poon, I.K.H. Disassembly of the Dying: Mechanisms and Functions. Trends Cell Biol. 2017, 27, 151–162. [Google Scholar] [CrossRef]
- El Andaloussi, S.; Mäger, I.; Breakefield, X.O.; Wood, M.J.A. Extracellular vesicles: Biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013, 12, 347–357. [Google Scholar] [CrossRef]
- Yim, N.; Ryu, S.-W.; Choi, K.; Lee, K.R.; Lee, S.; Choi, H.; Kim, J.; Shaker, M.R.; Sun, W.; Park, J.-H.; et al. Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein-protein interaction module. Nat. Commun. 2016, 7, 12277. [Google Scholar] [CrossRef]
- Staubach, S.; Razawi, H.; Hanisch, F.-G. Proteomics of MUC1-containing lipid rafts from plasma membranes and exosomes of human breast carcinoma cells MCF-7. Proteomics 2009, 9, 2820–2835. [Google Scholar] [CrossRef]
- Record, M.; Subra, C.; Silvente-Poirot, S.; Poirot, M. Exosomes as intercellular signalosomes and pharmacological effectors. Biochem. Pharmacol. 2011, 81, 1171–1182. [Google Scholar] [CrossRef]
- Lee, Y.; El Andaloussi, S.; Wood, M.J.A. Exosomes and microvesicles: Extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet. 2012, 21, R125–R134. [Google Scholar] [CrossRef]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef]
- Arroyo, J.D.; Chevillet, J.R.; Kroh, E.M.; Ruf, I.K.; Pritchard, C.C.; Gibson, D.F.; Mitchell, P.S.; Bennett, C.F.; Pogosova-Agadjanyan, E.L.; Stirewalt, D.L.; et al. Argonaute2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc. Natl. Acad. Sci. USA. 2011, 108, 5003–5008. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, S.; Weber, J.; Baxter, D.; Galas, D.J. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res. 2010, 38, 7248–7259. [Google Scholar] [CrossRef] [PubMed]
- Prud’homme, G.J.; Glinka, Y.; Lichner, Z.; Yousef, G.M. Neuropilin-1 is a receptor for extracellular miRNA and AGO2/miRNA complexes and mediates the internalization of miRNAs that modulate cell function. Oncotarget 2016, 7, 68057–68071. [Google Scholar] [CrossRef]
- Vickers, K.C.; Palmisano, B.T.; Shoucri, B.M.; Shamburek, R.D.; Remaley, A.T. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat. Cell Biol. 2011, 13, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Riwanto, M.; Besler, C.; Knau, A.; Fichtlscherer, S.; Röxe, T.; Zeiher, A.M.; Landmesser, U.; Dimmeler, S. Characterization of levels and cellular transfer of circulating lipoprotein-bound microRNAs. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, L.S.; Simionescu, N.; Sanda, G.M.; Carnuta, M.G.; Stancu, C.S.; Popescu, A.C.; Popescu, M.R.; Vlad, A.; Dimulescu, D.R.; Simionescu, M.; et al. MiR-486 and miR-92a Identified in Circulating HDL Discriminate between Stable and Vulnerable Coronary Artery Disease Patients. PLoS ONE 2015, 10, e0140958. [Google Scholar] [CrossRef] [PubMed]
- Tabet, F.; Vickers, K.C.; Cuesta Torres, L.F.; Wiese, C.B.; Shoucri, B.M.; Lambert, G.; Catherinet, C.; Prado-Lourenco, L.; Levin, M.G.; Thacker, S.; et al. HDL-transferred microRNA-223 regulates ICAM-1 expression in endothelial cells. Nat. Commun. 2014, 5, 3292. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, L.-A.; Murphy, P.R. MicroRNA: Biogenesis, Function and Role in Cancer. CG 2010, 11, 537–561. [Google Scholar] [CrossRef]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Bartel, D.P.; Chen, C.-Z. Micromanagers of gene expression: The potentially widespread influence of metazoan microRNAs. Nat. Rev. Genet. 2004, 5, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Li, W.-H. MicroRNA regulation of human protein protein interaction network. RNA 2007, 13, 1402–1408. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Yu, Z.; Pan, Y.; Purisima, E.O.; Wang, E. MicroRNAs preferentially target the genes with high transcriptional regulation complexity. Biochem. Biophys. Res. Commun. 2007, 352, 733–738. [Google Scholar] [CrossRef]
