Role of BET Inhibitors in Triple Negative Breast Cancers



Abstract
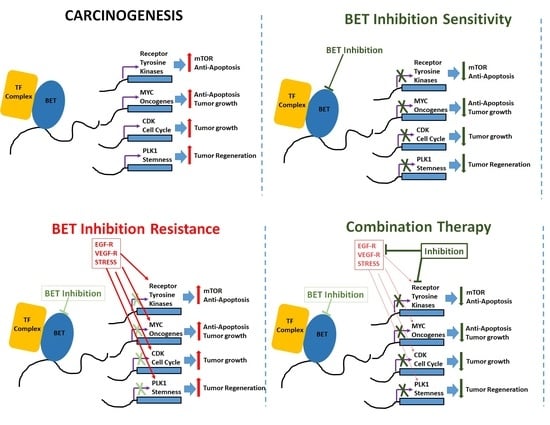
1. Introduction
2. Bromodomain and Extraterminal Domain
3. BET Inhibitors
4. Challenges
5. Future Role of Novel Drug Design and Combinatorial Therapy
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kim, M.; Costello, J. DNA methylation: An epigenetic mark of cellular memory. Exp. Mol. Med. 2017, 49, e322. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Epigenomics: Technologies and Applications. Circ. Res. 2018, 122, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, R.; Gupta, S. Epigenetic modifications in cancer. Clin. Genet. 2012, 81, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yue, D.; Lei, L.; Wang, H.; Lu, J.; Zhou, Y.; Liu, S.; Ding, T.; Guo, M.; Xu, L. Promoter-Operating Targeted Expression of Gene Therapy in Cancer: Current Stage and Prospect. Mol. Nucleic Acids 2018, 11, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA hypomethylation in cancer cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Barnes, C.E.; English, D.M.; Cowley, S.M. Acetylation & Co: An expanding repertoire of histone acylations regulates chromatin and transcription. Essays Biochem. 2019, 63, 97–107. [Google Scholar]
- Taniguchi, Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, R.; Zhou, M.M. The role of human bromodomains in chromatin biology and gene transcription. Curr. Opin. Drug Discov. Dev. 2009, 12, 659–665. [Google Scholar]
- Zaware, N.; Zhou, M.M. Bromodomain biology and drug discovery. Nat. Struct. Mol. Biol. 2019, 26, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef]
- Shi, J.; Vakoc, C.R. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol. Cell 2014, 54, 728–736. [Google Scholar] [CrossRef]
- Muller, S.; Filippakopoulos, P.; Knapp, S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13, e29. [Google Scholar] [CrossRef]
- Xu, Y.; Vakoc, C.R. Targeting Cancer Cells with BET Bromodomain Inhibitors. Cold Spring Harb. Perspect. Med. 2017, 7, a026674. [Google Scholar] [CrossRef]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef]
- Deeney, J.T.; Belkina, A.C.; Shirihai, O.S.; Corkey, B.E.; Denis, G.V. BET Bromodomain Proteins Brd2, Brd3 and Brd4 Selectively Regulate Metabolic Pathways in the Pancreatic beta-Cell. PLoS ONE 2016, 11, e0151329. [Google Scholar] [CrossRef]
- Lamonica, J.M.; Deng, W.; Kadauke, S.; Campbell, A.E.; Gamsjaeger, R.; Wang, H.; Cheng, Y.; Billin, A.N.; Hardison, R.C.; Mackay, J.P.; et al. Bromodomain protein Brd3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. Proc. Natl. Acad. Sci. USA 2011, 108, E159–E168. [Google Scholar] [CrossRef]
- Gamsjaeger, R.; Webb, S.R.; Lamonica, J.M.; Billin, A.; Blobel, G.A.; Mackay, J.P. Structural basis and specificity of acetylated transcription factor GATA1 recognition by BET family bromodomain protein Brd3. Mol. Cell Biol. 2011, 31, 2632–2640. [Google Scholar] [CrossRef] [PubMed]
- Wai, D.C.C.; Szyszka, T.N.; Campbell, A.E.; Kwong, C.; Wilkinson-White, L.E.; Silva, A.P.G.; Low, J.K.K.; Kwan, A.H.; Gamsjaeger, R.; Chalmers, J.D.; et al. The BRD3 ET domain recognizes a short peptide motif through a mechanism that is conserved across chromatin remodelers and transcriptional regulators. J. Biol. Chem. 2018, 293, 7160–7175. [Google Scholar] [CrossRef] [PubMed]
- French, C.A. Small-Molecule Targeting of BET Proteins in Cancer. Adv. Cancer Res. 2016, 131, 21–58. [Google Scholar] [PubMed]
- Itzen, F.; Greifenberg, A.K.; Bosken, C.A.; Geyer, M. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014, 42, 7577–7590. [Google Scholar] [CrossRef]
- Yan, J.; Li, Q.; Lievens, S.; Tavernier, J.; You, J. Abrogation of the Brd4-positive transcription elongation factor B complex by papillomavirus E2 protein contributes to viral oncogene repression. J. Virol. 2010, 84, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.C.; Debrosse, M.; Smith, M.; Dey, A.; Huynh, W.; Sarai, N.; Heightman, T.D.; Tamura, T.; Ozato, K. BRD4 coordinates recruitment of pause release factor P-TEFb and the pausing complex NELF/DSIF to regulate transcription elongation of interferon-stimulated genes. Mol. Cell Biol. 2013, 33, 2497–2507. [Google Scholar] [CrossRef]
