Are Synapse-Like Structures a Possible Way for Crosstalk of Cancer with Its Microenvironment?


{kind=link}
{kind=link}
Abstract
1. Introduction.
1.1. The Necessity of Changing the Paradigm in Cancer Therapy
1.2. A Brief Description of the TME and Its Importance for Cancer Progression
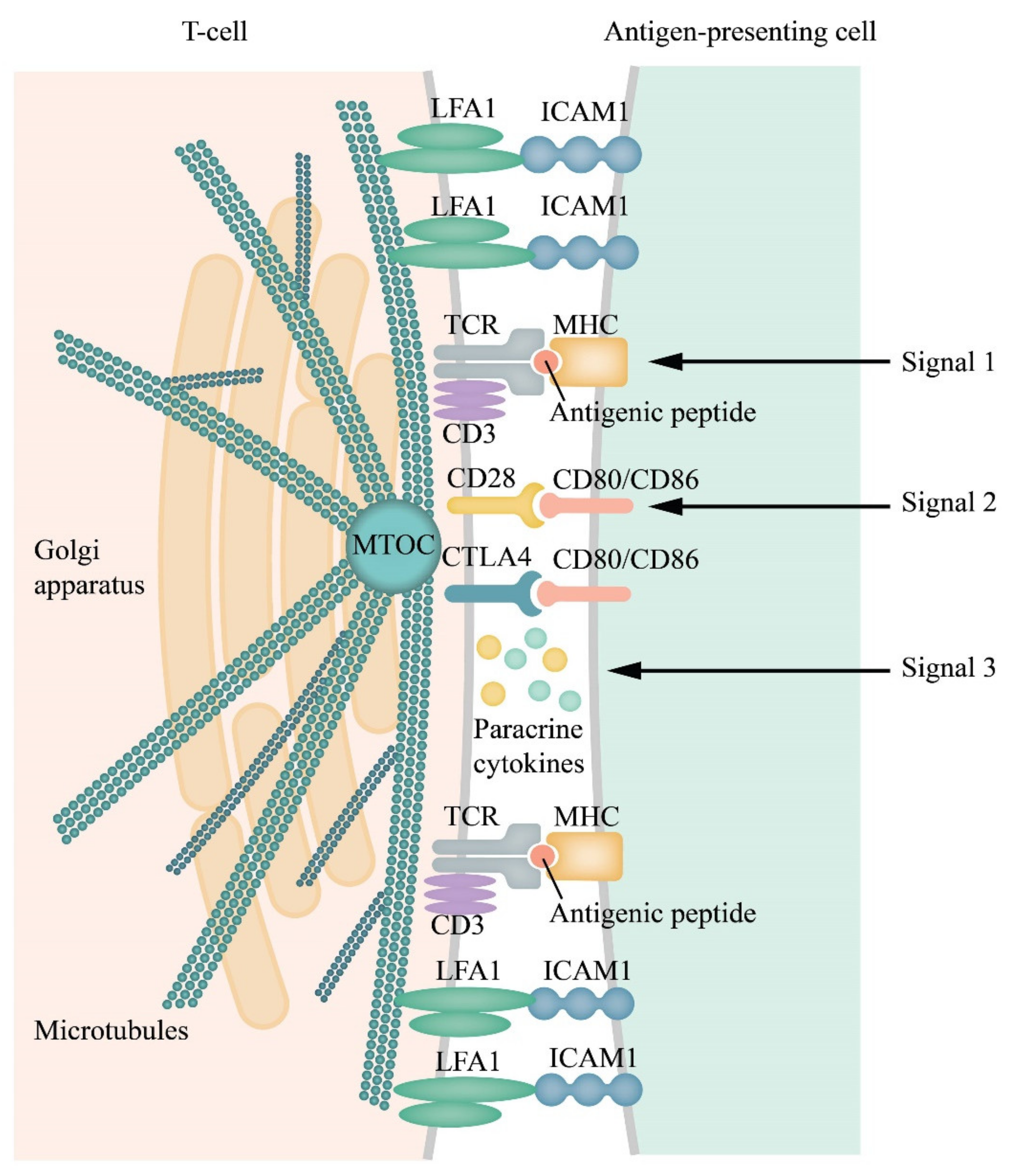
1.3. A Short Summary of the Formation of Immunological Synapses between T cells and the Activated Antigen-Presenting Cells
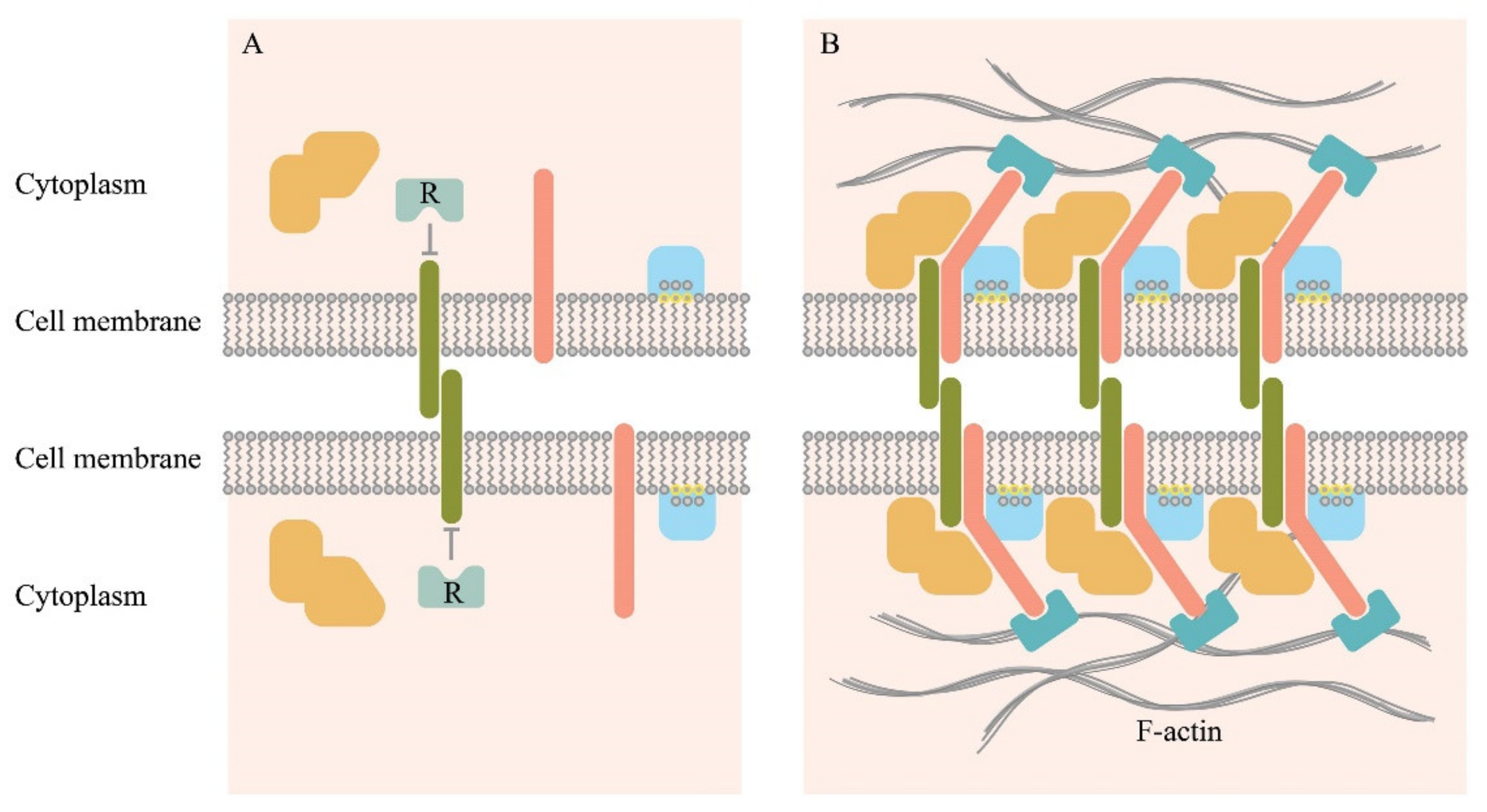
1.4. Clusterization of Receptors and Ligands is A Prerequisite and Signature of IS Formation
1.5. Remodeling of Cytoskeletons in Intercellular Interactions
1.6. Circulating Cancer Cells Form Clusters through Tomo- and Heterotypic Intercellular Adhesions That Are Responsible for Metastasis and Possess the Stemness Property
1.7. Why are CAFs “Chosen” for Cancer Cell Partners and Direct Contacts
2. Conclusions
The Power of Clusters in Signal Transmission, and Their Vulnerability to a Directed Disruption
- The proximity of the interacting cells.
- The presence of receptor clusters and corresponding ligands on the interacting cells.
- The presence of strong interactions that allow cancer cells to migrate together with the stromal cells within circulating clusters.
- A remodeled cytoskeleton in the interacting cells.
- Characteristic changes in the transcription regulation [81] and possible epigenetic changes.
