Proteomic Discovery of Biomarkers to Predict Prognosis of High-Grade Serous Ovarian Carcinoma

 , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Results
2.1. Patient Characteristics in the Proteomic Analysis
2.2. Results of Proteomic and Bioinformatic Analyses
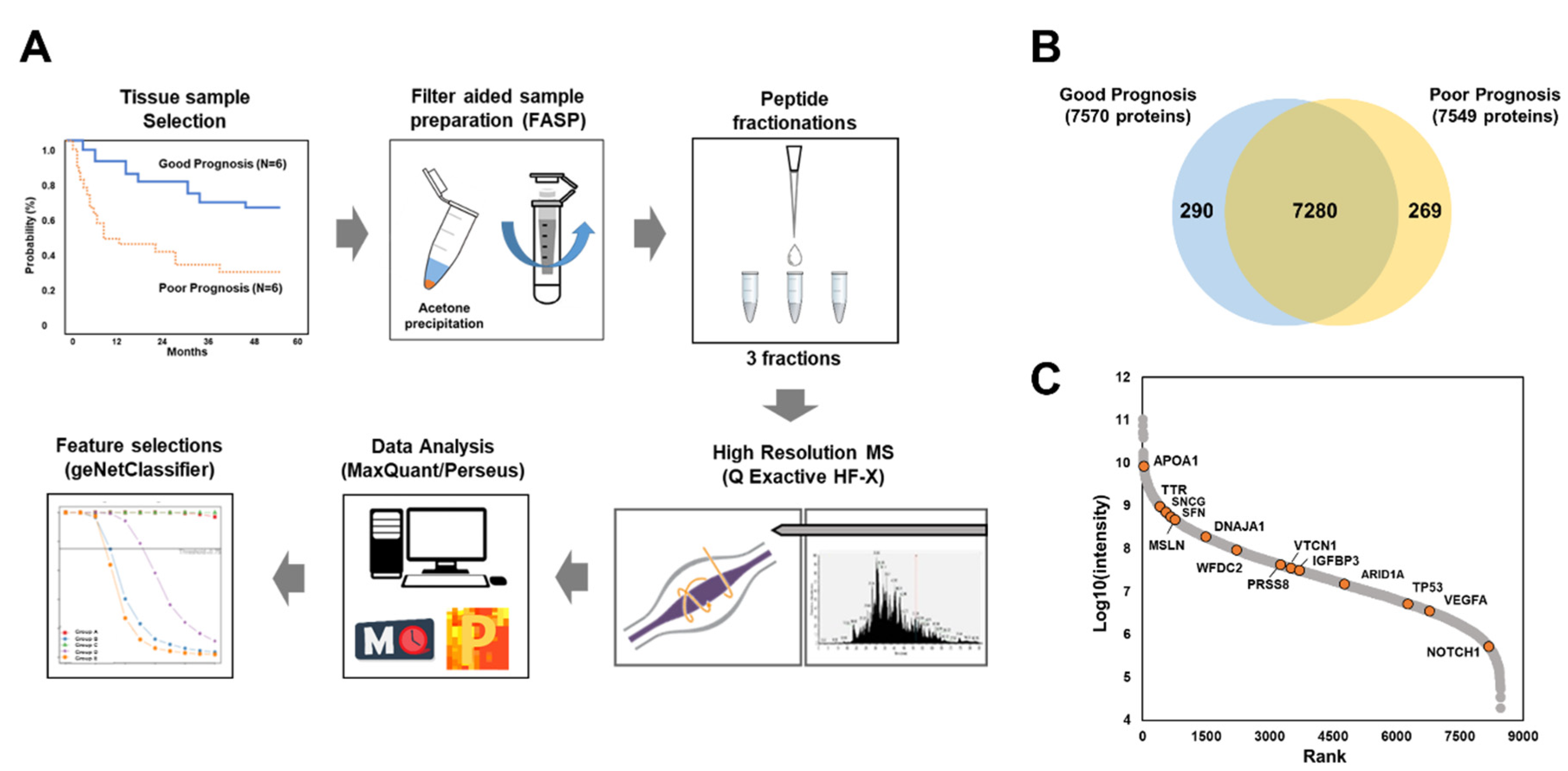
2.2.1. Global Proteomic Analysis of Ovarian Cancer Tissues
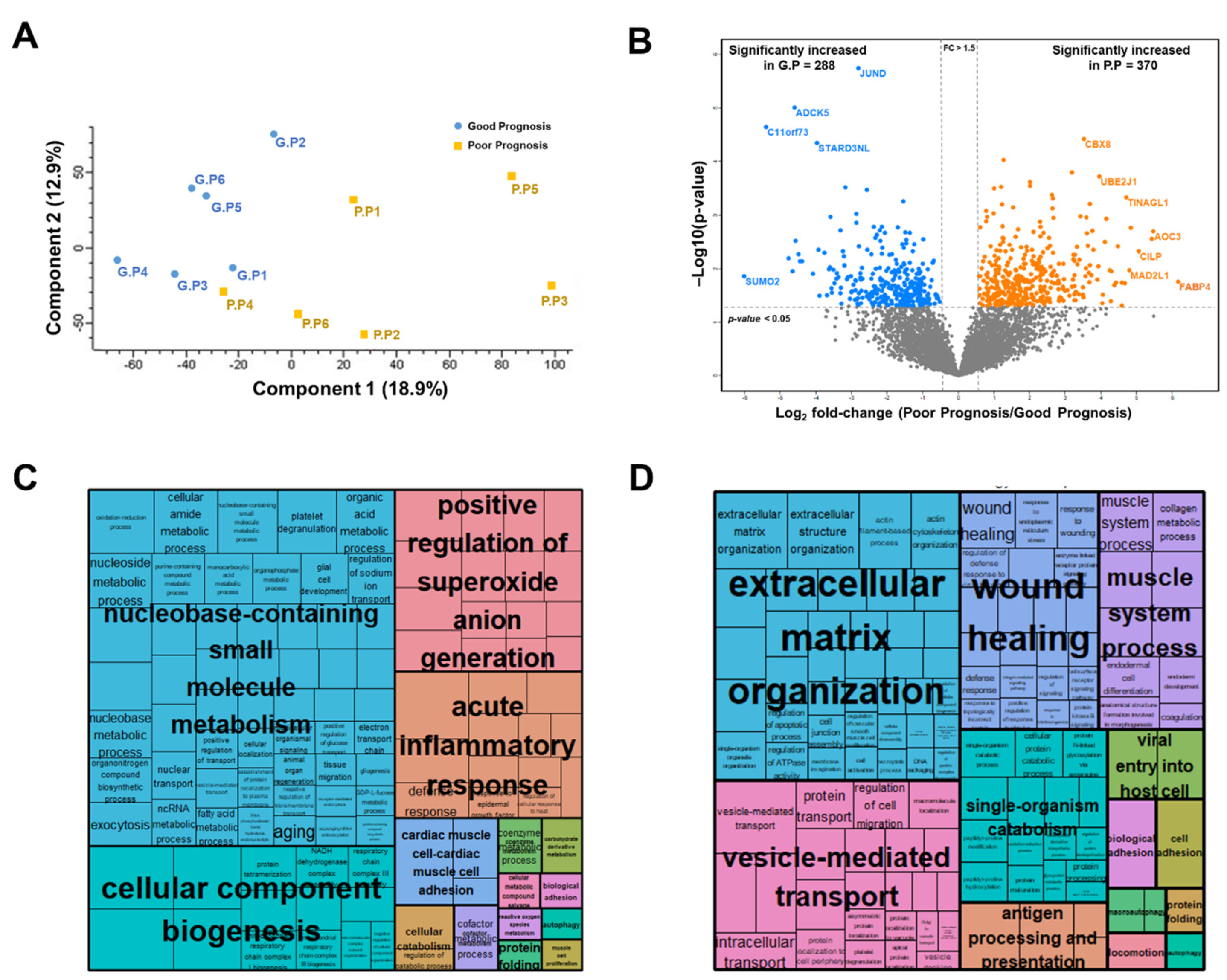
2.2.2. Label-Free Quantification
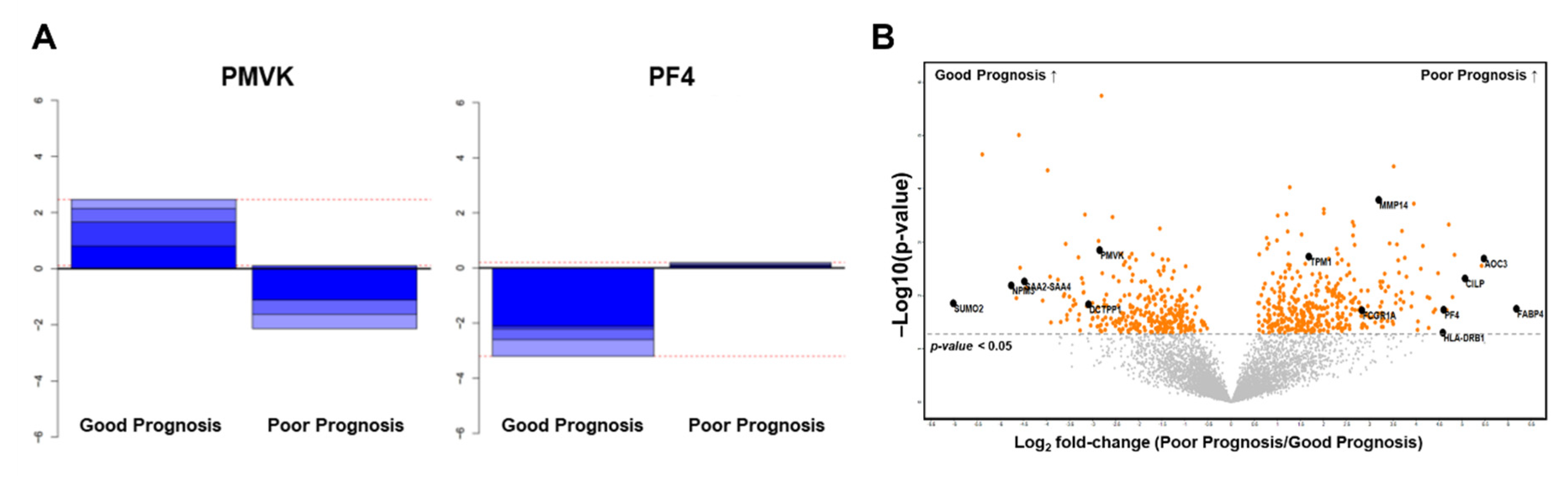
2.2.3. Selection of Candidate Prognostic Biomarkers
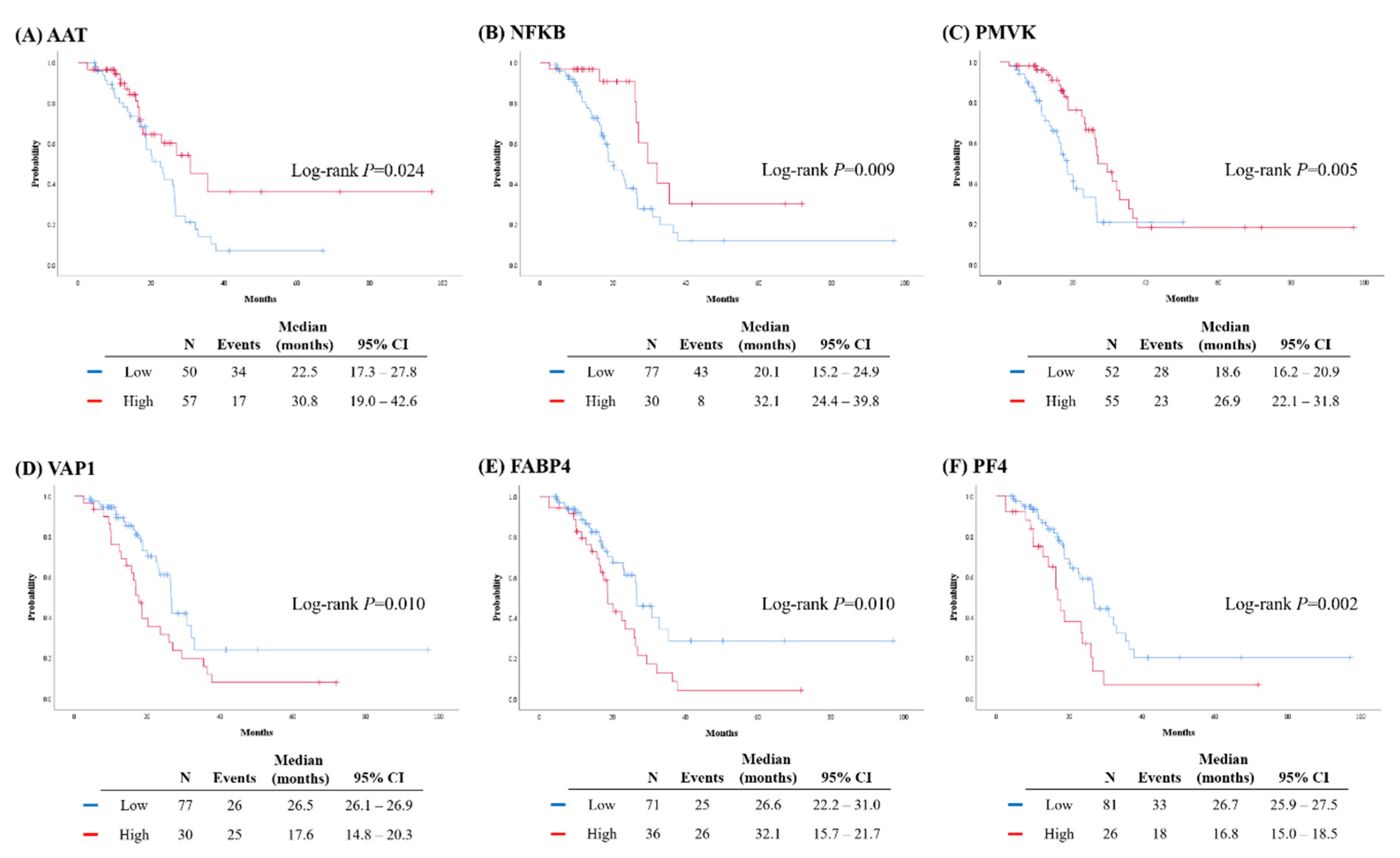
2.3. Validation of Protein Biomarkers through IHC Analysis
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Proteomic and Bioinformatic Analyses
4.2.1. Tissue Preparation
4.2.2. Desalting and Peptide Fractionation of Individual Samples
4.2.3. Offline High-pH Reversed-Peptide Fractionation for Library Construction
4.2.4. LC-MS/MS Analysis
4.2.5. Data Processing
4.2.6. Label-Free Quantification and Statistical Analysis
4.2.7. Bioinformatic Analysis
4.3. Validation via IHC Analysis
4.3.1. TMA Construction
4.3.2. IHC Staining
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.R.; Shih Ie, M. Ovarian cancer. Annu. Rev. Pathol. 2009, 4, 287–313. [Google Scholar] [CrossRef] [PubMed]
- Bristow, R.E.; Tomacruz, R.S.; Armstrong, D.K.; Trimble, E.L.; Montz, F.J. Survival effect of maximal cytoreductive surgery for advanced ovarian carcinoma during the platinum era: A meta-analysis. J. Clin. Oncol. 2002, 20, 1248–1259. [Google Scholar] [CrossRef]
