MiRNAs Targeting Double Strand DNA Repair Pathways Lurk in Genomically Unstable Rare Fragile Sites and Determine Cancer Outcomes


Abstract
1. Introduction
2. Results and Discussion
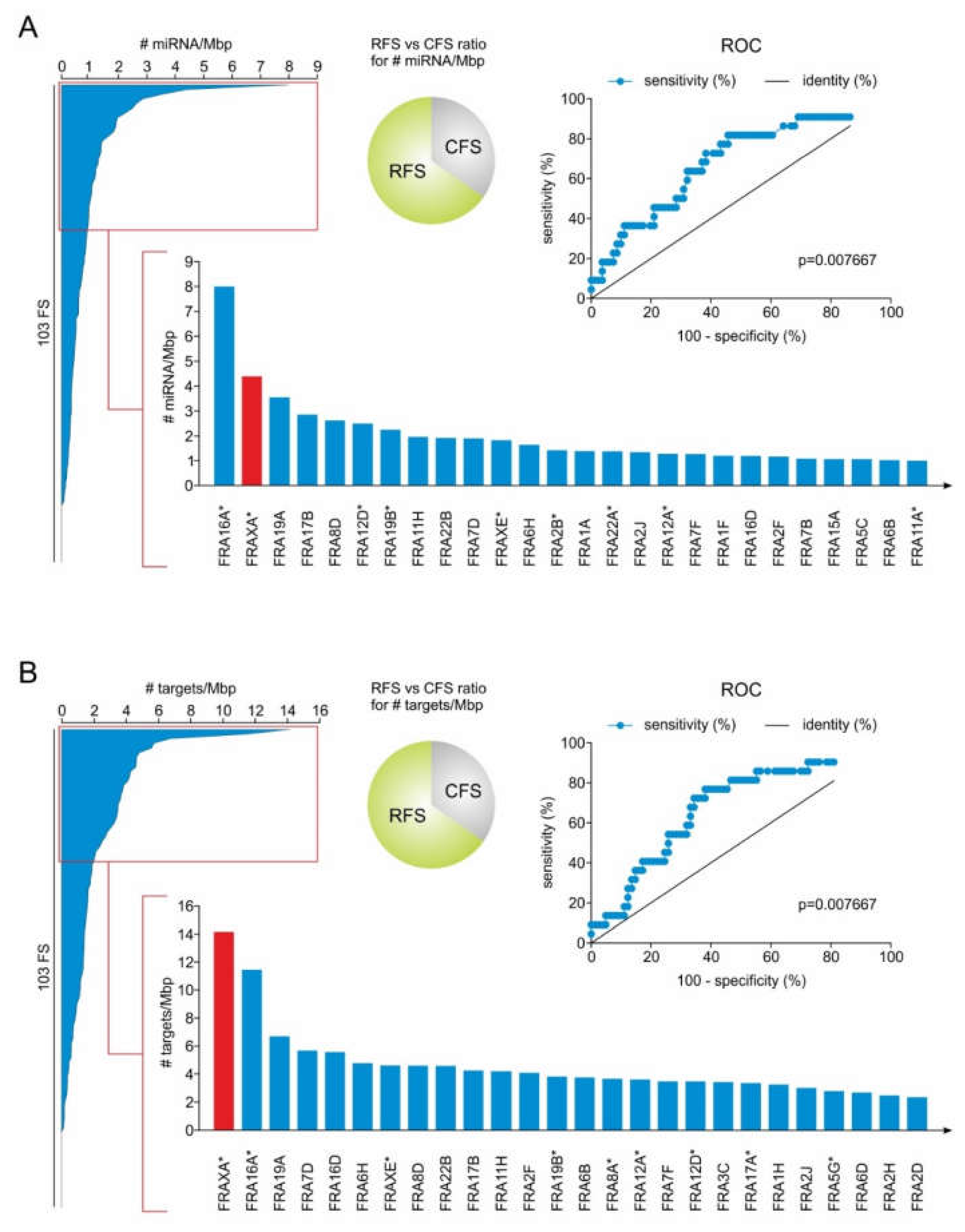
2.1. RFS Presents a Higher Density of miRNA Genes Compared to CFS
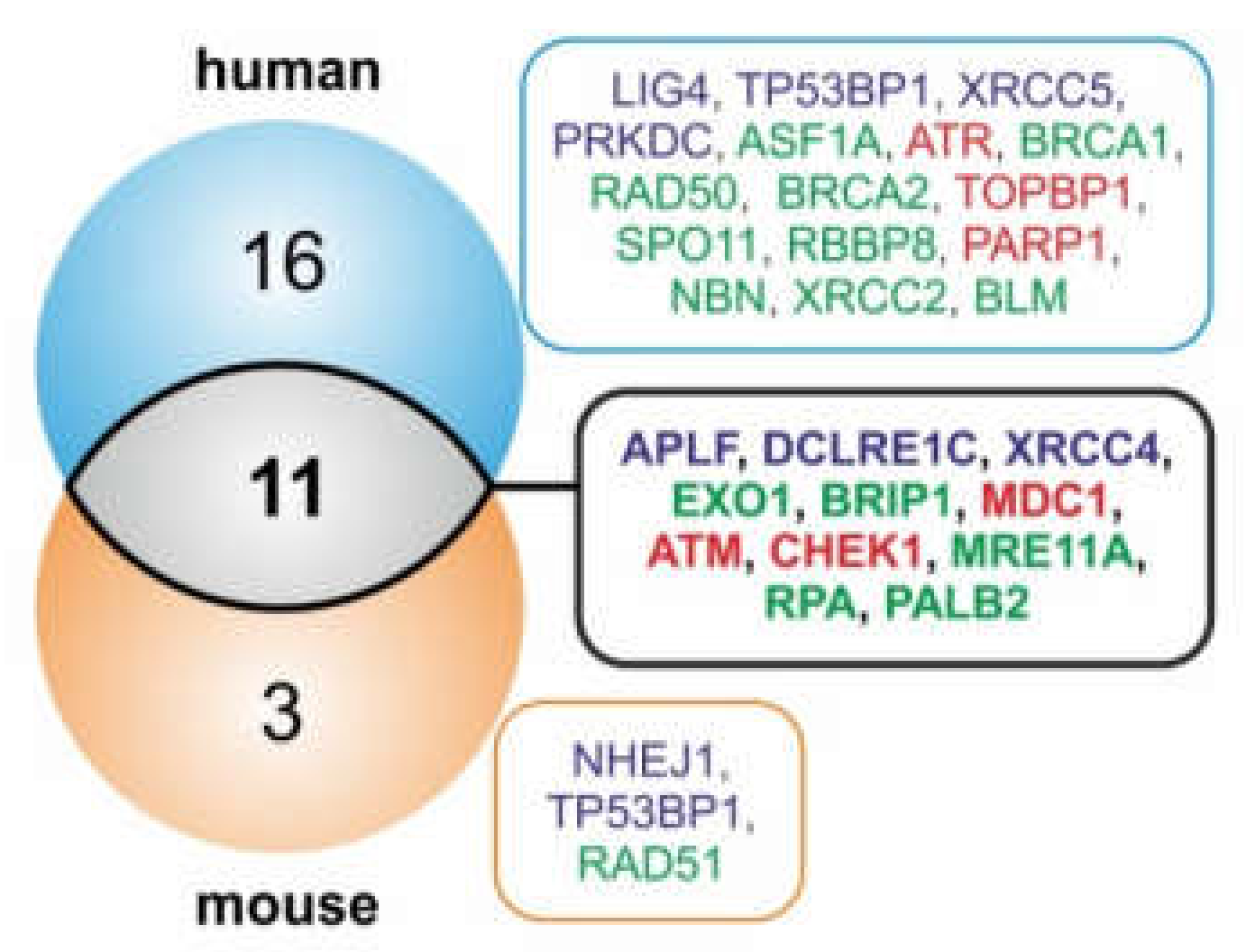
2.2. MiRNA Genes Targeting DSB Repair Components Over-Accumulate in RFS
2.3. FRAXA is a Conserved Hotspot for miRNA Genes Inhibiting Components of DSB Repair Pathways
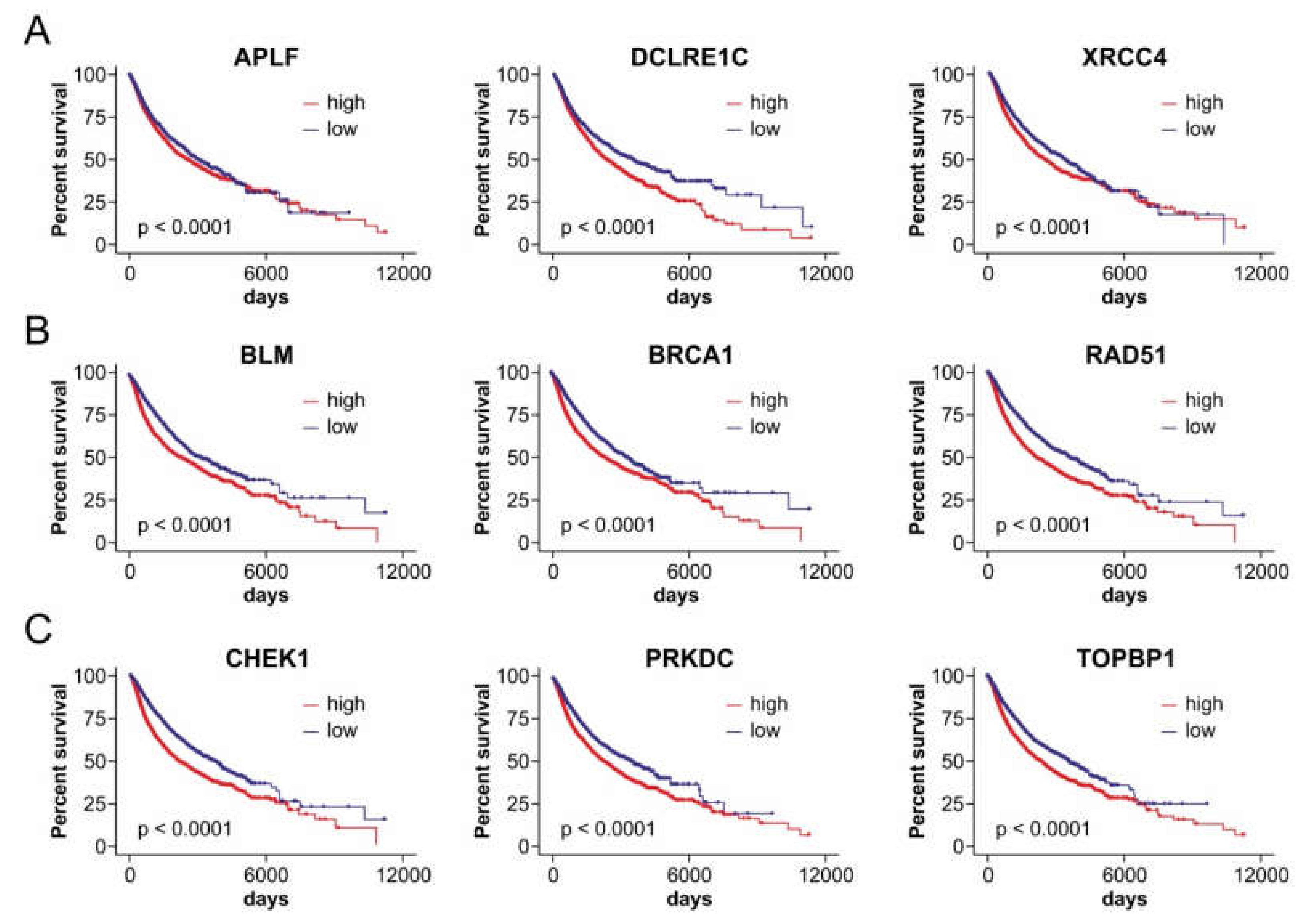
2.4. Axes of FRAXA-Residing miRNA Genes and Their DSB Repair Targets Affect Patient Outcomes in a Cancer Type-Dependent Manner
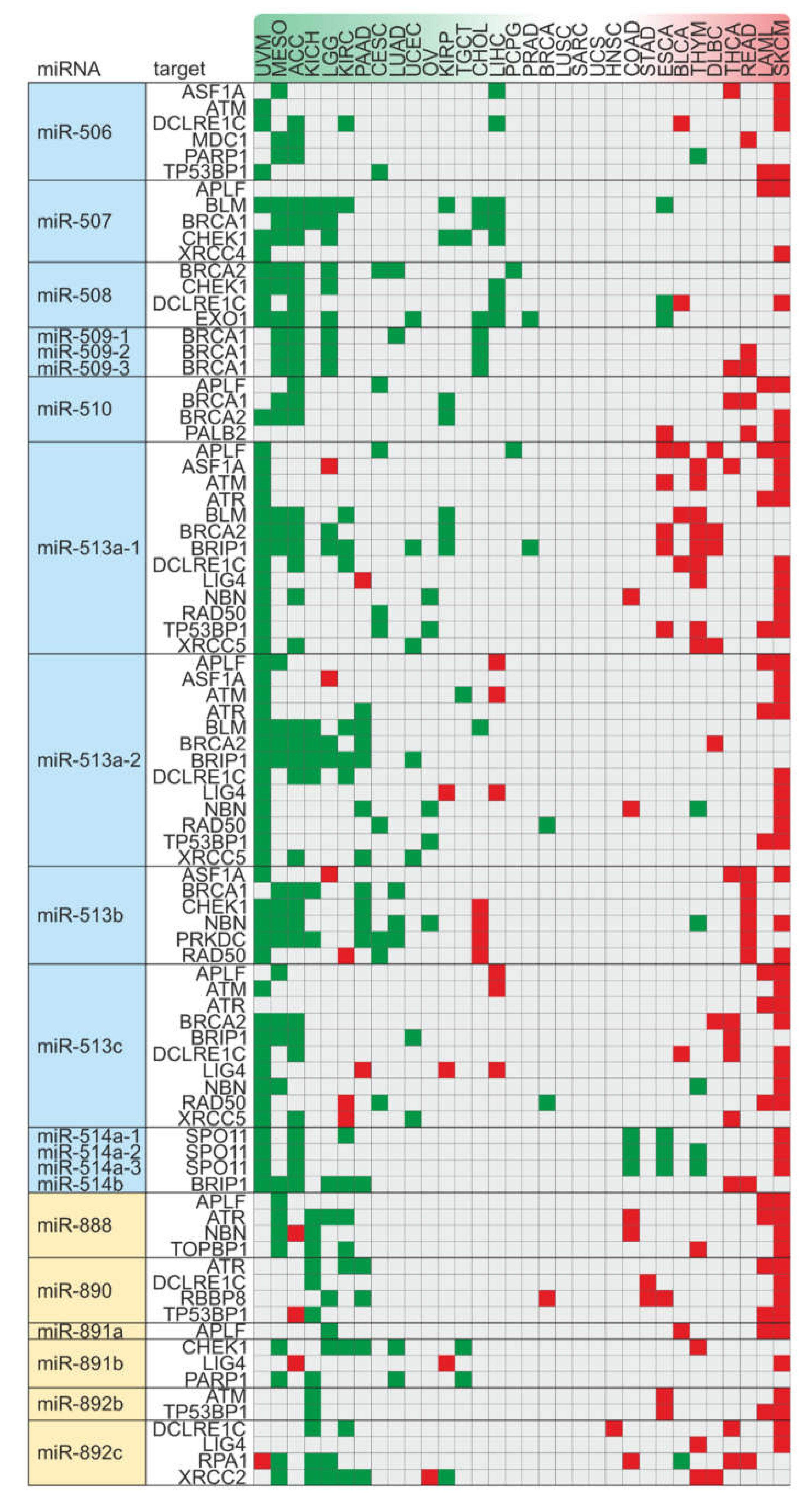
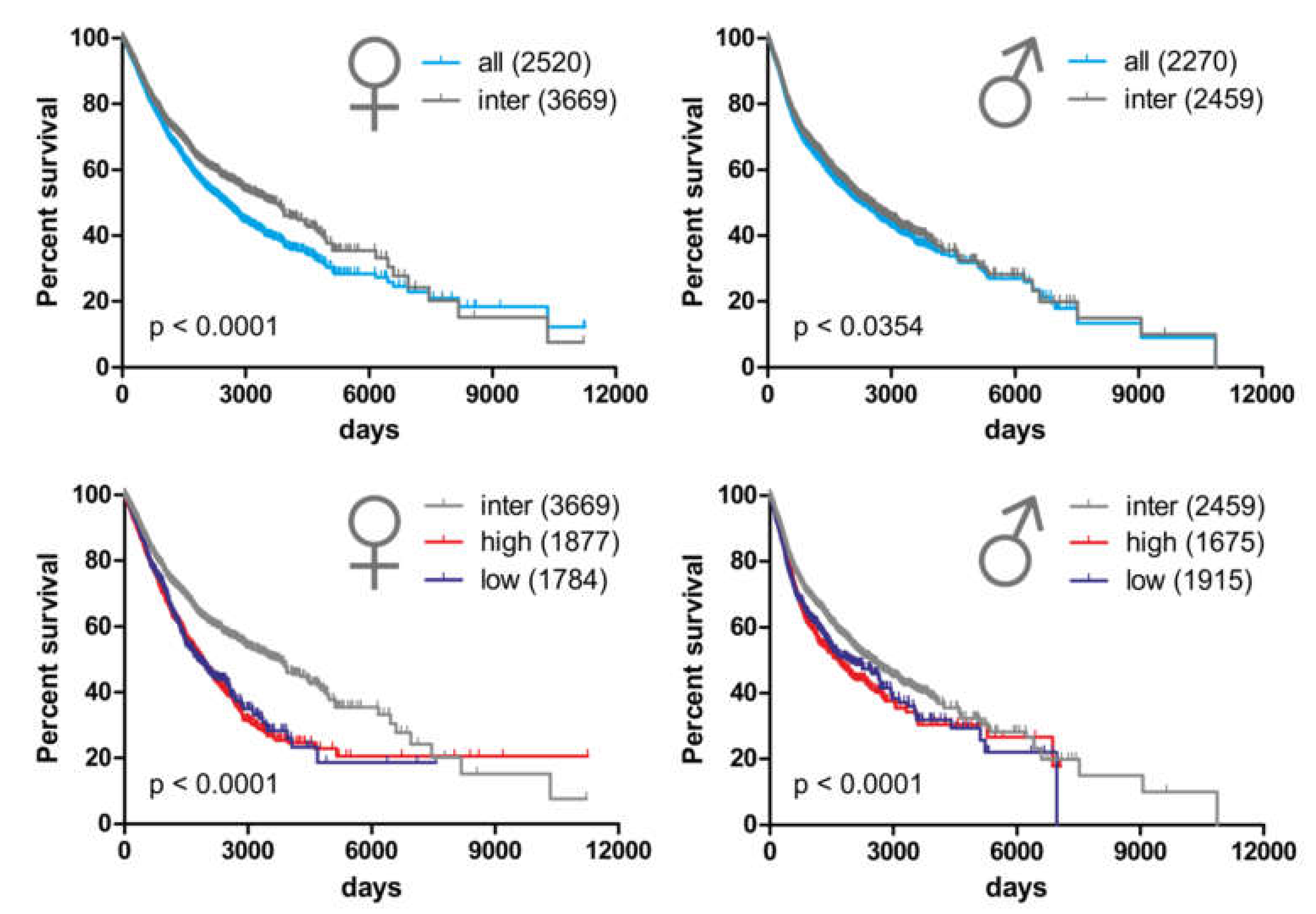
2.5. Alterations in Copy Number Variation of the miRNA-hosting FRAXA Regions Differentially Affect Outcomes in Male Versus Female Cancer Patients
3. Materials and Methods
3.1. Identification of miRNA Genes in Common and Rare Fragile Sites
3.2. MicroRNA Target Prediction
3.3. Kaplan–Meier Survival Analysis of TCGA Patient Data
3.4. Expression Correlation
3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.; McVey, M. Error-Prone Repair of DNA Double-Strand Breaks. J. Cell. Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.M.; Yates, L.A.; Zhang, X. Structures and regulations of ATM and ATR, master kinases in genome integrity. Curr. Opin. Struct. Biol. 2020, 61, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Ciardo, D.; Goldar, A.; Marheineke, K. On the Interplay of the DNA Replication Program and the Intra-S Phase Checkpoint Pathway. Genes (Basel) 2019, 10, 94. [Google Scholar] [CrossRef]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef]
- Lou, Z.; Chini, C.C.S.; Minter-Dykhouse, K.; Chen, J. Mediator of DNA damage checkpoint protein 1 regulates BRCA1 localization and phosphorylation in DNA damage checkpoint control. J. Biol. Chem. 2003, 278, 13599–13602. [Google Scholar] [CrossRef]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Beck, C.; Robert, I.; Reina-San-Martin, B.; Schreiber, V.; Dantzer, F. Poly(ADP-ribose) polymerases in double-strand break repair: Focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 2014, 329, 18–25. [Google Scholar] [CrossRef]
- Lee, K.Y.; Im, J.-S.; Shibata, E.; Dutta, A. ASF1a Promotes Non-homologous End Joining Repair by Facilitating Phosphorylation of MDC1 by ATM at Double-Strand Breaks. Mol. Cell 2017, 68, 61–75. [Google Scholar] [CrossRef]