- Huang, F.; Shao, W.; Fujinaga, K.; Peterlin, B.M. Bromodomain-containing protein 4-independent transcriptional activation by autoimmune regulator (AIRE) and NF-kappaB. J. Biol. Chem. 2018, 293, 4993–5004. [Google Scholar] [CrossRef]
- Chen, R.; Yik, J.H.; Lew, Q.J.; Chao, S.H. Brd4 and HEXIM1: Multiple roles in P-TEFb regulation and cancer. Biomed. Res. Int. 2014, 2014, 232870. [Google Scholar] [CrossRef]
- Togel, L.; Nightingale, R.; Chueh, A.C.; Jayachandran, A.; Tran, H.; Phesse, T.; Wu, R.; Sieber, O.M.; Arango, D.; Dhillon, A.S.; et al. Dual Targeting of Bromodomain and Extraterminal Domain Proteins, and WNT or MAPK Signaling, Inhibits c-MYC Expression and Proliferation of Colorectal Cancer Cells. Mol. Cancer 2016, 15, 1217–1226. [Google Scholar] [CrossRef]
- Sinha, A.; Faller, D.V.; Denis, G.V. Bromodomain analysis of Brd2-dependent transcriptional activation of cyclin A. Biochem. J. 2005, 387, 257–269. [Google Scholar] [CrossRef]
- LeRoy, G.; Rickards, B.; Flint, S.J. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol. Cell 2008, 30, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Mohan, M.; Lin, C.; Guest, E.; Shilatifard, A. Licensed to elongate: A molecular mechanism for MLL-based leukaemogenesis. Nat. Rev. Cancer 2010, 10, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Hajmirza, A.; Emadali, A.; Gauthier, A.; Casasnovas, O.; Gressin, R.; Callanan, M.B. BET Family Protein BRD4: An Emerging Actor in NFkappaB Signaling in Inflammation and Cancer. Biomedicines 2018, 6, 16. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, Y.; Zeng, L.; Wu, Y.; Deng, J.; Zhang, Q.; Lin, Y.; Li, J.; Kang, T.; Tao, M.; et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 2014, 25, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Cao, J.; Zhou, B.P. Twist-BRD4 complex: Potential drug target for basal-like breast cancer. Curr. Pharm. Des. 2015, 21, 1256–1261. [Google Scholar] [CrossRef]
- Huang, B.; Yang, X.D.; Zhou, M.M.; Ozato, K.; Chen, L.F. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol. Cell. Biol. 2009, 29, 1375–1387. [Google Scholar] [CrossRef]
- Lee, J.E.; Park, Y.K.; Park, S.; Jang, Y.; Waring, N.; Dey, A.; Ozato, K.; Lai, B.; Peng, W.; Ge, K. Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat. Commun. 2017, 8, 2217. [Google Scholar] [CrossRef]
- Roe, J.S.; Mercan, F.; Rivera, K.; Pappin, D.J.; Vakoc, C.R. BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Mol. Cell. 2015, 58, 1028–1039. [Google Scholar] [CrossRef]
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin. Epigenetics 2016, 8, 59. [Google Scholar] [CrossRef]
- Chan, H.M.; La Thangue, N.B. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J. Cell Sci. 2001, 114, 2363–2373. [Google Scholar]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Mirguet, O.; Gosmini, R.; Toum, J.; Clement, C.A.; Barnathan, M.; Brusq, J.M.; Mordaunt, J.E.; Grimes, R.M.; Crowe, M.; Pineau, O.; et al. Discovery of epigenetic regulator I-BET762: Lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J. Med. Chem. 2013, 56, 7501–7515. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- French, C.A.; Ramirez, C.L.; Kolmakova, J.; Hickman, T.T.; Cameron, M.J.; Thyne, M.E.; Kutok, J.L.; Toretsky, J.A.; Tadavarthy, A.K.; Kees, U.R.; et al. BRD-NUT oncoproteins: A family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene 2008, 27, 2237–2242. [Google Scholar] [CrossRef]
- Ott, C.J.; Kopp, N.; Bird, L.; Paranal, R.M.; Qi, J.; Bowman, T.; Rodig, S.J.; Kung, A.L.; Bradner, J.E.; Weinstock, D.M. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood 2012, 120, 2843–2852. [Google Scholar] [CrossRef]
- Mio, C.; Gerratana, L.; Bolis, M.; Caponnetto, F.; Zanello, A.; Barbina, M.; Di Loreto, C.; Garattini, E.; Damante, G.; Puglisi, F. BET proteins regulate homologous recombination-mediated DNA repair: BRCAness and implications for cancer therapy. Int. J. Cancer 2019, 144, 755–766. [Google Scholar] [CrossRef]