- Chi, D.S.; Franklin, C.C.; Levine, D.A.; Akselrod, F.; Sabbatini, P.; Jarnagin, W.R.; DeMatteo, R.; Poynor, E.A.; Abu-Rustum, N.R.; Barakat, R.R. Improved optimal cytoreduction rates for stages IIIC and IV epithelial ovarian, fallopian tube, and primary peritoneal cancer: A change in surgical approach. Gynecol. Oncol. 2004, 94, 650–654. [Google Scholar] [CrossRef] [PubMed]
- Cannistra, S.A. Cancer of the ovary. N. Engl. J. Med. 2004, 351, 2519–2529. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, J.; Wang, X.; Zhu, J.; Liu, Q.; Shi, Z.; Chambers, M.C.; Zimmerman, L.J.; Shaddox, K.F.; Kim, S.; et al. Proteogenomic characterization of human colon and rectal cancer. Nature 2014, 513, 382–387. [Google Scholar] [CrossRef]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef]
- Okayama, A.; Kimura, Y.; Miyagi, Y.; Oshima, T.; Oshita, F.; Ito, H.; Nakayama, H.; Nagashima, T.; Rino, Y.; Masuda, M.; et al. Relationship between phosphorylation of sperm-specific antigen and prognosis of lung adenocarcinoma. J. Proteomics 2016, 139, 60–66. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, T.; Zhang, Z.; Payne, S.H.; Zhang, B.; McDermott, J.E.; Zhou, J.Y.; Petyuk, V.A.; Chen, L.; Ray, D.; et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166, 755–765. [Google Scholar] [CrossRef]
- Coscia, F.; Watters, K.M.; Curtis, M.; Eckert, M.A.; Chiang, C.Y.; Tyanova, S.; Montag, A.; Lastra, R.R.; Lengyel, E.; Mann, M. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat. Commun. 2016, 7, 12645. [Google Scholar] [CrossRef]
- Dieters-Castator, D.Z.; Rambau, P.F.; Kelemen, L.E.; Siegers, G.M.; Lajoie, G.A.; Postovit, L.M.; Kobel, M. Proteomics-Derived Biomarker Panel Improves Diagnostic Precision to Classify Endometrioid and High-grade Serous Ovarian Carcinoma. Clin. Cancer Res. 2019, 25, 4309–4319. [Google Scholar] [CrossRef] [PubMed]