- Zimmermann, M.; Lottersberger, F.; Buonomo, S.B.; Sfeir, A.; Lange, T. de. 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science 2013, 339, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.F.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Grundy, G.J.; Moulding, H.A.; Caldecott, K.W.; Rulten, S.L. One ring to bring them all--the role of Ku in mammalian non-homologous end joining. DNA Repair (Amst) 2014, 17, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Iles, N.; Rulten, S.; El-Khamisy, S.F.; Caldecott, K.W. APLF (C2orf13) is a novel human protein involved in the cellular response to chromosomal DNA strand breaks. Mol. Cell. Biol. 2007, 27, 3793–3803. [Google Scholar] [CrossRef]
- Rulten, S.L.; Fisher, A.E.O.; Robert, I.; Zuma, M.C.; Rouleau, M.; Ju, L.; Poirier, G.; Reina-San-Martin, B.; Caldecott, K.W. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol. Cell 2011, 41, 33–45. [Google Scholar] [CrossRef]
- Ma, Y.; Pannicke, U.; Schwarz, K.; Lieber, M.R. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 2002, 108, 781–794. [Google Scholar] [CrossRef]
- Ma, Y.; Lu, H.; Tippin, B.; Goodman, M.F.; Shimazaki, N.; Koiwai, O.; Hsieh, C.-L.; Schwarz, K.; Lieber, M.R. A biochemically defined system for mammalian nonhomologous DNA end joining. Mol. Cell 2004, 16, 701–713. [Google Scholar] [CrossRef]
- Grawunder, U.; Wilm, M.; Wu, X.; Kulesza, P.; Wilson, T.E.; Mann, M.; Lieber, M.R. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature 1997, 388, 492–495. [Google Scholar] [CrossRef]
- Gu, J.; Lu, H.; Tippin, B.; Shimazaki, N.; Goodman, M.F.; Lieber, M.R. XRCC4:DNA ligase IV can ligate incompatible DNA ends and can ligate across gaps. EMBO J. 2007, 26, 1010–1023. [Google Scholar] [CrossRef]
- Ahnesorg, P.; Smith, P.; Jackson, S.P. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell 2006, 124, 301–313. [Google Scholar] [CrossRef]
- Tsai, C.J.; Kim, S.A.; Chu, G. Cernunnos/XLF promotes the ligation of mismatched and noncohesive DNA ends. Proc. Natl. Acad. Sci. USA 2007, 104, 7851–7856. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Buisson, R.; Dion-Côté, A.-M.; Coulombe, Y.; Launay, H.; Cai, H.; Stasiak, A.Z.; Stasiak, A.; Xia, B.; Masson, J.-Y. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat. Struct. Mol. Biol. 2010, 17, 1247–1254. [Google Scholar] [CrossRef]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef]
- Casari, E.; Rinaldi, C.; Marsella, A.; Gnugnoli, M.; Colombo, C.V.; Bonetti, D.; Longhese, M.P. Processing of DNA Double-Strand Breaks by the MRX Complex in a Chromatin Context. Front. Mol. Biosci. 2019, 6, 43. [Google Scholar] [CrossRef]