- Tang, R.; Li, L.; Zhu, D.; Hou, D.; Cao, T.; Gu, H.; Zhang, J.; Chen, J.; Zhang, C.-Y.; Zen, K. Mouse miRNA-709 directly regulates miRNA-15a/16-1 biogenesis at the posttranscriptional level in the nucleus: Evidence for a microRNA hierarchy system. Cell Res. 2012, 22, 504–515. [Google Scholar] [CrossRef]
- Wang, D.; Sun, X.; Wei, Y.; Liang, H.; Yuan, M.; Jin, F.; Chen, X.; Liu, Y.; Zhang, C.-Y.; Li, L.; et al. Nuclear miR-122 directly regulates the biogenesis of cell survival oncomiR miR-21 at the posttranscriptional level. Nucleic Acids Res. 2018, 46, 2012–2029. [Google Scholar] [CrossRef]
- Derti, A.; Garrett-Engele, P.; Macisaac, K.D.; Stevens, R.C.; Sriram, S.; Chen, R.; Rohl, C.A.; Johnson, J.M.; Babak, T. A quantitative atlas of polyadenylation in five mammals. Genome Res. 2012, 22, 1173–1183. [Google Scholar] [CrossRef]
- Tian, B.; Hu, J.; Zhang, H.; Lutz, C.S. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005, 33, 201–212. [Google Scholar] [CrossRef]
- Morris, A.R.; Bos, A.; Diosdado, B.; Rooijers, K.; Elkon, R.; Bolijn, A.S.; Carvalho, B.; Meijer, G.A.; Agami, R. Alternative cleavage and polyadenylation during colorectal cancer development. Clin. Cancer Res. 2012, 18, 5256–5266. [Google Scholar] [CrossRef]
- Mayr, C.; Bartel, D.P. Widespread shortening of 3′ UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 2009, 138, 673–684. [Google Scholar] [CrossRef]
- Andres, S.F.; Williams, K.N.; Plesset, J.B.; Headd, J.J.; Mizuno, R.; Chatterji, P.; Lento, A.A.; Klein-Szanto, A.J.; Mick, R.; Hamilton, K.E.; et al. IMP1 3′ UTR shortening enhances metastatic burden in colorectal cancer. Carcinogenesis 2019, 40, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Kim, J.K. Driving glioblastoma growth by alternative polyadenylation. Cell Res. 2014, 24, 1023–1024. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Masamha, C.P.; Xia, Z.; Yang, J.; Albrecht, T.R.; Li, M.; Shyu, A.-B.; Li, W.; Wagner, E.J. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 2014, 510, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Ding, J.; Li, D.; Yang, G.; Cheng, Z.; Zhu, Q. NUDT21 regulates 3′-UTR length and microRNA-mediated gene silencing in hepatocellular carcinoma. Cancer Lett. 2017, 410, 158–168. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Zhu, X.; Wang, H.; Jin, X. A dynamic interplay between alternative polyadenylation and microRNA regulation: Implications for cancer (Review). Int. J. Oncol. 2013, 43, 995–1001. [Google Scholar] [CrossRef]
- Chen, R.W.; Bemis, L.T.; Amato, C.M.; Myint, H.; Tran, H.; Birks, D.K.; Eckhardt, S.G.; Robinson, W.A. Truncation in CCND1 mRNA alters miR-16-1 regulation in mantle cell lymphoma. Blood 2008, 112, 822–829. [Google Scholar] [CrossRef]
- Sandberg, R.; Neilson, J.R.; Sarma, A.; Sharp, P.A.; Burge, C.B. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microRNA target sites. Science 2008, 320, 1643–1647. [Google Scholar] [CrossRef]
- Vasudevan, S.; Tong, Y.; Steitz, J.A. Switching from repression to activation: microRNAs can up-regulate translation. Science 2007, 318, 1931–1934. [Google Scholar] [CrossRef]
- Ma, F.; Liu, X.; Li, D.; Wang, P.; Li, N.; Lu, L.; Cao, X. MicroRNA-466l upregulates IL-10 expression in TLR-triggered macrophages by antagonizing RNA-binding protein tristetraprolin-mediated IL-10 mRNA degradation. J. Immunol. 2010, 184, 6053–6059. [Google Scholar] [CrossRef]