- Nolen, B.M.; Lokshin, A.E. Protein biomarkers of ovarian cancer: The forest and the trees. Future Oncol. 2012, 8, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Sirota, M.; Butte, A.J. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015, 6, 8971. [Google Scholar] [CrossRef] [PubMed]
- Aibar, S.; Fontanillo, C.; Droste, C.; Roson-Burgo, B.; Campos-Laborie, F.J.; Hernandez-Rivas, J.M.; De Las Rivas, J. Analyse multiple disease subtypes and build associated gene networks using genome-wide expression profiles. BMC Genomics 2015, 16 (Suppl. 5), S3. [Google Scholar] [CrossRef]
- Hao, D.; Li, J.; Jia, S.; Meng, Y.; Zhang, C.; Wang, L.; Di, L.J. Integrated Analysis Reveals Tubal- and Ovarian-Originated Serous Ovarian Cancer and Predicts Differential Therapeutic Responses. Clin. Cancer Res. 2017, 23, 7400–7411. [Google Scholar] [CrossRef]
- Normandin, K.; Peant, B.; Le Page, C.; de Ladurantaye, M.; Ouellet, V.; Tonin, P.N.; Provencher, D.M.; Mes-Masson, A.M. Protease inhibitor SERPINA1 expression in epithelial ovarian cancer. Clin. Exp. Metastasis 2010, 27, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Timms, J.F.; Arslan-Low, E.; Kabir, M.; Worthington, J.; Camuzeaux, S.; Sinclair, J.; Szaub, J.; Afrough, B.; Podust, V.N.; Fourkala, E.O.; et al. Discovery of serum biomarkers of ovarian cancer using complementary proteomic profiling strategies. Proteomics Clin. Appl. 2014, 8, 982–993. [Google Scholar] [CrossRef]
- Wu, W.; Wang, Q.; Yin, F.; Yang, Z.; Zhang, W.; Gabra, H.; Li, L. Identification of proteomic and metabolic signatures associated with chemoresistance of human epithelial ovarian cancer. Int. J. Oncol. 2016, 49, 1651–1665. [Google Scholar] [CrossRef]