- Wen, N.; Guo, B.; Zheng, H.; Xu, L.; Liang, H.; Wang, Q.; Wang, D.; Chen, X.; Zhang, S.; Li, Y.; et al. Bromodomain inhibitor jq1 induces cell cycle arrest and apoptosis of glioma stem cells through the VEGF/PI3K/AKT signaling pathway. Int. J. Oncol. 2019, 55, 879–895. [Google Scholar] [CrossRef]
- Jostes, S.; Nettersheim, D.; Fellermeyer, M.; Schneider, S.; Hafezi, F.; Honecker, F.; Schumacher, V.; Geyer, M.; Kristiansen, G.; Schorle, H. The bromodomain inhibitor JQ1 triggers growth arrest and apoptosis in testicular germ cell tumours in vitro and in vivo. J. Cell. Mol. Med. 2017, 21, 1300–1314. [Google Scholar] [CrossRef]
- Feng, Q.; Zhang, Z.; Shea, M.J.; Creighton, C.J.; Coarfa, C.; Hilsenbeck, S.G.; Lanz, R.; He, B.; Wang, L.; Fu, X.; et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014, 24, 809–819. [Google Scholar] [CrossRef]
- Sengupta, S.; Biarnes, M.C.; Clarke, R.; Jordan, V.C. Inhibition of BET proteins impairs estrogen-mediated growth and transcription in breast cancers by pausing RNA polymerase advancement. Breast Cancer Res. Treat. 2015, 150, 265–278. [Google Scholar] [CrossRef]
- Borbely, G.; Haldosen, L.A.; Dahlman-Wright, K.; Zhao, C. Induction of USP17 by combining BET and HDAC inhibitors in breast cancer cells. Oncotarget 2015, 6, 33623–33635. [Google Scholar] [CrossRef] [PubMed]
- da Motta, L.L.; Ledaki, I.; Purshouse, K.; Haider, S.; De Bastiani, M.A.; Baban, D.; Morotti, M.; Steers, G.; Wigfield, S.; Bridges, E.; et al. The BET inhibitor JQ1 selectively impairs tumour response to hypoxia and downregulates CA9 and angiogenesis in triple negative breast cancer. Oncogene 2017, 36, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, R.; Riveiro, M.E.; Astorgues-Xerri, L.; Odore, E.; Rezai, K.; Erba, E.; Panini, N.; Rinaldi, A.; Kwee, I.; Beltrame, L.; et al. The bromodomain inhibitor OTX015 (MK-8628) exerts anti-tumor activity in triple-negative breast cancer models as single agent and in combination with everolimus. Oncotarget 2017, 8, 7598–7613. [Google Scholar] [CrossRef] [PubMed]
- Coude, M.M.; Braun, T.; Berrou, J.; Dupont, M.; Bertrand, S.; Masse, A.; Raffoux, E.; Itzykson, R.; Delord, M.; Riveiro, M.E.; et al. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 2015, 6, 17698–17712. [Google Scholar] [CrossRef] [PubMed]
- Pastori, C.; Daniel, M.; Penas, C.; Volmar, C.H.; Johnstone, A.L.; Brothers, S.P.; Graham, R.M.; Allen, B.; Sarkaria, J.N.; Komotar, R.J.; et al. BET bromodomain proteins are required for glioblastoma cell proliferation. Epigenetics 2014, 9, 611–620. [Google Scholar] [CrossRef]
- Picaud, S.; Da Costa, D.; Thanasopoulou, A.; Filippakopoulos, P.; Fish, P.V.; Philpott, M.; Fedorov, O.; Brennan, P.; Bunnage, M.E.; Owen, D.R.; et al. PFI-1, a highly selective protein interaction inhibitor, targeting BET Bromodomains. Cancer Res. 2013, 73, 3336–3346. [Google Scholar] [CrossRef]
- Zhang, G.; Plotnikov, A.N.; Rusinova, E.; Shen, T.; Morohashi, K.; Joshua, J.; Zeng, L.; Mujtaba, S.; Ohlmeyer, M.; Zhou, M.M. Structure-guided design of potent diazobenzene inhibitors for the BET bromodomains. J. Med. Chem. 2013, 56, 9251–9264. [Google Scholar] [CrossRef]
- Perez-Salvia, M.; Esteller, M. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenetics 2017, 12, 323–339. [Google Scholar] [CrossRef]
- Gacias, M.; Gerona-Navarro, G.; Plotnikov, A.N.; Zhang, G.; Zeng, L.; Kaur, J.; Moy, G.; Rusinova, E.; Rodriguez, Y.; Matikainen, B.; et al. Selective chemical modulation of gene transcription favors oligodendrocyte lineage progression. Chem. Biol. 2014, 21, 841–854. [Google Scholar] [CrossRef]
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019, 30, 1051–1060. [Google Scholar] [CrossRef]
- Stuhlmiller, T.J.; Miller, S.M.; Zawistowski, J.S.; Nakamura, K.; Beltran, A.S.; Duncan, J.S.; Angus, S.P.; Collins, K.A.; Granger, D.A.; Reuther, R.A.; et al. Inhibition of Lapatinib-Induced Kinome Reprogramming in ERBB2-Positive Breast Cancer by Targeting BET Family Bromodomains. Cell Rep. 2015, 11, 390–404. [Google Scholar] [CrossRef] [PubMed]