Author Contributions
Funding
Conflicts of Interest
References
- Ledford, H. End of cancer-genome project prompts rethink. Nature 2015, 517, 128–129. [Google Scholar] [CrossRef] [PubMed]
- Sverdlov, E.D. Systems biology and personalized medicine: To be or not to be? Ross. Fiziol. Zhurnal Im. IM Sechenova 2014, 100, 505–541. [Google Scholar]
- Sverdlov, E.D. Genetic surgery—A right strategy to attack cancer. Curr. Gene Ther. 2011, 11, 501–531. [Google Scholar] [CrossRef] [PubMed]
- Janiszewska, M. The microcosmos of intratumor heterogeneity: The space-time of cancer evolution. Oncogene 2019, 39, 2031–2039. [Google Scholar] [CrossRef]
- Mallick, P. Physical Sciences and Engineering Advances in Life Sciences and Oncology; Springer International Publishing: New York, NY, USA, 2015; pp. 5–29. [Google Scholar]
- Noble, D. A theory of biological relativity: No privileged level of causation. Interface Focus 2012, 2, 55–64. [Google Scholar] [CrossRef]
- Noble, D. A biological relativity view of the relationships between genomes and phenotypes. Prog. Biophys. Mol. Biol. 2013, 111, 59–65. [Google Scholar] [CrossRef]
- Rickles, D.; Hawe, P.; Shiell, A. A simple guide to chaos and complexity. J. Epidemiol. Community Health 2007, 61, 933–937. [Google Scholar] [CrossRef]
- Suki, B.; Bates, J.H.; Frey, U. Complexity and emergent phenomena. Compr. Physiol. 2011, 1, 995–1029. [Google Scholar]
- Gershenson, C. Facing Complexity: Prediction vs. Adaptation. In Complexity Perspectives on Language, Communication and Society; Massip-Bonet, À., Bastardas-Boada, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 3–14. Available online: http://arxiv.org/ftp/arxiv/papers/1112/1112.3843.pdf (accessed on 2 February 2020).
- Sverdlov, E. Transcribed Junk Remains Junk If It Does Not Acquire A Selected Function in Evolution. Bioessays 2017, 39, 1700164. [Google Scholar] [CrossRef]
- Sverdlov, E.D. Multidimensional Complexity of Cancer. Simple Solutions Are Needed. Biochemistry (Mosc) 2016, 81, 731–738. [Google Scholar] [CrossRef]
- Frank, S.A. Evolution in health and medicine Sackler colloquium: Somatic evolutionary genomics: Mutations during development cause highly variable genetic mosaicism with risk of cancer and neurodegeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 1725–1730. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Bernards, R. Opportunities and challenges provided by crosstalk between signalling pathways in cancer. Oncogene 2015, 35, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Merlo, L.M.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Tannock, I.F.; Hickman, J.A. Limits to Personalized Cancer Medicine. N. Engl. J. Med. 2016, 375, 1289–1294. [Google Scholar] [CrossRef]
- Prasad, V. Perspective: The precision-oncology illusion. Nature 2016, 537, S63. [Google Scholar] [CrossRef]
- Gillies, R.J.; Verduzco, D.; Gatenby, R.A. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat. Rev. Cancer 2012, 12, 487–493. [Google Scholar] [CrossRef]
- Bhome, R.; Bullock, M.D.; Al Saihati, H.A.; Goh, R.W.; Primrose, J.N.; Sayan, A.E.; Mirnezami, A.H. A top-down view of the tumor microenvironment: Structure, cells and signaling. Front. Cell Dev. Biol. 2015, 3, 33. [Google Scholar] [CrossRef]
- Bhome, R.; Al Saihati, H.A.; Goh, R.W.; Bullock, M.D.; Primrose, J.N.; Thomas, G.J.; Sayan, A.E.; Mirnezami, A.H. Translational aspects in targeting the stromal tumour microenvironment: From bench to bedside. New Horiz. Transl. Med. 2016, 3, 9–21. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [PubMed]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Reina-Campos, M.; Moscat, J.; Diaz-Meco, M. Metabolism shapes the tumor microenvironment. Curr. Opin. Cell Biol. 2017, 48, 47–53. [Google Scholar] [CrossRef]
- Orimo, A.; Weinberg, R.A. Stromal fibroblasts in cancer: A novel tumor-promoting cell type. Cell Cycle 2006, 5, 1597–1601. [Google Scholar] [CrossRef]
- Sverdlov, E. Missed Druggable Cancer Hallmark: Cancer-Stroma Symbiotic Crosstalk as Paradigm and Hypothesis for Cancer Therapy. Bioessays 2018, 40, 1800079. [Google Scholar] [CrossRef]