- Yeo, S.K.; French, R.; Spada, F.; Clarkson, R. Opposing roles of Nfkb2 gene products p100 and p52 in the regulation of breast cancer stem cells. Breast Cancer Res. Treat. 2017, 162, 465–477. [Google Scholar] [CrossRef]
- Uno, M.; Saitoh, Y.; Mochida, K.; Tsuruyama, E.; Kiyono, T.; Imoto, I.; Inazawa, J.; Yuasa, Y.; Kubota, T.; Yamaoka, S. NF-kappaB inducing kinase, a central signaling component of the non-canonical pathway of NF-kappaB, contributes to ovarian cancer progression. PLoS ONE 2014, 9, e88347. [Google Scholar] [CrossRef]
- Shen, K.; Rice, S.D.; Gingrich, D.A.; Wang, D.; Mi, Z.; Tian, C.; Ding, Z.; Brower, S.L.; Ervin, P.R., Jr.; Gabrin, M.J.; et al. Distinct genes related to drug response identified in ER positive and ER negative breast cancer cell lines. PLoS ONE 2012, 7, e40900. [Google Scholar] [CrossRef] [PubMed]
- Irjala, H.; Salmi, M.; Alanen, K.; Grenman, R.; Jalkanen, S. Vascular adhesion protein 1 mediates binding of immunotherapeutic effector cells to tumor endothelium. J. Immunol. 2001, 166, 6937–6943. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.C.; Chang, S.J.; Kostoro, J.; Kwan, A.L.; Chai, C.Y. Vascular adhesion protein-1 as indicator of breast cancer tumor aggressiveness and invasiveness. APMIS 2018, 126, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Gharpure, K.M.; Pradeep, S.; Sans, M.; Rupaimoole, R.; Ivan, C.; Wu, S.Y.; Bayraktar, E.; Nagaraja, A.S.; Mangala, L.S.; Zhang, X.; et al. FABP4 as a key determinant of metastatic potential of ovarian cancer. Nat. Commun. 2018, 9, 2923. [Google Scholar] [CrossRef]