- Baumann, P.; Benson, F.E.; West, S.C. Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell 1996, 87, 757–766. [Google Scholar] [CrossRef]
- Bianco, P.R.; Tracy, R.B.; Kowalczykowski, S.C. DNA strand exchange proteins: A biochemical and physical comparison. Front. Biosci. 1998, 3, D570–D603. [Google Scholar] [CrossRef]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef]
- Keeney, S.; Giroux, C.N.; Kleckner, N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 1997, 88, 375–384. [Google Scholar] [CrossRef]
- Robert, T.; Vrielynck, N.; Mézard, C.; de Massy, B.; Grelon, M. A new light on the meiotic DSB catalytic complex. Semin. Cell Dev. Biol. 2016, 54, 165–176. [Google Scholar] [CrossRef]
- Cantor, S.B.; Bell, D.W.; Ganesan, S.; Kass, E.M.; Drapkin, R.; Grossman, S.; Wahrer, D.C.; Sgroi, D.C.; Lane, W.S.; Haber, D.A.; et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 2001, 105, 149–160. [Google Scholar] [CrossRef]
- Shim, K.S.; Schmutte, C.; Tombline, G.; Heinen, C.D.; Fishel, R. hXRCC2 enhances ADP/ATP processing and strand exchange by hRAD51. J. Biol. Chem. 2004, 279, 30385–30394. [Google Scholar] [CrossRef] [PubMed]
- Kowalczykowski, S.C. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Wold, M.S. Replication protein A: A heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997, 66, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Sleeth, K.M.; Sørensen, C.S.; Issaeva, N.; Dziegielewski, J.; Bartek, J.; Helleday, T. RPA mediates recombination repair during replication stress and is displaced from DNA by checkpoint signalling in human cells. J. Mol. Biol. 2007, 373, 38–47. [Google Scholar] [CrossRef]
- Franchitto, A. Genome instability at common fragile sites: Searching for the cause of their instability. Biomed Res. Int. 2013, 2013, 730714. [Google Scholar] [CrossRef]
- Cimprich, K.A. Fragile sites: Breaking up over a slowdown. Curr. Biol. 2003, 13, R231–R233. [Google Scholar] [CrossRef][Green Version]
- Dillon, L.W.; Burrow, A.A.; Wang, Y.-H. DNA instability at chromosomal fragile sites in cancer. Curr. Genomics 2010, 11, 326–337. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Tsantoulis, P.; Kotsinas, A.; Michalopoulos, I.; Townsend, P.; Gorgoulis, V.G. Are common fragile sites merely structural domains or highly organized “functional” units susceptible to oncogenic stress? Cell. Mol. Life Sci. 2014, 71, 4519–4544. [Google Scholar] [CrossRef]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Mathelier, A.; Carbone, A. Large scale chromosomal mapping of human microRNA structural clusters. Nucleic Acids Res. 2013, 41, 4392–4408. [Google Scholar] [CrossRef] [PubMed]