- Eiring, A.M.; Harb, J.G.; Neviani, P.; Garton, C.; Oaks, J.J.; Spizzo, R.; Liu, S.; Schwind, S.; Santhanam, R.; Hickey, C.J.; et al. miR-328 functions as an RNA decoy to modulate hnRNP E2 regulation of mRNA translation in leukemic blasts. Cell 2010, 140, 652–665. [Google Scholar] [CrossRef]
- Ørom, U.A.; Nielsen, F.C.; Lund, A.H. MicroRNA-10a binds the 5′ UTR of ribosomal protein mRNAs and enhances their translation. Molecular Cell 2008, 30, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Place, R.F.; Li, L.-C.; Pookot, D.; Noonan, E.J.; Dahiya, R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Majid, S.; Dar, A.A.; Saini, S.; Yamamura, S.; Hirata, H.; Tanaka, Y.; Deng, G.; Dahiya, R. MicroRNA-205-directed transcriptional activation of tumor suppressor genes in prostate cancer. Cancer 2010, 116, 5637–5649. [Google Scholar] [CrossRef]
- Matsui, M.; Chu, Y.; Zhang, H.; Gagnon, K.T.; Shaikh, S.; Kuchimanchi, S.; Manoharan, M.; Corey, D.R.; Janowski, B.A. Promoter RNA links transcriptional regulation of inflammatory pathway genes. Nucleic Acids Res. 2013, 41, 10086–10109. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Yao, Y.; Wang, Y.; Liu, B.; Wu, W.; Chen, J.; Su, F.; Yao, H.; Song, E. Up-regulation of miR-21 mediates resistance to trastuzumab therapy for breast cancer. J. Biol. Chem. 2011, 286, 19127–19137. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Xie, J.; Zhang, M.; Zhao, Z.; Wan, Y.; Yao, Y. miRNA-21 promotes proliferation and invasion of triple-negative breast cancer cells through targeting PTEN. Am. J. Transl. Res. 2017, 9, 953–961. [Google Scholar] [PubMed]
- Yu, X.; Li, R.; Shi, W.; Jiang, T.; Wang, Y.; Li, C.; Qu, X. Silencing of MicroRNA-21 confers the sensitivity to tamoxifen and fulvestrant by enhancing autophagic cell death through inhibition of the PI3K-AKT-mTOR pathway in breast cancer cells. Biomed. Pharmacother. 2016, 77, 37–44. [Google Scholar] [CrossRef]
- Li, N.; Miao, Y.; Shan, Y.; Liu, B.; Li, Y.; Zhao, L.; Jia, L. MiR-106b and miR-93 regulate cell progression by suppression of PTEN via PI3K/Akt pathway in breast cancer. Cell Death Dis. 2017, 8, e2796. [Google Scholar] [CrossRef]
- Shi, W.; Gerster, K.; Alajez, N.M.; Tsang, J.; Waldron, L.; Pintilie, M.; Hui, A.B.; Sykes, J.; P’ng, C.; Miller, N.; et al. MicroRNA-301 mediates proliferation and invasion in human breast cancer. Cancer Res. 2011, 71, 2926–2937. [Google Scholar] [CrossRef]
- Ho, J.J.D.; Metcalf, J.L.; Yan, M.S.; Turgeon, P.J.; Wang, J.J.; Chalsev, M.; Petruzziello-Pellegrini, T.N.; Tsui, A.K.Y.; He, J.Z.; Dhamko, H.; et al. Functional importance of Dicer protein in the adaptive cellular response to hypoxia. J. Biol. Chem. 2012, 287, 29003–29020. [Google Scholar] [CrossRef]
- Shen, J.; Xia, W.; Khotskaya, Y.B.; Huo, L.; Nakanishi, K.; Lim, S.-O.; Du, Y.; Wang, Y.; Chang, W.-C.; Chen, C.-H.; et al. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature 2013, 497, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Pampalakis, G.; Diamandis, E.P.; Katsaros, D.; Sotiropoulou, G. Down-regulation of dicer expression in ovarian cancer tissues. Clin. Biochem. 2010, 43, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Dedes, K.J.; Natrajan, R.; Lambros, M.B.; Geyer, F.C.; Lopez-Garcia, M.A.; Savage, K.; Jones, R.L.; Reis-Filho, J.S. Down-regulation of the miRNA master regulators Drosha and Dicer is associated with specific subgroups of breast cancer. Eur. J. Cancer 2011, 47, 138–150. [Google Scholar] [CrossRef]
- Karube, Y.; Tanaka, H.; Osada, H.; Tomida, S.; Tatematsu, Y.; Yanagisawa, K.; Yatabe, Y.; Takamizawa, J.; Miyoshi, S.; Mitsudomi, T.; et al. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci. 2005, 96, 111–115. [Google Scholar] [CrossRef]