- Poruk, K.E.; Firpo, M.A.; Huerter, L.M.; Scaife, C.L.; Emerson, L.L.; Boucher, K.M.; Jones, K.A.; Mulvihill, S.J. Serum platelet factor 4 is an independent predictor of survival and venous thromboembolism in patients with pancreatic adenocarcinoma. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2605–2610. [Google Scholar] [CrossRef]
- Siamakpour-Reihani, S.; Owzar, K.; Jiang, C.; Turner, T.; Deng, Y.; Bean, S.M.; Horton, J.K.; Berchuck, A.; Marks, J.R.; Dewhirst, M.W.; et al. Prognostic significance of differential expression of angiogenic genes in women with high-grade serous ovarian carcinoma. Gynecol. Oncol. 2015, 139, 23–29. [Google Scholar] [CrossRef]
- Piver, M.S.; Moyer, M.; Diakun, K.; Lele, S.B.; Chu, T.M. Serum alpha 1-acid glycoprotein in epithelial ovarian cancer. Gynecol. Oncol. 1988, 29, 305–308. [Google Scholar] [CrossRef]
- Moore, L.E.; Pfeiffer, R.M.; Zhang, Z.; Lu, K.H.; Fung, E.T.; Bast, R.C., Jr. Proteomic biomarkers in combination with CA 125 for detection of epithelial ovarian cancer using prediagnostic serum samples from the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial. Cancer 2012, 118, 91–100. [Google Scholar] [CrossRef]
- Collinson, F.; Hutchinson, M.; Craven, R.A.; Cairns, D.A.; Zougman, A.; Wind, T.C.; Gahir, N.; Messenger, M.P.; Jackson, S.; Thompson, D.; et al. Predicting response to bevacizumab in ovarian cancer: A panel of potential biomarkers informing treatment selection. Clin. Cancer Res. 2013, 19, 5227–5239. [Google Scholar] [CrossRef]
- Moore, R.G.; Blackman, A.; Miller, M.C.; Robison, K.; DiSilvestro, P.A.; Eklund, E.E.; Strongin, R.; Messerlian, G. Multiple biomarker algorithms to predict epithelial ovarian cancer in women with a pelvic mass: Can additional makers improve performance? Gynecol. Oncol. 