- Ozeri-Galai, E.; Schwartz, M.; Rahat, A.; Kerem, B. Interplay between ATM and ATR in the regulation of common fragile site stability. Oncogene 2008, 27, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Casper, A.M.; Nghiem, P.; Arlt, M.F.; Glover, T.W. ATR regulates fragile site stability. Cell 2002, 111, 779–789. [Google Scholar] [CrossRef]
- Kumari, D.; Somma, V.; Nakamura, A.J.; Bonner, W.M.; D’Ambrosio, E.; Usdin, K. The role of DNA damage response pathways in chromosome fragility in Fragile X syndrome. Nucleic Acids Res. 2009, 37, 4385–4392. [Google Scholar] [CrossRef]
- Schwartz, M.; Zlotorynski, E.; Goldberg, M.; Ozeri, E.; Rahat, A.; Le Sage, C.; Chen, B.P.C.; Chen, D.J.; Agami, R.; Kerem, B. Homologous recombination and nonhomologous end-joining repair pathways regulate fragile site stability. Genes Dev. 2005, 19, 2715–2726. [Google Scholar] [CrossRef]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef]
- Debacker, K.; Kooy, R.F. Fragile sites and human disease. Hum. Mol. Genet. 2007, 16, R150–R158. [Google Scholar] [CrossRef]
- Thys, R.G.; Lehman, C.E.; Pierce, L.C.T.; Wang, Y.-H. DNA secondary structure at chromosomal fragile sites in human disease. Curr. Genomics 2015, 16, 60–70. [Google Scholar] [CrossRef]
- Zlotorynski, E.; Rahat, A.; Skaug, J.; Ben-Porat, N.; Ozeri, E.; Hershberg, R.; Levi, A.; Scherer, S.W.; Margalit, H.; Kerem, B. Molecular basis for expression of common and rare fragile sites. Mol. Cell. Biol. 2003, 23, 7143–7151. [Google Scholar] [CrossRef]
- Lucá, R.; Averna, M.; Zalfa, F.; Vecchi, M.; Bianchi, F.; La Fata, G.; Del Nonno, F.; Nardacci, R.; Bianchi, M.; Nuciforo, P.; et al. The fragile X protein binds mRNAs involved in cancer progression and modulates metastasis formation. EMBO Mol. Med. 2013, 5, 1523–1536. [Google Scholar] [CrossRef] [PubMed]
- Zalfa, F.; Panasiti, V.; Carotti, S.; Zingariello, M.; Perrone, G.; Sancillo, L.; Pacini, L.; Luciani, F.; Roberti, V.; D’Amico, S.; et al. The fragile X mental retardation protein regulates tumor invasiveness-related pathways in melanoma cells. Cell Death Dis. 2017, 8, e3169. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.; Marquardt, S.; Li, F.; Spitschak, A.; Murr, N.; Edelhäuser, B.A.H.; Iliakis, G.; Pützer, B.M.; Logotheti, S. Rewiring E2F1 with classical NHEJ via APLF suppression promotes bladder cancer invasiveness. J. Exp. Clin. Cancer Res. 2019, 38, 292. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Herrera, A.; Castresana, J.; Robinson, T.J. Is mammalian chromosomal evolution driven by regions of genome fragility? Genome Biol. 2006, 7, R115. [Google Scholar] [CrossRef]