- Gonda, T.A.; Varro, A.; Wang, T.C.; Tycko, B. Molecular biology of cancer-associated fibroblasts: Can these cells be targeted in anti-cancer therapy? Semin. Cell Dev. Biol. 2010, 21, 2–10. [Google Scholar] [CrossRef]
- De Palma, M.; Hanahan, D. The biology of personalized cancer medicine: Facing individual complexities underlying hallmark capabilities. Mol. Oncol. 2012, 6, 111–127. [Google Scholar] [CrossRef]
- Chen, F.; Zhuang, X.; Lin, L.; Yu, P.; Wang, Y.; Shi, Y.; Hu, G.; Sun, Y. New horizons in tumor microenvironment biology: Challenges and opportunities. BMC Med. 2015, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- Gandellini, P.; Andriani, F.; Merlino, G.; D’Aiuto, F.; Roz, L.; Callari, M. Complexity in the tumour microenvironment: Cancer associated fibroblast gene expression patterns identify both common and unique features of tumour-stroma crosstalk across cancer types. Semin. Cancer Biol. 2015, 35, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Stadler, M.; Walter, S.; Walzl, A.; Kramer, N.; Unger, C.; Scherzer, M.; Unterleuthner, D.; Hengstschlager, M.; Krupitza, G.; Dolznig, H. Increased complexity in carcinomas: Analyzing and modeling the interaction of human cancer cells with their microenvironment. Semin. Cancer Biol. 2015, 35, 107–124. [Google Scholar] [CrossRef]
- Zi, F.; He, J.; He, D.; Li, Y.; Yang, L.; Cai, Z. Fibroblast activation protein alpha in tumor microenvironment: Recent progression and implications (review). Mol. Med. Rep. 2015, 11, 3203–3211. [Google Scholar] [CrossRef] [PubMed]
- Raffaghello, L.; Dazzi, F. Classification and biology of tumour associated stromal cells. Immunol. Lett. 2015, 168, 175–182. [Google Scholar] [CrossRef]
- Perrimon, N.; Pitsouli, C.; Shilo, B.Z. Signaling mechanisms controlling cell fate and embryonic patterning. Cold Spring Harb. Perspect. Biol. 2012, 4, a005975. [Google Scholar] [CrossRef]
- Bizzarri, M.; Cucina, A. Tumor and the microenvironment: A chance to reframe the paradigm of carcinogenesis? BioMed Res. Int. 2014, 2014, 934038. [Google Scholar] [CrossRef]
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/CXCR4: A symbiotic bridge linking cancer cells and their stromal neighbors in oncogenic communication networks. Oncogene 2016, 35, 816–826. [Google Scholar] [CrossRef]
- Sluka, P.; Davis, I.D. Cell mates: Paracrine and stromal targets for prostate cancer therapy. Nat. Rev. Urol. 2013, 10, 441–451. [Google Scholar] [CrossRef]
- Baluska, F.M.S. Synaptic view of eukaryotic cell. Int. J. Gen. Syst. 2014, 43, 740–756. [Google Scholar] [CrossRef]
- Sadelain, M.; Riviere, I.; Brentjens, R. Targeting tumours with genetically enhanced T lymphocytes. Nat. Rev. Cancer 2003, 3, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef] [PubMed]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Marcucci, F.; Rumio, C.; Corti, A. Tumor cell-associated immune checkpoint molecules—Drivers of malignancy and stemness. Biochim. Biophys. Acta 2017, 1868, 571–583. [Google Scholar] [CrossRef]
- Meissner, J.M.; Sikorski, A.F.; Nawara, T.; Grzesiak, J.; Marycz, K.; Boguslawska, D.M.; Michalczyk, I.; Lecomte, M.C.; Machnicka, B. alphaII-spectrin in T cells is involved in the regulation of cell-cell contact leading to immunological synapse formation? PLoS ONE 2017, 12, e0189545. [Google Scholar] [CrossRef]
- Angus, K.L.; Griffiths, G.M. Cell polarisation and the immunological synapse. Curr. Opin. Cell Biol. 2013, 25, 85–91. [Google Scholar] [CrossRef]
- Ritter, A.T.; Angus, K.L.; Griffiths, G.M. The role of the cytoskeleton at the immunological synapse. Immunol. Rev. 2013, 256, 107–117. [Google Scholar] [CrossRef]
- Dustin, M.L. The immunological synapse. Cancer Immunol. Res. 2014, 2, 1023–1033. [Google Scholar] [CrossRef]
- Bertrand, F.; Muller, S.; Roh, K.H.; Laurent, C.; Dupre, L.; Valitutti, S. An initial and rapid step of lytic granule secretion precedes microtubule organizing center polarization at the cytotoxic T lymphocyte/target cell synapse. Proc. Natl. Acad. Sci. USA 2013, 110, 6073–6078. [Google Scholar] [CrossRef]