2019, 154, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, K.; Woo, J.; Park, J.; Kim, H.; Lee, K.E.; Kim, Y.; Moon, K.C.; Kim, J.Y.; Park, I.A.; et al. Quantitative Proteomic Analysis Identifies AHNAK (Neuroblast Differentiation-associated Protein AHNAK) as a Novel Candidate Biomarker for Bladder Urothelial Carcinoma Diagnosis by Liquid-based Cytology. Mol. Cell. Proteomics 2018, 17, 1788–1802. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Gaugaz, F.Z. Fast and sensitive total protein and Peptide assays for proteomic analysis. Anal. Chem. 2015, 87, 4110–4116. [Google Scholar] [CrossRef] [PubMed]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote) omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Deeb, S.J.; Tyanova, S.; Hummel, M.; Schmidt-Supprian, M.; Cox, J.; Mann, M. Machine Learning-based Classification of Diffuse Large B-cell Lymphoma Patients by Their Protein Expression Profiles. Mol. Cell. Proteomics 2015, 14, 2947–2960. [Google Scholar] [CrossRef]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | All (n = 107, %) |
|---|---|
| Age, years | |
| Mean ± SD | 55.6 ± 10.1 |
| Menopause | 69 (71.9) |
| Personal history of breast cancer | 16 (15.0) |
| Family history of breast cancer | 4 (3.7) |
| Family history of ovarian cancer | 5 (4.7) |
| Serum CA-125, IU/ml | |
| Median (range) | 677.5 (5.1–11,630.0) |
| FIGO stage | |
| I-II | 9 (8.4) |
| III | 68 (63.6) |
| IV | 30 (28.0) |
| Primary treatment strategy | |
| PDS | 102 (95.3) |
| NAC | 5 (4.7) |
| Residual tumor after PDS/IDS | |
| No gross | 73 (68.2) |
| <1 cm | 21 (19.6) |
| ≥1 and <2 cm | 7 (6.5) |
| ≥2 cm | 6 (5.6) |
| Recurrence | |
| No | 56 (52.3) |
| Yes | 51 (47.7) |
| No post-operative chemotherapy (within recurrent disease) | 1 (0.9) |
| PSR 1 (within recurrent disease) | 38 (35.5) |