- Klauber-DeMore, N.; Schulte, B.A.; Wang, G.Y. Targeting MYC for triple-negative breast cancer treatment. Oncoscience 2018, 5, 120–121. [Google Scholar] [PubMed]
- Bihani, T.; Ezell, S.A.; Ladd, B.; Grosskurth, S.E.; Mazzola, A.M.; Pietras, M.; Reimer, C.; Zinda, M.; Fawell, S.; D’Cruz, C.M. Resistance to everolimus driven by epigenetic regulation of MYC in ER+ breast cancers. Oncotarget 2015, 6, 2407–2420. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J., 3rd. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Khandekar, D.; Amara, S.; Tiriveedhi, V. Immunogenicity of Tumor Initiating Stem Cells: Potential Applications in Novel Anticancer Therapy. Front. Oncol. 2019, 9, 315. [Google Scholar] [CrossRef]
- Horne, G.A.; Stewart, H.J.; Dickson, J.; Knapp, S.; Ramsahoye, B.; Chevassut, T. Nanog requires BRD4 to maintain murine embryonic stem cell pluripotency and is suppressed by bromodomain inhibitor JQ1 together with Lefty1. Stem Cells Dev. 2015, 24, 879–891. [Google Scholar] [CrossRef]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sanchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef]
- Kroon, P.; Berry, P.A.; Stower, M.J.; Rodrigues, G.; Mann, V.M.; Simms, M.; Bhasin, D.; Chettiar, S.; Li, C.; Li, P.K.; et al. JAK-STAT blockade inhibits tumor initiation and clonogenic recovery of prostate cancer stem-like cells. Cancer Res. 2013, 73, 5288–5298. [Google Scholar] [CrossRef]
- Chan, C.H.; Fang, C.; Yarilina, A.; Prinjha, R.K.; Qiao, Y.; Ivashkiv, L.B. BET bromodomain inhibition suppresses transcriptional responses to cytokine-Jak-STAT signaling in a gene-specific manner in human monocytes. Eur. J. Immunol. 2015, 45, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Hu, G.; Wang, H.; Li, Z.; Guo, Z. PLK1 inhibitor facilitates the suppressing effect of temozolomide on human brain glioma stem cells. J. Cell. Mol. Med. 2018, 22, 5300–5310. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Nie, Y.; Yue, F.; Kong, Y.; Gu, L.; Gavin, T.P.; Liu, X.; Kuang, S. A requirement of Polo-like kinase 1 in murine embryonic myogenesis and adult muscle regeneration. eLife 2019, 8, e47097. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Li, J.; Luo, Q.; Wang, R.; Kong, Y.; Carlock, C.; Liu, Z.; Elzey, B.D.; Liu, X. Plk1 Inhibition Enhances the Efficacy of BET Epigenetic Reader Blockade in Castration-Resistant Prostate Cancer. Mol. Cancer 2018, 17, 1554–1565. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Jimenez, C.; Alcaraz-Sanabria, A.; Perez-Pena, J.; Corrales-Sanchez, V.; Serrano-Heras, G.; Galan-Moya, E.M.; Serrano-Oviedo, L.; Montero, J.C.; Burgos, M.; Llopis, J.; et al. Targeting basal-like breast tumors with bromodomain and extraterminal domain (BET) and polo-like kinase inhibitors. Oncotarget 2017, 8, 19478–19490. [Google Scholar] [CrossRef] [PubMed]
- Shu, S.; Lin, C.Y.; He, H.H.; Witwicki, R.M.; Tabassum, D.P.; Roberts, J.M.; Janiszewska, M.; Huh, S.J.; Liang, Y.; Ryan, J.; et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016, 529, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.M. Phospho-BRD4: Transcription plasticity and drug targeting. Drug Discov. Today Technol. 2016, 19, 17–22. [Google Scholar] [CrossRef]
- Abedin, S.M.; Boddy, C.S.; Munshi, H.G. BET inhibitors in the treatment of hematologic malignancies: Current insights and future prospects. Oncol. Targets 2016, 9, 5943–5953. [Google Scholar]
- Bhattacharya, S.; Piya, S.; Borthakur, G. Bromodomain inhibitors: What does the future hold? Clin. Adv. Hematol. Oncol. 2018, 16, 504–515. [Google Scholar]
- Ocana, A.; Nieto-Jimenez, C.; Pandiella, A. BET inhibitors as novel therapeutic agents in breast cancer. Oncotarget 2017, 8, 71285–71291. [Google Scholar] [CrossRef]
- Marcotte, R.; Sayad, A.; Brown, K.R.; Sanchez-Garcia, F.; Reimand, J.; Haider, M.; Virtanen, C.; Bradner, J.E.; Bader, G.D.; Mills, G.B.; et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell 2016, 164, 293–309. [Google Scholar] [CrossRef]