- Hafner, A.E.; Rieger, H. Spatial Cytoskeleton Organization Supports Targeted Intracellular Transport. Biophys. J. 2018, 114, 1420–1432. [Google Scholar] [CrossRef]
- Xie, J.; Tato, C.M.; Davis, M.M. How the immune system talks to itself: The varied role of synapses. Immunol. Rev. 2013, 251, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Sanchez-Madrid, F. Role of the cytoskeleton during leukocyte responses. Nat. Rev. Immunol. 2004, 4, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L.; Baldari, C.T. The Immune Synapse: Past, Present, and Future. Methods Mol. Biol. 2017, 1584, 1–5. [Google Scholar] [PubMed]
- Dustin, M.L.; Choudhuri, K. Signaling and Polarized Communication across the T Cell Immunological Synapse. Annu. Rev. Cell Dev. Biol. 2016, 32, 303–325. [Google Scholar] [CrossRef]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef]
- Pettmann, J.; Santos, A.M.; Dushek, O.; Davis, S.J. Membrane Ultrastructure and T Cell Activation. Front. Immunol. 2018, 9, 2152. [Google Scholar] [CrossRef]
- Fooksman, D.R.; Vardhana, S.; Vasiliver-Shamis, G.; Liese, J.; Blair, D.A.; Waite, J.; Sacristan, C.; Victora, G.D.; Zanin-Zhorov, A.; Dustin, M.L. Functional anatomy of T cell activation and synapse formation. Annu. Rev. Immunol. 2010, 28, 79–105. [Google Scholar] [CrossRef]
- Mayya, V.; Judokusumo, E.; Abu Shah, E.; Peel, C.G.; Neiswanger, W.; Depoil, D.; Blair, D.A.; Wiggins, C.H.; Kam, L.C.; Dustin, M.L. Durable Interactions of T Cells with T Cell Receptor Stimuli in the Absence of a Stable Immunological Synapse. Cell Rep. 2018, 22, 340–349. [Google Scholar] [CrossRef]
- Dustin, M.L. A dynamic view of the immunological synapse. Semin. Immunol. 2005, 17, 400–410. [Google Scholar] [CrossRef]
- Calvo, V.; Izquierdo, M. Imaging Polarized Secretory Traffic at the Immune Synapse in Livin T Lymphocytes. Front. Immunol. 2018, 9, 684. [Google Scholar] [CrossRef]
- Shi, Y. To forge a solid immune recognition. Protein Cell 2012, 3, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.J. Signal initiation in biological systems: The properties and detection of transient extracellular protein interactions. Mol. Biosyst. 2009, 5, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Cebecauer, M.; Spitaler, M.; Serge, A.; Magee, A.I. Signalling complexes and clusters: Functional advantages and methodological hurdles. J. Cell Sci. 2010, 123, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Mege, R.M.; Ishiyama, N. Integration of Cadherin Adhesion and Cytoskeleton at Adherens Junctions. Cold Spring Harb. Perspect. Biol. 2017, 9, a028738. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Parajo, M.F.; Cambi, A.; Torreno-Pina, J.A.; Thompson, N.; Jacobson, K. Nanoclustering as a dominant feature of plasma membrane organization. J. Cell Sci. 2014, 127, 4995–5005. [Google Scholar] [CrossRef]
- Hartman, N.C.; Groves, J.T. Signaling clusters in the cell membrane. Curr. Opin. Cell Biol. 2011, 23, 370–376. [Google Scholar] [CrossRef]
- Yap, A.S.; Gomez, G.A.; Parton, R.G. Adherens Junctions Revisualized: Organizing Cadherins as Nanoassemblies. Dev. Cell 2015, 35, 12–20. [Google Scholar] [CrossRef]
- Nussinov, R.; Jang, H. Dynamic multiprotein assemblies shape the spatial structure of cell signaling. Prog. Biophys. Mol. Biol. 2014, 116, 158–164. [Google Scholar] [CrossRef]
- Nussinov, R.; Jang, H.; Tsai, C.J. Oligomerization and nanocluster organization render specificity. Biol. Rev. Camb. Philos. Soc. 2015, 90, 587–598. [Google Scholar] [CrossRef]
- Hatherley, D.; Lea, S.M.; Johnson, S.; Barclay, A.N. Structures of CD200/CD200 receptor family and implications for topology, regulation, and evolution. Structure 2013, 21, 820–832. [Google Scholar] [CrossRef]
- Wright, G.J.; Puklavec, M.J.; Willis, A.C.; Hoek, R.M.; Sedgwick, J.D.; Brown, M.H.; Barclay, A.N. Lymphoid/neuronal cell surface OX2 glycoprotein recognizes a novel receptor on macrophages implicated in the control of their function. Immunity 2000, 13, 233–242. [Google Scholar] [CrossRef]