| PRR (within recurrent disease) | 12 (11.2) |
| Platinum sensitivity | |
| Platinum-sensitive 2 | 72 (67.3) |
| Platinum-resistant | 12 (11.2) |
| Germline BRCA mutation | |
| BRCA1 | 37 (34.6) |
| BRCA2 | 17 (15.9) |
| Both | 0 |
| Characteristics | Multivariate Analysis | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aHR | 95% CI | aHR | 95% CI | aHR | 95% CI | aHR | 95% CI | aHR | 95% CI | aHR | 95% CI | |
| Age, years | p = 0.072 | p = 0.667 | p = 0.879 | p = 0.417 | p = 0.332 | p = 0.365 | ||||||
| ≥55 vs. <55 | 1.755 | 0.951-3.238 | 1.133 | 0.641–2.005 | 1.046 | 0.586-1.866 | 1.265 | 0.717-2.230 | 1.327 | 0.750-2.349 | 1.300 | 0.737-2.295 |
| CA-125, IU/ml | p = 0.028 | p = 0.061 | p = 0.103 | p = 0.066 | p = 0.124 | p = 0.157 | ||||||
| ≥700 vs. <700 | 1.911 | 1.073-3.405 | 1.720 | 0.976–3.031 | 1.603 | 0.909–2.826 | 1.695 | 0.965–2.977 | 1.553 | 0.886–2.721 | 1.500 | 0.856–2.628 |
| FIGO stage | p = 0.470 | p = 0.182 | p = 0.260 | p = 0.304 | p = 0.281 | p = 0.255 | ||||||
| III–IV vs. I–II | 2.149 | 0.270–17.098 | 4.010 | 0.522–30.829 | 3.220 | 0.421–24.652 | 2.920 | 0.379–22.499 | 3.066 | 0.400–23.498 | 3.227 | 0.429–24.274 |
| Residual tumor after PDS/IDS | p = 0.057 | p = 0.183 | p = 0.019 | p = 0.142 | p = 0.118 | p = 0.137 | ||||||
| Gross vs. No gross | 1.732 | 0.985–3.048 | 1.474 | 0.833–2.608 | 2.020 | 1.124–3.630 | 1.531 | 0.868–2.703 | 1.578 | 0.891–2.794 | 1.538 | 0.872–2.711 |
| Germline BRCA status | p = 0.085 | p = 0.101 | p = 0.425 | p = 0.094 | p = 0.088 | p = 0.162 | ||||||
| Mutation vs. WT | 0.598 | 0.333–1.073 | 0.614 | 0.343–1.099 | 0.780 | 0.424–1.436 | 0.600 | 0.329–1.091 | 0.594 | 0.326–1.081 | 0.654 | 0.361–1.186 |