- Stratikopoulos, E.E.; Dendy, M.; Szabolcs, M.; Khaykin, A.J.; Lefebvre, C.; Zhou, M.M.; Parsons, R. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer Cell 2015, 27, 837–851. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, T.; Chen, Z.; Soucheray, M.; Carretero, J.; Kikuchi, E.; Tchaicha, J.H.; Gao, Y.; Cheng, K.A.; Cohoon, T.J.; Qi, J.; et al. Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 6183–6192. [Google Scholar] [CrossRef] [PubMed]
- Jauset, T.; Masso-Valles, D.; Martinez-Martin, S.; Beaulieu, M.E.; Foradada, L.; Fiorentino, F.P.; Yokota, J.; Haendler, B.; Siegel, S.; Whitfield, J.R.; et al. BET inhibition is an effective approach against KRAS-driven PDAC and NSCLC. Oncotarget 2018, 9, 18734–18746. [Google Scholar] [CrossRef] [PubMed]
- Alqahtani, A.; Choucair, K.; Ashraf, M.; Hammouda, D.M.; Alloghbi, A.; Khan, T.; Senzer, N.; Nemunaitis, J. Bromodomain and extra-terminal motif inhibitors: A review of preclinical and clinical advances in cancer therapy. Future Sci. OA 2019, 5, FSO372. [Google Scholar] [CrossRef] [PubMed]
- Steegmaier, M.; Hoffmann, M.; Baum, A.; Lenart, P.; Petronczki, M.; Krssak, M.; Gurtler, U.; Garin-Chesa, P.; Lieb, S.; Quant, J.; et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 2007, 17, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Wells, C.; Felletar, I.; Brotherton, D.; Martin, S.; Savitsky, P.; Diez-Dacal, B.; Philpott, M.; Bountra, C.; Lingard, H.; et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. USA 2013, 110, 19754–19759. [Google Scholar] [CrossRef]
- McLure, K.G.; Gesner, E.M.; Tsujikawa, L.; Kharenko, O.A.; Attwell, S.; Campeau, E.; Wasiak, S.; Stein, A.; White, A.; Fontano, E.; et al. RVX-208, an inducer of ApoA-I in humans, is a BET bromodomain antagonist. PLoS ONE 2013, 8, e83190. [Google Scholar] [CrossRef]
- Kharenko, O.A.; Gesner, E.M.; Patel, R.G.; Norek, K.; White, A.; Fontano, E.; Suto, R.K.; Young, P.R.; McLure, K.G.; Hansen, H.C. RVX-297—A novel BD2 selective inhibitor of BET bromodomains. Biochem. Biophys. Res. Commun. 2016, 477, 62–67. [Google Scholar] [CrossRef]
- Martin, M.P.; Olesen, S.H.; Georg, G.I.; Schonbrunn, E. Cyclin-dependent kinase inhibitor dinaciclib interacts with the acetyl-lysine recognition site of bromodomains. ACS Chem. Biol. 2013, 8, 2360–2365. [Google Scholar] [CrossRef]
- Ember, S.W.; Zhu, J.Y.; Olesen, S.H.; Martin, M.P.; Becker, A.; Berndt, N.; Georg, G.I.; Schonbrunn, E. Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem. Biol. 2014, 9, 1160–1171. [Google Scholar] [CrossRef]
- Ciceri, P.; Muller, S.; O’Mahony, A.; Fedorov, O.; Filippakopoulos, P.; Hunt, J.P.; Lasater, E.A.; Pallares, G.; Picaud, S.; Wells, C.; et al. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat. Chem. Biol. 2014, 10, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Lewin, J.; Soria, J.C.; Stathis, A.; Delord, J.P.; Peters, S.; Awada, A.; Aftimos, P.G.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. Phase Ib Trial with Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients with Selected Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 3007–3014. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; You, J. Mechanistic analysis of the role of bromodomain-containing protein 4 (BRD4) in BRD4-NUT oncoprotein-induced transcriptional activation. J. Biol. Chem. 2015, 290, 2744–2758. [Google Scholar] [CrossRef] [PubMed]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef]
- Li, N.; Yang, L.; Qi, X.K.; Lin, Y.X.; Xie, X.; He, G.P.; Feng, Q.S.; Liu, L.R.; Xie, X.; Zeng, Y.X.; et al. BET bromodomain inhibitor JQ1 preferentially suppresses EBV-positive nasopharyngeal carcinoma cells partially through repressing c-Myc. Cell Death Dis. 2018, 9, 761. [Google Scholar] [CrossRef]
- Wadhwa, E.; Nicolaides, T. Bromodomain Inhibitor Review: Bromodomain and Extra-terminal Family Protein Inhibitors as a Potential New Therapy in Central Nervous System Tumors. Cureus 2016, 8, e620. [Google Scholar] [CrossRef]