- Serge, A. The Molecular Architecture of Cell Adhesion: Dynamic Remodeling Revealed by Videonanoscopy. Front. Cell Dev. Biol. 2016, 4, 413. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.D.; Yap, A.S. Towards a Dynamic Understanding of Cadherin-Based Mechanobiology. Trends Cell Biol. 2015, 25, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Jeanes, A.; Gottardi, C.J.; Yap, A.S. Cadherins and cancer: How does cadherin dysfunction promote tumor progression? Oncogene 2008, 27, 6920–6929. [Google Scholar] [CrossRef] [PubMed]
- McKayed, K.K.; Simpson, J.C. Actin in action: Imaging approaches to study cytoskeleton structure and function. Cells 2013, 2, 715–731. [Google Scholar] [CrossRef]
- Wu, K.; Zhang, X.; Li, F.; Xiao, D.; Hou, Y.; Zhu, S.; Liu, D.; Ye, X.; Ye, M.; Yang, J.; et al. Frequent alterations in cytoskeleton remodelling genes in primary and metastatic lung adenocarcinomas. Nat. Commun. 2015, 6, 10131. [Google Scholar] [CrossRef]
- Soini, T.; Eloranta, K.; Pihlajoki, M.; Kyronlahti, A.; Akinrinade, O.; Andersson, N.; Lohi, J.; Pakarinen, M.P.; Wilson, D.B.; Heikinheimo, M. Transcription factor GATA4 associates with mesenchymal-like gene expression in human hepatoblastoma cells. Tumour. Biol. 2018, 40, 1010428318785498. [Google Scholar] [CrossRef]
- Gallegos, L.L.; Ng, M.R.; Sowa, M.E.; Selfors, L.M.; White, A.; Zervantonakis, I.K.; Singh, P.; Dhakal, S.; Harper, J.W.; Brugge, J.S. A protein interaction map for cell-cell adhesion regulators identifies DUSP23 as a novel phosphatase for beta-catenin. Sci. Rep. 2016, 6, 1–15. [Google Scholar]
- Santos Guasch, G.L.; Beeler, J.S.; Marshall, C.B.; Shaver, T.M.; Sheng, Q.; Johnson, K.N.; Boyd, K.L.; Venters, B.J.; Cook, R.S.; Pietenpol, J.A. p73 Is Required for Ovarian Follicle Development and Regulates a Gene Network Involved in Cell-to-Cell Adhesion. iScience 2018, 8, 236–249. [Google Scholar] [CrossRef]
- Al Tanoury, Z.; Piskunov, A.; Andriamoratsiresy, D.; Gaouar, S.; Lutzing, R.; Ye, T.; Jost, B.; Keime, C.; Rochette-Egly, C. Genes involved in cell adhesion and signaling: A new repertoire of retinoic acid receptor target genes in mouse embryonic fibroblasts. J. Cell Sci. 2014, 127, 521–533. [Google Scholar] [CrossRef]
- Minsky, N.; Roeder, R.G. Inhibition of Adhesion Molecule Gene Expression and Cell Adhesion by the Metabolic Regulator PGC-1alpha. PLoS ONE 2016, 11, e0165598. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Mayor, R. Tuning Collective Cell Migration by Cell–Cell Junction Regulation. Cold Spring Harb. Perspect. Biol. 2017, 9, a029199. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer. 2019, 19, 716–732. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef]
- Harney, A.S.; Arwert, E.N.; Entenberg, D.; Wang, Y.; Guo, P.; Qian, B.Z.; Oktay, M.H.; Pollard, J.W.; Jones, J.G.; Condeelis, J.S. Real-Time Imaging Reveals Local, Transient Vascular Permeability, and Tumor Cell Intravasation Stimulated by TIE2hi Macrophage-Derived VEGFA. Cancer Discov. 2015, 5, 932–943. [Google Scholar] [CrossRef]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112. [Google Scholar] [CrossRef]
- Hong, Y.; Fang, F.; Zhang, Q. Circulating tumor cell clusters: What we know and what we expect (Review). Int. J. Oncol. 2016, 49, 2206–2216. [Google Scholar] [CrossRef]
- Yu, M. Metastasis Stemming from Circulating Tumor Cell Clusters. Trends Cell Biol. 2019, 29, 275–276. [Google Scholar] [CrossRef]
- Giuliano, M.; Shaikh, A.; Lo, H.C.; Arpino, G.; De Placido, S.; Zhang, X.H.; Cristofanilli, M.; Schiff, R.; Trivedi, M.V. Perspective on Circulating Tumor Cell Clusters: Why It Takes a Village to Metastasize. Cancer Res. 2018, 78, 845–852. [Google Scholar] [CrossRef]
- Wang, W.C.; Zhang, X.F.; Peng, J.; Li, X.F.; Wang, A.L.; Bie, Y.Q.; Shi, L.H.; Lin, M.B.; Zhang, X.F. Survival Mechanisms and Influence Factors of Circulating Tumor Cells. BioMed Res. Int. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.C.; Martin, S.S. Insights on CTC Biology and Clinical Impact Emerging from Advances in Capture Technology. Cells 2019, 8, 553. [Google Scholar] [CrossRef] [PubMed]