- Pelliccia, F.; Bosco, N.; Rocchi, A. Breakages at common fragile sites set boundaries of amplified regions in two leukemia cell lines K562-Molecular characterization of FRA2H and localization of a new CFS FRA2S. Cancer Lett. 2010, 299, 37–44. [Google Scholar] [CrossRef]
- Fechter, A.; Buettel, I.; Kuehnel, E.; Savelyeva, L.; Schwab, M. Common fragile site FRA11G and rare fragile site FRA11B at 11q23.3 encompass distinct genomic regions. Genes Chromosomes Cancer 2007, 46, 98–106. [Google Scholar] [CrossRef]
- Fechter, A.; Buettel, I.; Kuehnel, E.; Schwab, M.; Savelyeva, L. Cloning of genetically tagged chromosome break sequences reveals new fragile sites at 6p21 and 13q22. Int. J. Cancer 2007, 120, 2359–2367. [Google Scholar] [CrossRef]
- Helmrich, A.; Stout-Weider, K.; Matthaei, A.; Hermann, K.; Heiden, T.; Schrock, E. Identification of the human/mouse syntenic common fragile site FRA7K/Fra12C1–relation of FRA7K and other human common fragile sites on chromosome 7 to evolutionary breakpoints. Int. J. Cancer 2007, 120, 48–54. [Google Scholar] [CrossRef]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef]
- Paraskevopoulou, M.D.; Georgakilas, G.; Kostoulas, N.; Vlachos, I.S.; Vergoulis, T.; Reczko, M.; Filippidis, C.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-microT web server v5.0: Service integration into miRNA functional analysis workflows. Nucleic Acids Res. 2013, 41, W169–W173. [Google Scholar] [CrossRef]
- Reczko, M.; Maragkakis, M.; Alexiou, P.; Grosse, I.; Hatzigeorgiou, A.G. Functional microRNA targets in protein coding sequences. Bioinformatics 2012, 28, 771–776. [Google Scholar] [CrossRef]
- Qing, X.; Shi, J.; Dong, T.; Wu, C.; Hu, L.; Li, H. Dysregulation of an X-linked primate-specific epididymal microRNA cluster in unexplained asthenozoospermia. Oncotarget 2017, 8, 56839–56849. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Peng, Y.; Wang, W.; Su, B. Rapid evolution of an X-linked microRNA cluster in primates. Genome Res. 2007, 17, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhang, Y.; Zhang, R.; Qi, X.; Su, B. Functional divergence of the rapidly evolving miR-513 subfamily in primates. BMC Evol. Biol. 2013, 13, 255. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, Y.; Dong, D.; Zhang, Z. Evolution of an X-linked primate-specific micro RNA cluster. Mol. Biol. Evol. 2010, 27, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, M.; Tan, K.; Plank, T.-D.M.; Song, H.-W.; Dumdie, J.N.; Jones, S.; Shum, E.Y.; Sheridan, S.D.; Peterson, K.J.; Gromoll, J.; et al. A microRNA cluster in the Fragile-X region expressed during spermatogenesis targets FMR1. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Amaral, P.P.; Leonardi, T.; Han, N.; Viré, E.; Gascoigne, D.K.; Arias-Carrasco, R.; Büscher, M.; Pandolfini, L.; Zhang, A.; Pluchino, S.; et al. Genomic positional conservation identifies topological anchor point RNAs linked to developmental loci. Genome Biol. 2018, 19, 32. [Google Scholar] [CrossRef]
- Kiwerska, K.; Szyfter, K. DNA repair in cancer initiation, progression, and therapy-a double-edged sword. J. Appl. Genet. 2019, 60, 329–334. [Google Scholar] [CrossRef]
- Biau, J.; Chautard, E.; Verrelle, P.; Dutreix, M. Altering DNA Repair to Improve Radiation Therapy: Specific and Multiple Pathway Targeting. Front. Oncol. 2019, 9, 1009. [Google Scholar] [CrossRef]
- Kun, S.; Duan, Q.; Liu, G.; Lu, J.-M. Prognostic value of DNA repair genes based on stratification of glioblastomas. Oncotarget 2017, 8, 58222–58230. [Google Scholar] [CrossRef]
- Santarpia, L.; Iwamoto, T.; Di Leo, A.; Hayashi, N.; Bottai, G.; Stampfer, M.; André, F.; Turner, N.C.; Symmans, W.F.; Hortobágyi, G.N.; et al. DNA repair gene patterns as prognostic and predictive factors in molecular breast cancer subtypes. Oncologist 2013, 18, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Rosales-Reynoso, M.A.; Barros-Núñez, P.; Peprah, E. DNA repair/replication transcripts are down regulated in patients with Fragile X Syndrome. BMC Res. Notes 2013, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Bjerregaard, V.A.; Garribba, L.; McMurray, C.T.; Hickson, I.D.; Liu, Y. Folate deficiency drives mitotic missegregation of the human FRAXA locus. Proc. Natl. Acad. Sci. USA 2018, 115, 13003–13008. [Google Scholar] [CrossRef] [PubMed]
- Bjerregaard, V.A.; Özer, Ö.; Hickson, I.D.; Liu, Y. The Detection and Analysis of Chromosome Fragile Sites. Methods Mol. Biol. 2018, 1672, 471–482. [Google Scholar] [CrossRef]
- Koufaris, C. Human and primate-specific microRNAs in cancer: Evolution, and significance in comparison with more distantly-related research models: The great potential of evolutionary young microRNA in cancer research. Bioessays 2016, 38, 286–294. [Google Scholar] [CrossRef]