- Karreth, F.A.; Pandolfi, P.P. ceRNA cross-talk in cancer: When ce-bling rivalries go awry. Cancer Discov. 2013, 3, 1113–1121. [Google Scholar] [CrossRef]
- Li, L.M.; Luo, F.J.; Song, X. MicroRNA-370-3p inhibits cell proliferation and induces chronic myelogenous leukaemia cell apoptosis by suppressing PDLIM1/Wnt/β-catenin signalling. Neoplasma 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-F.; Zhang, J.-G.; Zhou, H.; Dai, T.-T.; Guo, F.-B.; Xu, S.-Y.; Chen, Y. MicroRNA-425-5p promotes breast cancer cell growth by inducing PI3K/AKT signaling. Kaohsiung J. Med. Sci. 2019. [Google Scholar] [CrossRef]
- Bahena-Ocampo, I.; Espinosa, M.; Ceballos-Cancino, G.; Lizarraga, F.; Campos-Arroyo, D.; Schwarz, A.; Maldonado, V.; Melendez-Zajgla, J.; Garcia-Lopez, P. miR-10b expression in breast cancer stem cells supports self-renewal through negative PTEN regulation and sustained AKT activation. EMBO Rep. 2016, 17, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Zheng, W.; Li, N.; Su, Z.; Zhao, L.; Zhou, H.; Jia, L. MicroRNA-130b targets PTEN to mediate drug resistance and proliferation of breast cancer cells via the PI3K/Akt signaling pathway. Sci. Rep. 2017, 7, 41942. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Zhu, Q.; Tang, L. MiR-99a antitumor activity in human breast cancer cells through targeting of mTOR expression. PLoS ONE 2014, 9, e92099. [Google Scholar] [CrossRef]
- Wang, B.; Wang, H.; Yang, Z. MiR-122 inhibits cell proliferation and tumorigenesis of breast cancer by targeting IGF1R. PLoS ONE 2012, 7, e47053. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Barry, L.; Lin, S.S.H.; Huang, V.; Li, L.-C. RNAa in action: From the exception to the norm. RNA Biol. 2014, 11, 1221–1225. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huang, V.; Place, R.F.; Portnoy, V.; Wang, J.; Qi, Z.; Jia, Z.; Yu, A.; Shuman, M.; Yu, J.; Li, L.-C. Upregulation of Cyclin B1 by miRNA and its implications in cancer. Nucleic Acids Res. 2012, 40, 1695–1707. [Google Scholar] [CrossRef]
- Toscano-Garibay, J.D.; Aquino-Jarquin, G. Transcriptional regulation mechanism mediated by miRNA-DNA•DNA triplex structure stabilized by Argonaute. Biochim. Biophys. Acta 2014, 1839, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Paugh, S.W.; Coss, D.R.; Bao, J.; Laudermilk, L.T.; Grace, C.R.; Ferreira, A.M.; Waddell, M.B.; Ridout, G.; Naeve, D.; Leuze, M.; et al. MicroRNAs Form Triplexes with Double Stranded DNA at Sequence-Specific Binding Sites; a Eukaryotic Mechanism via which microRNAs Could Directly Alter Gene Expression. PLoS Comput. Biol. 2016, 12, e1004744. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Molecular Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Portnoy, V.; Huang, V.; Place, R.F.; Li, L.-C. Small RNA and transcriptional upregulation. Wiley Interdiscip. Rev. RNA 2011, 2, 748–760. [Google Scholar] [CrossRef]
- Perrotti, D.; Cesi, V.; Trotta, R.; Guerzoni, C.; Santilli, G.; Campbell, K.; Iervolino, A.; Condorelli, F.; Gambacorti-Passerini, C.; Caligiuri, M.A.; et al. BCR-ABL suppresses C/EBPalpha expression through inhibitory action of hnRNP E2. Nat. Genet. 2002, 30, 48–58. [Google Scholar] [CrossRef]