- Agnoletto, C.; Corra, F.; Minotti, L.; Baldassari, F.; Crudele, F.; Cook, W.J.J.; Di Leva, G.; d’Adamo, A.P.; Gasparini, P.; Volinia, S. Heterogeneity in Circulating Tumor Cells: The Relevance of the Stem-Cell Subset. Cancers 2019, 11, 483. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Jolly, M.K.; Woodward, W.A.; Levine, H.; Deem, M.W. Analysis of Hierarchical Organization in Gene Expression Networks Reveals Underlying Principles of Collective Tumor Cell Dissemination and Metastatic Aggressiveness of Inflammatory Breast Cancer. Front. Oncol. 2018, 8, 244. [Google Scholar] [CrossRef]
- Liu, X.; Taftaf, R.; Kawaguchi, M.; Chang, Y.F.; Chen, W.; Entenberg, D.; Zhang, Y.; Gerratana, L.; Huang, S.; Patel, D.B.; et al. Homophilic CD44 Interactions Mediate Tumor Cell Aggregation and Polyclonal Metastasis in Patient-Derived Breast Cancer Models. Cancer Discov. 2019, 9, 96–113. [Google Scholar] [CrossRef]
- Rodrigues, P.; Vanharanta, S. Circulating Tumor Cells: Come Together, Right Now, Over Metastasis. Cancer Discov. 2019, 9, 22–24. [Google Scholar] [CrossRef]
- Basu, S.; Cheriyamundath, S.; Ben-Ze’ev, A. Cell-cell adhesion: Linking Wnt/beta-catenin signaling with partial EMT and stemness traits in tumorigenesis. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Santi, A.; Kugeratski, F.G.; Zanivan, S. Cancer Associated Fibroblasts: The Architects of Stroma Remodeling. Proteomics 2018, 18, 1700167. [Google Scholar] [CrossRef]
- Marsh, T.; Pietras, K.; McAllister, S.S. Fibroblasts as architects of cancer pathogenesis. Biochim. Biophys. Acta 2013, 1832, 1070–1078. [Google Scholar] [CrossRef]
- Kwa, M.Q.; Herum, K.M.; Brakebusch, C. Cancer-associated fibroblasts: How do they contribute to metastasis? Clin. Exp. Metastasis 2019, 36, 71–86. [Google Scholar] [CrossRef]
- Matsumura, Y.; Ito, Y.; Mezawa, Y.; Sulidan, K.; Daigo, Y.; Hiraga, T.; Mogushi, K.; Wali, N.; Suzuki, H.; Itoh, T.; et al. Stromal fibroblasts induce metastatic tumor cell clusters via epithelial-mesenchymal plasticity. Life Sci. Alliance 2019, 2, e201900425. [Google Scholar] [CrossRef] [PubMed]
- Goulet, C.R.; Champagne, A.; Bernard, G.; Vandal, D.; Chabaud, S.; Pouliot, F.; Bolduc, S. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of bladder cancer cells through paracrine IL-6 signalling. BMC Cancer 2019, 19, 137. [Google Scholar] [CrossRef] [PubMed]
- Labernadie, A.; Kato, T.; Brugues, A.; Serra-Picamal, X.; Derzsi, S.; Arwert, E.; Weston, A.; Gonzalez-Tarrago, V.; Elosegui-Artola, A.; Albertazzi, L.; et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat. Cell Biol. 2017, 19, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Wang, B. Cancer cells exploit the Eph-ephrin system to promote invasion and metastasis: Tales of unwitting partners. Sci. Signal. 2011, 4, pe28. [Google Scholar] [CrossRef]
- Mattes, B.; Scholpp, S. Emerging role of contact-mediated cell communication in tissue development and diseases. Histochem. Cell Biol. 2018, 150, 431–442. [Google Scholar] [CrossRef]
- Liao, J.; Qian, F.; Tchabo, N.; Mhawech-Fauceglia, P.; Beck, A.; Qian, Z.; Wang, X.; Huss, W.J.; Lele, S.B.; Morrison, C.D.; et al. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PLoS ONE 2014, 9, e84941. [Google Scholar] [CrossRef]
- Lupia, M.; Cavallaro, U. Ovarian cancer stem cells: Still an elusive entity? Mol. Cancer 2017, 16, 64. [Google Scholar] [CrossRef]
- Ghoneum, A.; Afify, H.; Salih, Z.; Kelly, M.; Said, N. Role of tumor microenvironment in the pathobiology of ovarian cancer: Insights and therapeutic opportunities. Cancer Med. 2018, 7, 5047–5056. [Google Scholar] [CrossRef]
- Motohara, T.; Masuda, K.; Morotti, M.; Zheng, Y.; El-Sahhar, S.; Chong, K.Y.; Wietek, N.; Alsaadi, A.; Karaminejadranjbar, M.; Hu, Z.; et al. An evolving story of the metastatic voyage of ovarian cancer cells: Cellular and molecular orchestration of the adipose-rich metastatic microenvironment. Oncogene 2019, 38, 2885–2898. [Google Scholar] [CrossRef]
- Azimian-Zavareh, V.; Hossein, G.; Ebrahimi, M.; Dehghani-Ghobadi, Z. Wnt11 alters integrin and cadherin expression by ovarian cancer spheroids and inhibits tumorigenesis and metastasis. Exp. Cell Res. 2018, 369, 90–104. [Google Scholar] [CrossRef]