| AAT | p = 0.006 | |||||||||||
| High vs. Low | 0.398 | 0.207–0.768 | ||||||||||
| NFKB | p = 0.030 | |||||||||||
| High vs. Low | 0.424 | 0.196–0.920 | ||||||||||
| PMVK | p = 0.009 | |||||||||||
| High vs. Low | 0.430 | 0.228–0.809 | ||||||||||
| VAP1 | p = 0.024 | |||||||||||
| High vs. Low | 1.911 | 1.089–3.354 | ||||||||||
| FABP4 | p = 0.023 | |||||||||||
| High vs. Low | 1.908 | 1.093–3.331 | ||||||||||
| PF4 | p = 0.017 | |||||||||||
| High vs. Low | 2.071 | 1.139–3.765 | ||||||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.I.; Jung, M.; Dan, K.; Lee, S.; Lee, C.; Kim, H.S.; Chung, H.H.; Kim, J.-W.; Park, N.H.; Song, Y.-S.; et al. Proteomic Discovery of Biomarkers to Predict Prognosis of High-Grade Serous Ovarian Carcinoma. Cancers 2020, 12, 790. https://doi.org/10.3390/cancers12040790
Kim SI, Jung M, Dan K, Lee S, Lee C, Kim HS, Chung HH, Kim J-W, Park NH, Song Y-S, et al. Proteomic Discovery of Biomarkers to Predict Prognosis of High-Grade Serous Ovarian Carcinoma. Cancers. 2020; 12(4):790. https://doi.org/10.3390/cancers12040790
Chicago/Turabian StyleKim, Se Ik, Minsun Jung, Kisoon Dan, Sungyoung Lee, Cheol Lee, Hee Seung Kim, Hyun Hoon Chung, Jae-Weon Kim, Noh Hyun Park, Yong-Sang Song, and et al. 2020. "Proteomic Discovery of Biomarkers to Predict Prognosis of High-Grade Serous Ovarian Carcinoma" Cancers 12, no. 4: 790. https://doi.org/10.3390/cancers12040790
APA StyleKim, S. I., Jung, M., Dan, K., Lee, S., Lee, C., Kim, H. S., Chung, H. H., Kim, J.-W., Park, N. H., Song, Y.-S., Han, D., & Lee, M. (2020). Proteomic Discovery of Biomarkers to Predict Prognosis of High-Grade Serous Ovarian Carcinoma. Cancers, 12(4), 790. https://doi.org/10.3390/cancers12040790