- Hasegawa, T.; Glavich, G.J.; Pahuski, M.; Short, A.; Semmes, O.J.; Yang, L.; Galkin, V.; Drake, R.; Esquela-Kerscher, A. Characterization and Evidence of the miR-888 Cluster as a Novel Cancer Network in Prostate. Mol. Cancer Res. 2018, 16, 669–681. [Google Scholar] [CrossRef]
- Logotheti, S.; Marquardt, S.; Pützer, B.M. p73-Governed miRNA Networks: Translating Bioinformatics Approaches to Therapeutic Solutions for Cancer Metastasis. Methods Mol. Biol. 2019, 1912, 33–52. [Google Scholar] [CrossRef]
- Ren, L.-L.; Yan, T.-T.; Shen, C.-Q.; Tang, J.-Y.; Kong, X.; Wang, Y.-C.; Chen, J.; Liu, Q.; He, J.; Zhong, M.; et al. The distinct role of strand-specific miR-514b-3p and miR-514b-5p in colorectal cancer metastasis. Cell Death Dis. 2018, 9, 687. [Google Scholar] [CrossRef]
- Patel, V.D.; Capra, J.A. Ancient human miRNAs are more likely to have broad functions and disease associations than young miRNAs. BMC Genomics 2017, 18, 672. [Google Scholar] [CrossRef]
- Else, T.; Kim, A.C.; Sabolch, A.; Raymond, V.M.; Kandathil, A.; Caoili, E.M.; Jolly, S.; Miller, B.S.; Giordano, T.J.; Hammer, G.D. Adrenocortical carcinoma. Endocr. Rev. 2014, 35, 282–326. [Google Scholar] [CrossRef]
- Kaliki, S.; Shields, C.L. Uveal melanoma: Relatively rare but deadly cancer. Eye (London) 2017, 31, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Dawson, L. A Rare Tumor with a Very Rare Initial Presentation: Thymic Carcinoma as Bone Marrow Metastasis. Case Rep. Pathol. 2017, 2017, 6497376. [Google Scholar] [CrossRef] [PubMed]
- The, L.R.M. Pleural mesothelioma: Tackling a deadly cancer. Lancet Respir. Med. 2019, 7, 99. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Clocchiatti, A.; Cora, E.; Zhang, Y.; Dotto, G.P. Sexual dimorphism in cancer. Nat. Rev. Cancer 2016, 16, 330–339. [Google Scholar] [CrossRef]
- Arnold, A.P.; Disteche, C.M. Sexual Inequality in the Cancer Cell. Cancer Res. 2018, 78, 5504–5505. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Repair Component | Repair Function | Ref. |
|---|---|---|
| Common/upstream factors | ||
| ATM | activation of checkpoint signaling, orchestrating cell cycle with DNA repair | [3] |
| ATR | activation of checkpoint signaling, orchestrating cell cycle with DNA repair | [3] |
| CHEK1 | checkpoint-mediated cell cycle arrest | [4] |
| CHEK2 | checkpoint-mediated cell cycle arrest | [4] |
| TOPBP1 | checkpoint control | [5] |
| MDC1 | checkpoint control | [6] |
| H2AX/H2afx | DNA double-strand break sensor | [7] |
| PARP1 | DNA double-strand break sensor, recruitment of DNA repair proteins | [8,9] |