- Rathert, P.; Roth, M.; Neumann, T.; Muerdter, F.; Roe, J.S.; Muhar, M.; Deswal, S.; Cerny-Reiterer, S.; Peter, B.; Jude, J.; et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 2015, 525, 543–547. [Google Scholar] [CrossRef]
- Anand, P.; Brown, J.D.; Lin, C.Y.; Qi, J.; Zhang, R.; Artero, P.C.; Alaiti, M.A.; Bullard, J.; Alazem, K.; Margulies, K.B.; et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell 2013, 154, 569–582. [Google Scholar] [CrossRef]
- Matzuk, M.M.; McKeown, M.R.; Filippakopoulos, P.; Li, Q.; Ma, L.; Agno, J.E.; Lemieux, M.E.; Picaud, S.; Yu, R.N.; Qi, J.; et al. Small-molecule inhibition of BRDT for male contraception. Cell 2012, 150, 673–684. [Google Scholar] [CrossRef]
- Berkovits, B.D.; Wolgemuth, D.J. The role of the double bromodomain-containing BET genes during mammalian spermatogenesis. Curr. Top. Dev. Biol. 2013, 102, 293–326. [Google Scholar] [PubMed]
- Sullivan, J.M.; Badimon, A.; Schaefer, U.; Ayata, P.; Gray, J.; Chung, C.W.; von Schimmelmann, M.; Zhang, F.; Garton, N.; Smithers, N.; et al. Autism-like syndrome is induced by pharmacological suppression of BET proteins in young mice. J. Exp. Med. 2015, 212, 1771–1781. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A.C.; Denis, G.V.; Bradner, J.; Sebastiani, P.; Margolis, D.M.; Montano, M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc. Biol. 2012, 92, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Amorim, S.; Stathis, A.; Gleeson, M.; Iyengar, S.; Magarotto, V.; Leleu, X.; Morschhauser, F.; Karlin, L.; Broussais, F.; Rezai, K.; et al. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: A dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–e204. [Google Scholar] [CrossRef]
- Rhyasen, G.W.; Hattersley, M.M.; Yao, Y.; Dulak, A.; Wang, W.; Petteruti, P.; Dale, I.L.; Boiko, S.; Cheung, T.; Zhang, J.; et al. AZD5153: A Novel Bivalent BET Bromodomain Inhibitor Highly Active against Hematologic Malignancies. Mol. Cancer 2016, 15, 2563–2574. [Google Scholar] [CrossRef]
- Tanaka, M.; Roberts, J.M.; Seo, H.S.; Souza, A.; Paulk, J.; Scott, T.G.; DeAngelo, S.L.; Dhe-Paganon, S.; Bradner, J.E. Design and characterization of bivalent BET inhibitors. Nat. Chem. Biol. 2016, 12, 1089–1096. [Google Scholar] [CrossRef]
- Lu, J.; Qian, Y.; Altieri, M.; Dong, H.; Wang, J.; Raina, K.; Hines, J.; Winkler, J.D.; Crew, A.P.; Coleman, K.; et al. Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chem. Biol. 2015, 22, 755–763. [Google Scholar] [CrossRef]
- Sakamoto, K.M.; Kim, K.B.; Kumagai, A.; Mercurio, F.; Crews, C.M.; Deshaies, R.J. Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 8554–8559. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, B.; Yang, C.Y.; Ji, J.; McEachern, D.; Przybranowski, S.; Jiang, H.; Hu, J.; Xu, F.; Zhao, Y.; et al. Targeted Degradation of BET Proteins in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 2476–2487. [Google Scholar] [CrossRef]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef]
- Karakashev, S.; Zhu, H.; Yokoyama, Y.; Zhao, B.; Fatkhutdinov, N.; Kossenkov, A.V.; Wilson, A.J.; Simpkins, F.; Speicher, D.; Khabele, D.; et al. BET Bromodomain Inhibition Synergizes with PARP Inhibitor in Epithelial Ovarian Cancer. Cell Rep. 2017, 21, 3398–3405. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, Y.; Shan, W.; Hu, Z.; Yuan, J.; Pi, J.; Wang, Y.; Fan, L.; Tang, Z.; Li, C.; et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med. 2017, 9, eaal1645. [Google Scholar] [CrossRef] [PubMed]
- Bui, M.H.; Lin, X.; Albert, D.H.; Li, L.; Lam, L.T.; Faivre, E.J.; Warder, S.E.; Huang, X.; Wilcox, D.; Donawho, C.K.; et al. Preclinical Characterization of BET Family Bromodomain Inhibitor ABBV-075 Suggests Combination Therapeutic Strategies. Cancer Res. 2017, 77, 2976–2989. [Google Scholar] [CrossRef] [PubMed]
- Ember, S.W.; Lambert, Q.T.; Berndt, N.; Gunawan, S.; Ayaz, M.; Tauro, M.; Zhu, J.Y.; Cranfill, P.J.; Greninger, P.; Lynch, C.C.; et al. Potent Dual BET Bromodomain-Kinase Inhibitors as Value-Added Multitargeted Chemical Probes and Cancer Therapeutics. Mol. Cancer 2017, 16, 1054–1067. [Google Scholar] [CrossRef]