- Guil, S.; Cáceres, J.F. The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nat. Struct. Mol. Biol. 2007, 14, 591–596. [Google Scholar] [CrossRef]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.-K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liang, H.; Zhang, J.; Zen, K.; Zhang, C.-Y. microRNAs are ligands of Toll-like receptors. RNA 2013, 19, 737–739. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Croce, C.M. A new role for microRNAs, as ligands of Toll-like receptors. RNA Biol. 2013, 10, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.’e.; Ding, J.; Yang, J.; Guo, X.; Zheng, Y. MicroRNA Roles in the Nuclear Factor Kappa B Signaling Pathway in Cancer. Front. Immunol. 2018, 9, 546. [Google Scholar] [CrossRef] [PubMed]
- Özata, D.M.; Li, X.; Lee, L.; Liu, J.; Warsito, D.; Hajeri, P.; Hultman, I.; Fotouhi, O.; Marklund, S.; Ährlund-Richter, L.; et al. Loss of miR-514a-3p regulation of PEG3 activates the NF-kappa B pathway in human testicular germ cell tumors. Cell Death Dis. 2017, 8, e2759. [Google Scholar] [CrossRef]
- Jeon, Y.-J.; Middleton, J.; Kim, T.; Laganà, A.; Piovan, C.; Secchiero, P.; Nuovo, G.J.; Cui, R.; Joshi, P.; Romano, G.; et al. A set of NF-κB-regulated microRNAs induces acquired TRAIL resistance in lung cancer. Proc. Natl. Acad. Sci. USA 2015, 112, E3355–E3364. [Google Scholar] [CrossRef]
- Ma, X.; Becker Buscaglia, L.E.; Barker, J.R.; Li, Y. MicroRNAs in NF-kappaB signaling. J. Mol. Cell Biol. 2011, 3, 159–166. [Google Scholar] [CrossRef]
- Guo, L.-M.; Pu, Y.; Han, Z.; Liu, T.; Li, Y.-X.; Liu, M.; Li, X.; Tang, H. MicroRNA-9 inhibits ovarian cancer cell growth through regulation of NF-kappaB1. FEBS J. 2009, 276, 5537–5546. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, H.; Li, P.; Zhao, Y.; Liu, M.; Tang, H. NF-κB-modulated miR-130a targets TNF-α in cervical cancer cells. J. Transl. Med. 2014, 12, 155. [Google Scholar] [CrossRef]
- Zhang, S.; Shan, C.; Kong, G.; Du, Y.; Ye, L.; Zhang, X. MicroRNA-520e suppresses growth of hepatoma cells by targeting the NF-κB-inducing kinase (NIK). Oncogene 2012, 31, 3607–3620. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Palsson-McDermott, E.; Hennessy, E.J.; Martin, C.; O’Leary, J.J.; Ruan, Q.; Johnson, D.S.; Chen, Y.; O’Neill, L.A.J. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat. Immunol. 2010, 11, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Zhang, C.; Li, G.; Jin, G.; Liu, C. MiR-940 inhibited pancreatic ductal adenocarcinoma growth by targeting MyD88. Cell. Physiol. Biochem. 2015, 35, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- d’Adhemar, C.J.; Spillane, C.D.; Gallagher, M.F.; Bates, M.; Costello, K.M.; Barry-O’Crowley, J.; Haley, K.; Kernan, N.; Murphy, C.; Smyth, P.C.; et al. The MyD88+ phenotype is an adverse prognostic factor in epithelial ovarian cancer. PLoS ONE 2014, 9, e100816. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Morgan, M.J.; Choksi, S.; Zhang, Y.; Kim, Y.-S.; Liu, Z.-G. MicroRNAs modulate the noncanonical transcription factor NF-kappaB pathway by regulating expression of the kinase IKKalpha during macrophage differentiation. Nat. Immunol. 2010, 11, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Shin, V.Y.; Jin, H.; Ng, E.K.O.; Cheng, A.S.L.; Chong, W.W.S.; Wong, C.Y.P.; Leung, W.K.; Sung, J.J.Y.; Chu, K.-M. NF-κB targets miR-16 and miR-21 in gastric cancer: Involvement of prostaglandin E receptors. Carcinogenesis 2011, 32, 240–245. [Google Scholar] [CrossRef]
- Wang, H.; Garzon, R.; Sun, H.; Ladner, K.J.; Singh, R.; Dahlman, J.; Cheng, A.; Hall, B.M.; Qualman, S.J.; Chandler, D.S.; et al. NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 2008, 14, 369–381. [Google Scholar] [CrossRef]