| Non-Homologous End Joining (NHEJ) | ||
| ASF1A | facilitates ATM-MDC1 interaction (checkpoint control), recruitment of TP53BP1 | [10] |
| TP53BP1/Trp53bp1 | pathway choice: DNA end resection inhibitor | [11,12] |
| Ku70/Ku80 (XRCC6/5) | DNA-binding, end-protection recruitment of NHEJ complex | [13] |
| APLF | DNA-end processing, facilitate DNA ligation | [14,15] |
| Artemis (DCLRE1C) | DNA-end processing | [16] |
| PRKDC | DNA-end protection and activation of Artemis | [16,17] |
| LIG4 | ligation of DNA ends | [18,19] |
| XRCC4 | facilitates LIG4 activity | [18,19] |
| NHEJ1 (XLF) | facilitates LIG4 activity | [20,21] |
| Homologous recombination (HR) | ||
| BRCA1 | pathway choice: antagonizes TP53BP1 (DNA-end resection activator) | [12] |
| BRCA2 | mediator of strand invasion | [22] |
| EXO1 | pathway choice: DNA-end resection | [23] |
| PALB2 | recruitment and stabilization of BRCA2 and RAD51 | [24,25] |
| RAD50-MRE11-NBN | MRN complex: DNA-end resection | [26] |
| RAD51 | strand invasion and exchange of homologous DNA sequences | [27,28] |
| RBBP8 (CtIP) | DNA-end resection | [12,29] |
| SPO11 | induction of programmed meiotic DNA double-strand break | [30,31] |
| BRIP1 (BACH1) | co-factor of BRCA1 | [32] |
| XRCC2 | paralog of RAD51, facilitates strand invasion | [33,34] |
| BLM | pathway choice: DNA-end resection | [23] |
| RPA | stabilization of ssDNA | [34,35,36] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marquardt, S.; Richter, C.; Pützer, B.M.; Logotheti, S. MiRNAs Targeting Double Strand DNA Repair Pathways Lurk in Genomically Unstable Rare Fragile Sites and Determine Cancer Outcomes. Cancers 2020, 12, 876. https://doi.org/10.3390/cancers12040876
Marquardt S, Richter C, Pützer BM, Logotheti S. MiRNAs Targeting Double Strand DNA Repair Pathways Lurk in Genomically Unstable Rare Fragile Sites and Determine Cancer Outcomes. Cancers. 2020; 12(4):876. https://doi.org/10.3390/cancers12040876
Chicago/Turabian StyleMarquardt, Stephan, Christin Richter, Brigitte M. Pützer, and Stella Logotheti. 2020. "MiRNAs Targeting Double Strand DNA Repair Pathways Lurk in Genomically Unstable Rare Fragile Sites and Determine Cancer Outcomes" Cancers 12, no. 4: 876. https://doi.org/10.3390/cancers12040876
APA StyleMarquardt, S., Richter, C., Pützer, B. M., & Logotheti, S. (2020). MiRNAs Targeting Double Strand DNA Repair Pathways Lurk in Genomically Unstable Rare Fragile Sites and Determine Cancer Outcomes. Cancers, 12(4), 876. https://doi.org/10.3390/cancers12040876