- Hogg, S.J.; Vervoort, S.J.; Deswal, S.; Ott, C.J.; Li, J.; Cluse, L.A.; Beavis, P.A.; Darcy, P.K.; Martin, B.P.; Spencer, A.; et al. BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep. 2017, 18, 2162–2174. [Google Scholar] [CrossRef]
- Lai, X.; Stiff, A.; Duggan, M.; Wesolowski, R.; Carson, W.E., 3rd; Friedman, A. Modeling combination therapy for breast cancer with BET and immune checkpoint inhibitors. Proc. Natl. Acad. Sci. USA 2018, 115, 5534–5539. [Google Scholar] [CrossRef]
- Sammons, S.; Kornblum, N.S.; Blackwell, K.L. Fulvestrant-Based Combination Therapy for Second-Line Treatment of Hormone Receptor-Positive Advanced Breast Cancer. Target Oncol. 2019, 14, 1–12. [Google Scholar] [CrossRef]
- Fiskus, W.; Sharma, S.; Qi, J.; Shah, B.; Devaraj, S.G.; Leveque, C.; Portier, B.P.; Iyer, S.; Bradner, J.E.; Bhalla, K.N. BET protein antagonist JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (TKI) and overcomes resistance to FLT3-TKI in AML cells expressing FLT-ITD. Mol. Cancer 2014, 13, 2315–2327. [Google Scholar] [CrossRef]
- Bauer, K.; Berger, D.; Zielinski, C.C.; Valent, P.; Grunt, T.W. Hitting two oncogenic machineries in cancer cells: Cooperative effects of the multi-kinase inhibitor ponatinib and the BET bromodomain blockers JQ1 or dBET1 on human carcinoma cells. Oncotarget 2018, 9, 26491–26506. [Google Scholar] [CrossRef]
- Heinemann, A.; Cullinane, C.; De Paoli-Iseppi, R.; Wilmott, J.S.; Gunatilake, D.; Madore, J.; Strbenac, D.; Yang, J.Y.; Gowrishankar, K.; Tiffen, J.C.; et al. Combining BET and HDAC inhibitors synergistically induces apoptosis of melanoma and suppresses AKT and YAP signaling. Oncotarget 2015, 6, 21507–21521. [Google Scholar] [CrossRef]
- Shahbazi, J.; Liu, P.Y.; Atmadibrata, B.; Bradner, J.E.; Marshall, G.M.; Lock, R.B.; Liu, T. The Bromodomain Inhibitor JQ1 and the Histone Deacetylase Inhibitor Panobinostat Synergistically Reduce N-Myc Expression and Induce Anticancer Effects. Clin. Cancer Res. 2016, 22, 2534–2544. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Qi, J.; Bradner, J.E.; Said, J.W.; Doan, N.B.; Forscher, C.; Yang, H.; Koeffler, H.P. Synergistic effect of JQ1 and rapamycin for treatment of human osteosarcoma. Int. J. Cancer 2015, 136, 2055–2064. [Google Scholar] [CrossRef]
- Wong, C.; Laddha, S.V.; Tang, L.; Vosburgh, E.; Levine, A.J.; Normant, E.; Sandy, P.; Harris, C.R.; Chan, C.S.; Xu, E.Y. The bromodomain and extra-terminal inhibitor CPI203 enhances the antiproliferative effects of rapamycin on human neuroendocrine tumors. Cell Death Dis. 2014, 5, e1450. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Zhang, Z.; Ma, P.; An, S.; Shen, Y.; Zhu, L.; Zhuang, G. Concomitant BET and MAPK blockade for effective treatment of ovarian cancer. Oncotarget 2016, 7, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Paoluzzi, L.; Hanniford, D.; Sokolova, E.; Osman, I.; Darvishian, F.; Wang, J.; Bradner, J.E.; Hernando, E. BET and BRAF inhibitors act synergistically against BRAF-mutant melanoma. Cancer Med. 2016, 5, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Matkar, S.; Sharma, P.; Gao, S.; Gurung, B.; Katona, B.W.; Liao, J.; Muhammad, A.B.; Kong, X.C.; Wang, L.; Jin, G.; et al. An Epigenetic Pathway Regulates Sensitivity of Breast Cancer Cells to HER2 Inhibition via FOXO/c-Myc Axis. Cancer Cell 2015, 28, 472–485. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, R.; Matta, H.; Tolani, B.; Triche, T., Jr.; Chaudhary, P.M. Immunomodulatory drugs target IKZF1-IRF4-MYC axis in primary effusion lymphoma in a cereblon-dependent manner and display synergistic cytotoxicity with BRD4 inhibitors. Oncogene 2016, 35, 1797–1810. [Google Scholar] [CrossRef]
- Adeegbe, D.O.; Liu, S.; Hattersley, M.M.; Bowden, M.; Zhou, C.W.; Li, S.; Vlahos, R.; Grondine, M.; Dolgalev, I.; Ivanova, E.V.; et al. BET Bromodomain Inhibition Cooperates with PD-1 Blockade to Facilitate Antitumor Response in Kras-Mutant Non-Small Cell Lung Cancer. Cancer Immunol. Res. 2018, 6, 1234–1245. [Google Scholar] [CrossRef]