- Kalluri, R.; Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Belli, C.; Trapani, D.; Viale, G.; D’Amico, P.; Duso, B.A.; Della Vigna, P.; Orsi, F.; Curigliano, G. Targeting the microenvironment in solid tumors. Cancer Treat. Rev. 2018, 65, 22–32. [Google Scholar] [CrossRef] [PubMed]
- De Wever, O.; Van Bockstal, M.; Mareel, M.; Hendrix, A.; Bracke, M. Carcinoma-associated fibroblasts provide operational flexibility in metastasis. Semin. Cancer Biol. 2014, 25, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Heneberg, P. Paracrine tumor signaling induces transdifferentiation of surrounding fibroblasts. Crit. Rev. Oncol. Hematol. 2016, 97, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef]
- Tao, L.; Huang, G.; Song, H.; Chen, Y.; Chen, L. Cancer associated fibroblasts: An essential role in the tumor microenvironment. Oncol. Lett. 2017, 14, 2611–2620. [Google Scholar] [CrossRef]
- Semba, S.; Kodama, Y.; Ohnuma, K.; Mizuuchi, E.; Masuda, R.; Yashiro, M.; Hirakawa, K.; Yokozaki, H. Direct cancer-stromal interaction increases fibroblast proliferation and enhances invasive properties of scirrhous-type gastric carcinoma cells. Br. J. Cancer 2009, 101, 1365–1373. [Google Scholar] [CrossRef]
- Choe, C.; Shin, Y.S.; Kim, S.H.; Jeon, M.J.; Choi, S.J.; Lee, J.; Kim, J. Tumor-stromal interactions with direct cell contacts enhance motility of non-small cell lung cancer cells through the hedgehog signaling pathway. Anticancer. Res. 2013, 33, 3715–3723. [Google Scholar]
- He, X.J.; Tao, H.Q.; Hu, Z.M.; Ma, Y.Y.; Xu, J.; Wang, H.J.; Xia, Y.J.; Li, L.; Fei, B.Y.; Li, Y.Q.; et al. Expression of galectin-1 in carcinoma-associated fibroblasts promotes gastric cancer cell invasion through upregulation of integrin beta1. Cancer Sci. 2014, 105, 1402–1410. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Sakai, R. Direct Interaction between Carcinoma Cells and Cancer Associated Fibroblasts for the Regulation of Cancer Invasion. Cancers 2015, 7, 2054–2062. [Google Scholar] [CrossRef]
- Theveneau, E.; Linker, C. Leaders in collective migration: Are front cells really endowed with a particular set of skills? F1000Research 2017, 6, 1899. [Google Scholar] [CrossRef] [PubMed]
- Jaqaman, K.; Grinstein, S. Regulation from within: The cytoskeleton in transmembrane signaling. Trends Cell Biol. 2012, 22, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Guidolin, D.; Marcoli, M.; Tortorella, C.; Maura, G.; Agnati, L. Receptor-Receptor Interactions as a Widespread Phenomenon: Novel Targets for Drug Development? Front. Endocrinol. (Lausanne) 2019, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Berque-Bestel, I.; Lezoualc’h, F.; Jockers, R. Bivalent ligands as specific pharmacological tools for G protein-coupled receptor dimers. Curr. Drug Discov. Technol. 2008, 5, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Cirri, P.; Chiarugi, P. Cancer-associated-fibroblasts and tumour cells: A diabolic liaison driving cancer progression. Cancer Metastasis Rev. 2012, 31, 195–208. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alekseenko, I.V.; Chernov, I.P.; Kostrov, S.V.; Sverdlov, E.D. Are Synapse-Like Structures a Possible Way for Crosstalk of Cancer with Its Microenvironment? Cancers 2020, 12, 806. https://doi.org/10.3390/cancers12040806
Alekseenko IV, Chernov IP, Kostrov SV, Sverdlov ED. Are Synapse-Like Structures a Possible Way for Crosstalk of Cancer with Its Microenvironment? Cancers. 2020; 12(4):806. https://doi.org/10.3390/cancers12040806
Chicago/Turabian StyleAlekseenko, Irina V, Igor P Chernov, Sergei V Kostrov, and Eugene D Sverdlov. 2020. "Are Synapse-Like Structures a Possible Way for Crosstalk of Cancer with Its Microenvironment?" Cancers 12, no. 4: 806. https://doi.org/10.3390/cancers12040806
APA StyleAlekseenko, I. V., Chernov, I. P., Kostrov, S. V., & Sverdlov, E. D. (2020). Are Synapse-Like Structures a Possible Way for Crosstalk of Cancer with Its Microenvironment? Cancers, 12(4), 806. https://doi.org/10.3390/cancers12040806