- Bazzoni, F.; Rossato, M.; Fabbri, M.; Gaudiosi, D.; Mirolo, M.; Mori, L.; Tamassia, N.; Mantovani, A.; Cassatella, M.A.; Locati, M. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc. Natl. Acad. Sci. USA 2009, 106, 5282–5287. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, S.; Hu, T.; Liu, S.; He, Y.; Sun, S. Up-regulated microRNA-143 transcribed by nuclear factor kappa B enhances hepatocarcinoma metastasis by repressing fibronectin expression. Hepatology 2009, 50, 490–499. [Google Scholar] [CrossRef]
- Taganov, K.D.; Boldin, M.P.; Chang, K.-J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef]
- Scisciani, C.; Vossio, S.; Guerrieri, F.; Schinzari, V.; de Iaco, R.; D’Onorio de Meo, P.; Cervello, M.; Montalto, G.; Pollicino, T.; Raimondo, G.; et al. Transcriptional regulation of miR-224 upregulated in human HCCs by NFκB inflammatory pathways. J. Hepatol. 2012, 56, 855–861. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Hirsch, H.A.; Struhl, K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009, 139, 693–706. [Google Scholar] [CrossRef]
- Mott, J.L.; Kurita, S.; Cazanave, S.C.; Bronk, S.F.; Werneburg, N.W.; Fernandez-Zapico, M.E. Transcriptional suppression of mir-29b-1/mir-29a promoter by c-Myc, hedgehog, and NF-kappaB. J. Cell. Biochem. 2010, 110, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, D.; Scott, G.K.; Schokrpur, S.; Patil, C.K.; Campisi, J.; Benz, C.C. Expression of microRNA-146 suppresses NF-kappaB activity with reduction of metastatic potential in breast cancer cells. Oncogene 2008, 27, 5643–5647. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Cao, J.; Liu, S.; Pan, H.; Liu, X.; Sui, A.; Wang, L.; Yao, R.; Liu, Z.; Liang, J. Upregulated microRNA-429 inhibits the migration of HCC cells by targeting TRAF6 through the NF-κB pathway. Oncol. Rep. 2017, 37, 2883–2890. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhou, B.; Xu, L.; Fan, H.; Xie, J.; Wang, D. MicroRNA-146a promotes gastric cancer cell apoptosis by targeting transforming growth factor β-activated kinase 1. Mol. Med. Rep. 2017, 16, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Wang, R.; Zhou, L.; Zhu, Y.; Gong, J.; Zhuang, S.-M. MicroRNA-26b suppresses the NF-κB signaling and enhances the chemosensitivity of hepatocellular carcinoma cells by targeting TAK1 and TAB3. Mol. Cancer 2014, 13, 35. [Google Scholar] [CrossRef]
- Liu, W.; Wang, X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019, 20, 18. [Google Scholar] [CrossRef]
- Pla, A.; Zhong, X.; Rayner, S. miRAW: A deep learning-based approach to predict microRNA targets by analyzing whole microRNA transcripts. PLoS Comput. Biol. 2018, 14, e1006185. [Google Scholar] [CrossRef]
- Zhao, B.; Xue, B. Significant improvement of miRNA target prediction accuracy in large datasets using meta-strategy based on comprehensive voting and artificial neural networks. BMC Genomics 2019, 20, 158. [Google Scholar] [CrossRef]
- Ebisuya, M.; Yamamoto, T.; Nakajima, M.; Nishida, E. Ripples from neighbouring transcription. Nat. Cell Biol. 2008, 10, 1106–1113. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Syed, S.N.; Brüne, B. MicroRNAs as Emerging Regulators of Signaling in the Tumor Microenvironment. Cancers 2020, 12, 911. https://doi.org/10.3390/cancers12040911
Syed SN, Brüne B. MicroRNAs as Emerging Regulators of Signaling in the Tumor Microenvironment. Cancers. 2020; 12(4):911. https://doi.org/10.3390/cancers12040911
Chicago/Turabian StyleSyed, Shahzad Nawaz, and Bernhard Brüne. 2020. "MicroRNAs as Emerging Regulators of Signaling in the Tumor Microenvironment" Cancers 12, no. 4: 911. https://doi.org/10.3390/cancers12040911
APA StyleSyed, S. N., & Brüne, B. (2020). MicroRNAs as Emerging Regulators of Signaling in the Tumor Microenvironment. Cancers, 12(4), 911. https://doi.org/10.3390/cancers12040911