- Muralidharan, S.V.; Bhadury, J.; Nilsson, L.M.; Green, L.C.; McLure, K.G.; Nilsson, J.A. BET bromodomain inhibitors synergize with ATR inhibitors to induce DNA damage, apoptosis, senescence-associated secretory pathway and ER stress in Myc-induced lymphoma cells. Oncogene 2016, 35, 4689–4697. [Google Scholar] [CrossRef]



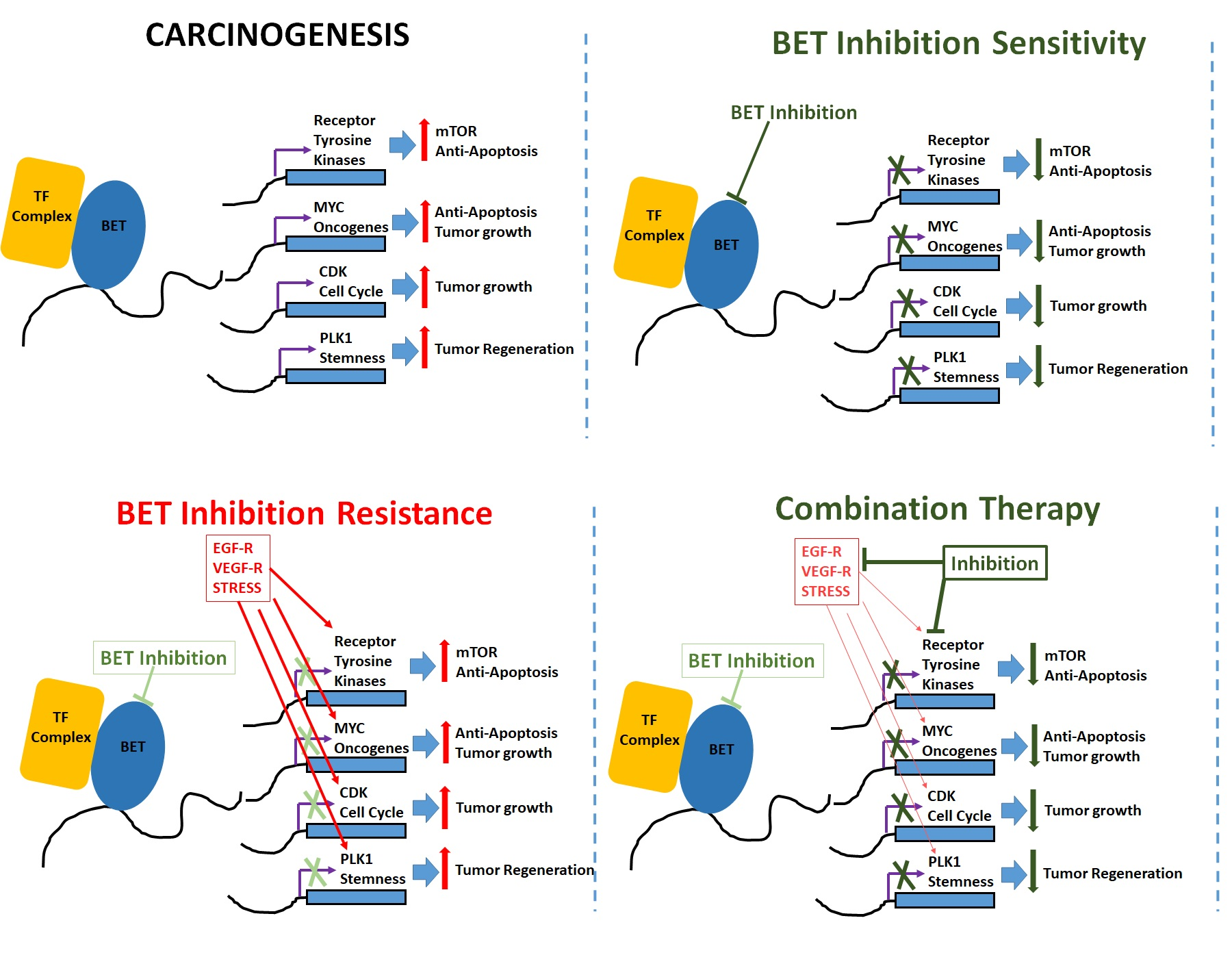{kind=link}
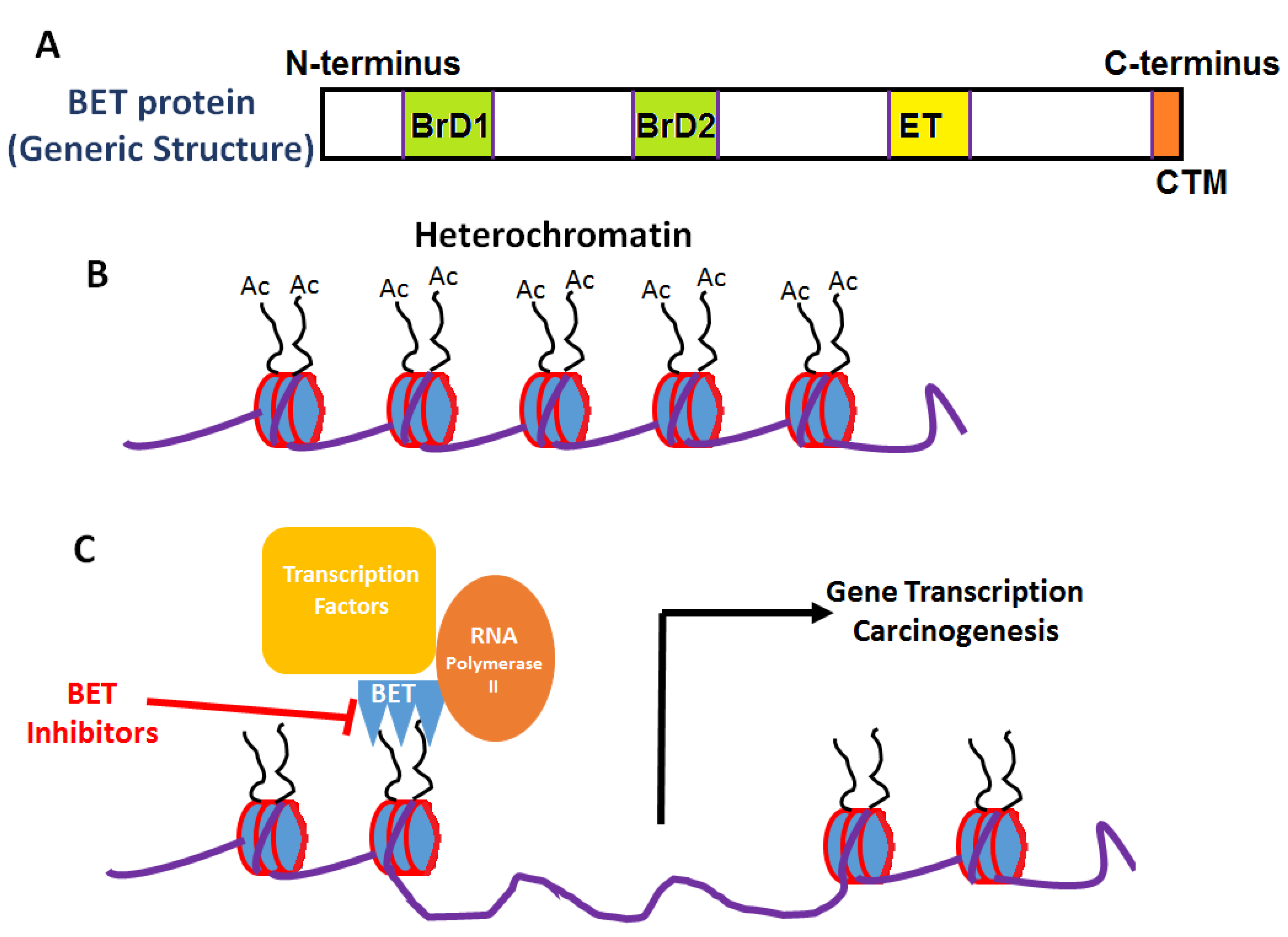{kind=link}
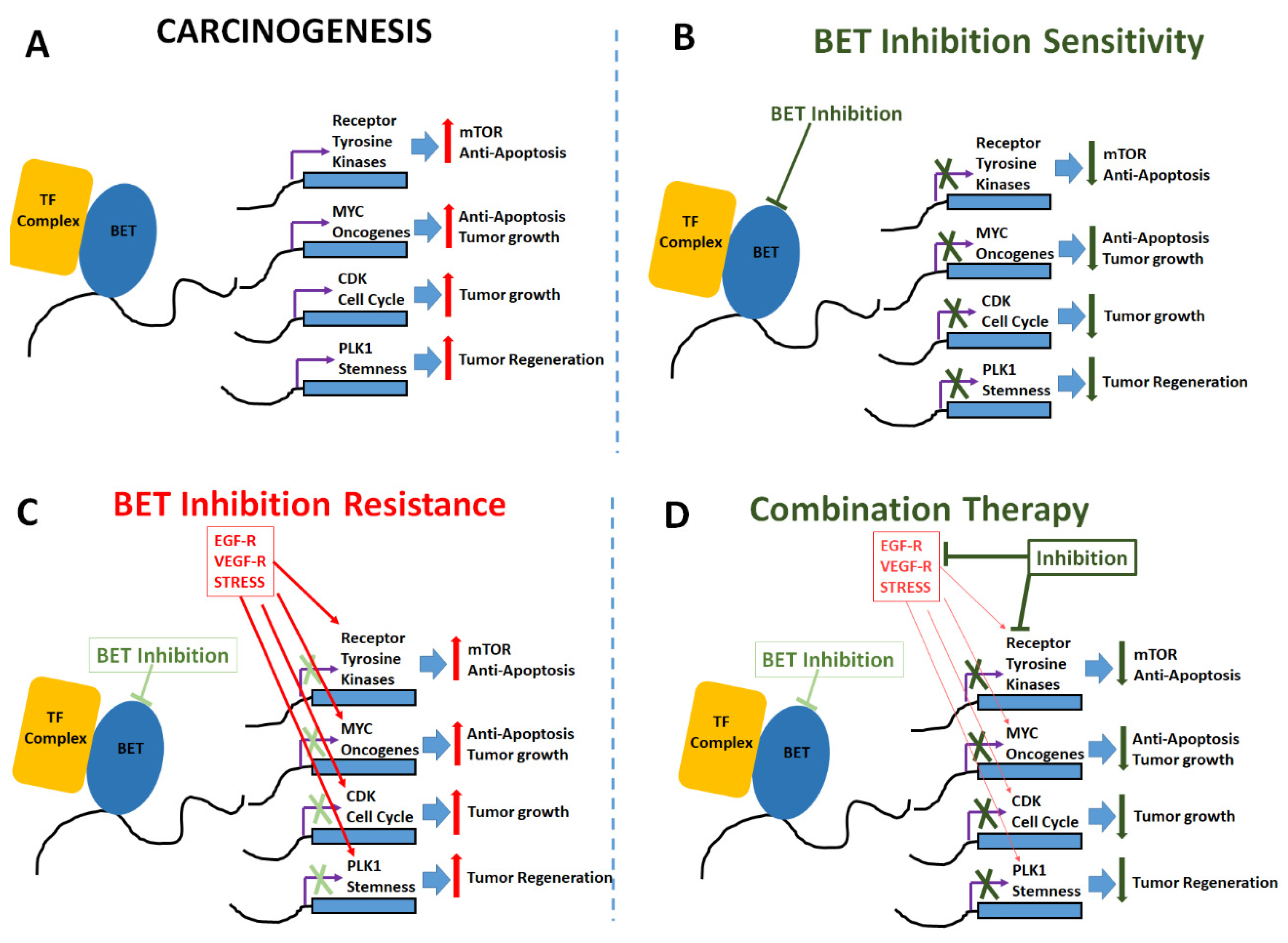{kind=link}
| Drug | Identifier # | Tumor Type | Clinical Phase | Status |
|---|---|---|---|---|
| MK-8628/OTX105 | NCT02259114 | NUT Midline Carcinoma | Phase IB | Completed [93] |
| Non-small Cell Lung Cancer | ||||
| Castrate-resistant Prostate Cancer | ||||
| Pancreatic Ductal Adenocarcinoma | ||||
| GSK525762 | NCT01587703 | All solid tumors, Midline | Phase 1 | Active, not recruiting |
| MK-8628 | NCT02698176 | NUT Midline Carcinoma | Phase 1 | Terminated |
| Non-small Cell Lung Cancer | ||||
| Castrate-resistant Prostate Cancer | ||||
| Pancreatic Ductal Adenocarcinoma | ||||
| INCB054329 | NCT02431260 | Solid Tumors and Hematologic Malignancy | Phase 1 Phase 2 | Terminated |
| Therapeutic Agents | Malignancies | Toxicities |
|---|---|---|
| MK-8268/OTX-015 [88] | Relapsed/refractory leukemia | diarrhea, fatigue, anorexia. Toxicities also included dysgeusia, abdominal pain, decreased clotting factor VII |
| MK-8628/OTX015 [89] | Relapsed/refractory lymphoma or multiple myeloma | thrombocytopenia, neutropenia, hyponatremia; diarrhea, dysgeusia, fatigue, anemia |
| MK-8628/OTX-015 [90] | NUT midline carcinoma | thrombocytopenia, nausea, dysgeusia, hyperglycemia, fatigue |
| BAY1238097 [91] | Advanced solid tumors or NHL | headache, vomiting, low back pain, Recurrent headaches |
| Combination Therapy | Pre-Clinical Models Tested |
|---|---|
| JQ1 and FLT3-TK1 [118] | Immunodeficient mice injected with OCIAML3 or MOLM13 cells |
| JQ1/dBET1 and Ponatinib [119] | Colon (HCT116, HT29), breast (MCF-7, SKBR3) and ovarian (A2780, SKOV3) cancer cells |
| I-BET151 and panobinostat [120] | Immunodeficient mice injected with patient-derived melanoma cells resistant to vemurafenib |
| JQ1 and panobinostat [121] | Syngeneic orthotopic murine tumors, SK-N-BE (2) neuroblastoma cells |
| JQ1 and romidepsin [48] | Murine tumor models of NT2/D1 and NCCIT embryonal carcinoma |
| JQ1 and rapamycin [122] | Immunodeficient mice injected with MNNG/HOS osteosarcoma cells |
| CPI203 and rapamycin [123] | Immunodeficient mice injected with BON-1 pancreatic neuroendocrine tumor cells |
| JQ1 and trametinib [124] | Immunodeficient mice injected with ES2 ovarian clear cell carcinoma cells |
| JQ1 and vemurafenib [125] | Immunodeficient mice injected with A375 melanoma cells |
| JQ1 and fulvestrant [49] | Immunodeficient mice injected with tamoxifen-resistant MCF7 breast cancer cells |
| I-BET151 and lapatinib [126] | Immunodeficient mice injected with Her2þ BT474 breast cancer cells |
| JQ1 and lenalidomide [127] | Immunodeficient mice injected with BC-3 lymphoma cells |
| JQ1 and unidentified PD-1 inhibitor [128] | KRASmt NSCLC murine tumor model |
| RVX2135 and ATR inhibitor AZ20 [129] | Syngeneic λ820 and λ2749 murine Myc-induced lymphoma xenografts |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khandekar, D.; Tiriveedhi, V. Role of BET Inhibitors in Triple Negative Breast Cancers. Cancers 2020, 12, 784. https://doi.org/10.3390/cancers12040784
Khandekar D, Tiriveedhi V. Role of BET Inhibitors in Triple Negative Breast Cancers. Cancers. 2020; 12(4):784. https://doi.org/10.3390/cancers12040784
Chicago/Turabian StyleKhandekar, Durga, and Venkataswarup Tiriveedhi. 2020. "Role of BET Inhibitors in Triple Negative Breast Cancers" Cancers 12, no. 4: 784. https://doi.org/10.3390/cancers12040784
APA StyleKhandekar, D., & Tiriveedhi, V. (2020). Role of BET Inhibitors in Triple Negative Breast Cancers. Cancers, 12(4), 784. https://doi.org/10.3390/cancers12040784