β-Caryophyllene Inhibits Cell Proliferation through a Direct Modulation of CB2 Receptors in Glioblastoma Cells

 ,
,  ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Results
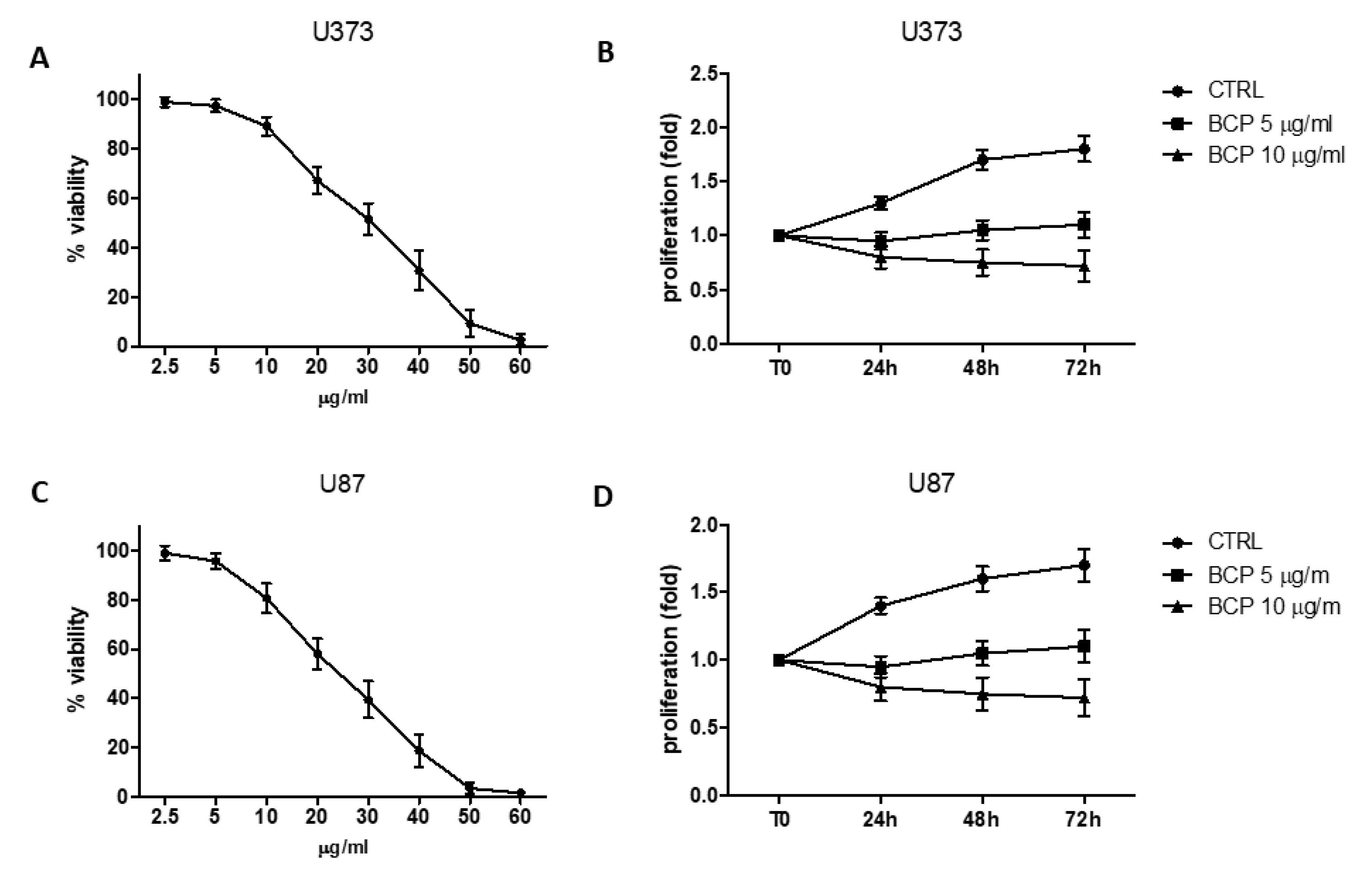
2.1. BCP Reduces Cell Viability
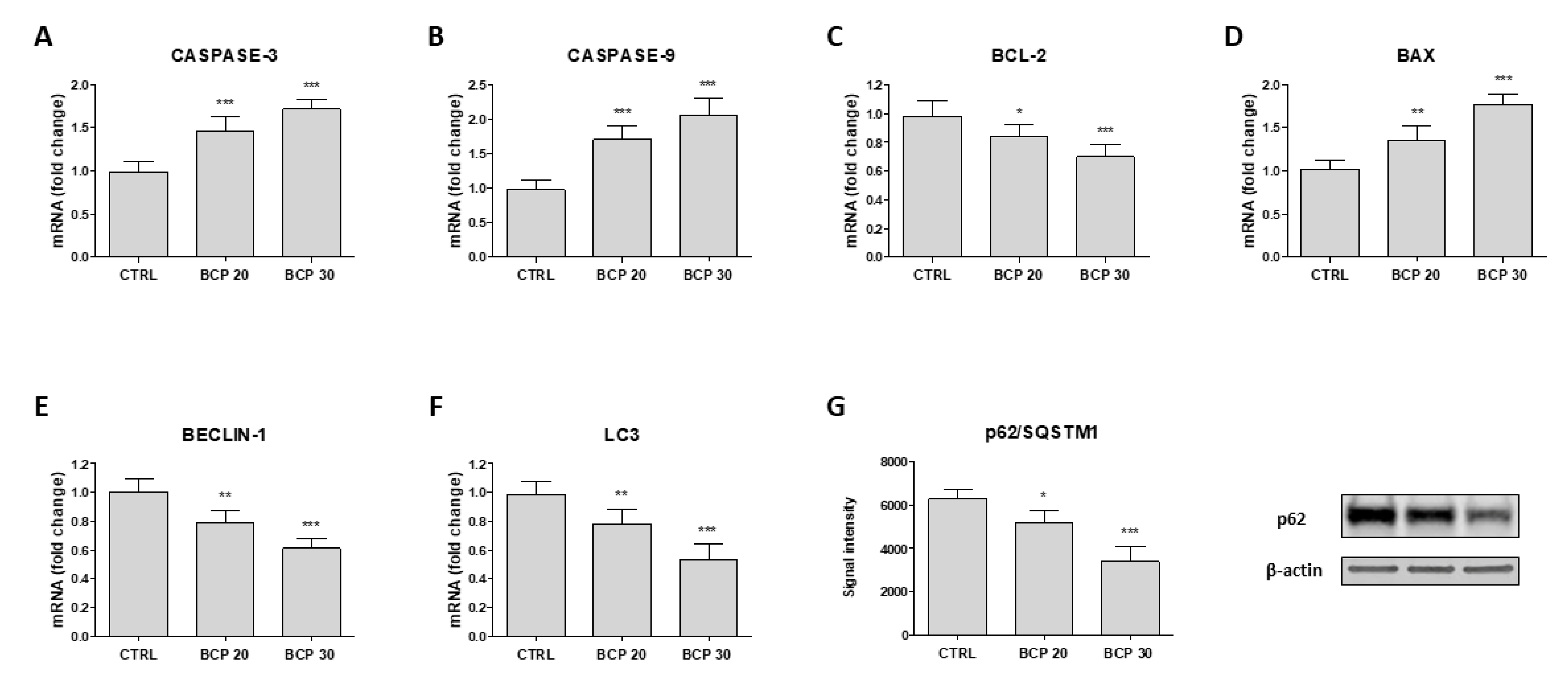
2.2. BCP Induces Apoptotic Cell Death
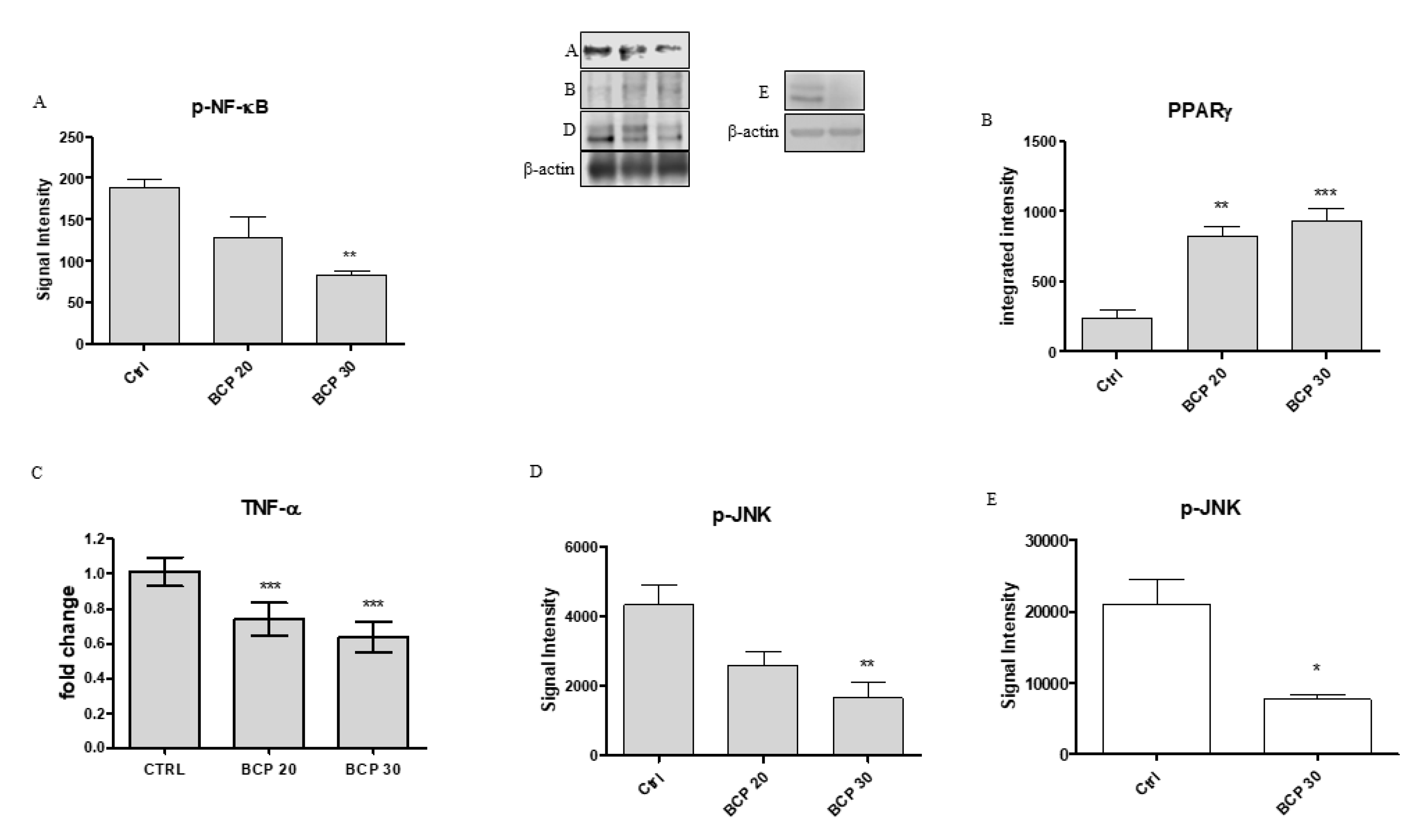
2.3. BCP Plays an Anti-Inflammatory Activity through the Modulation of NF-κB and PPAR-γ
2.4. BCP Carries Out an Anti-Proliferative Effect through Jun N-Terminal Kinase (JNK) Reduction
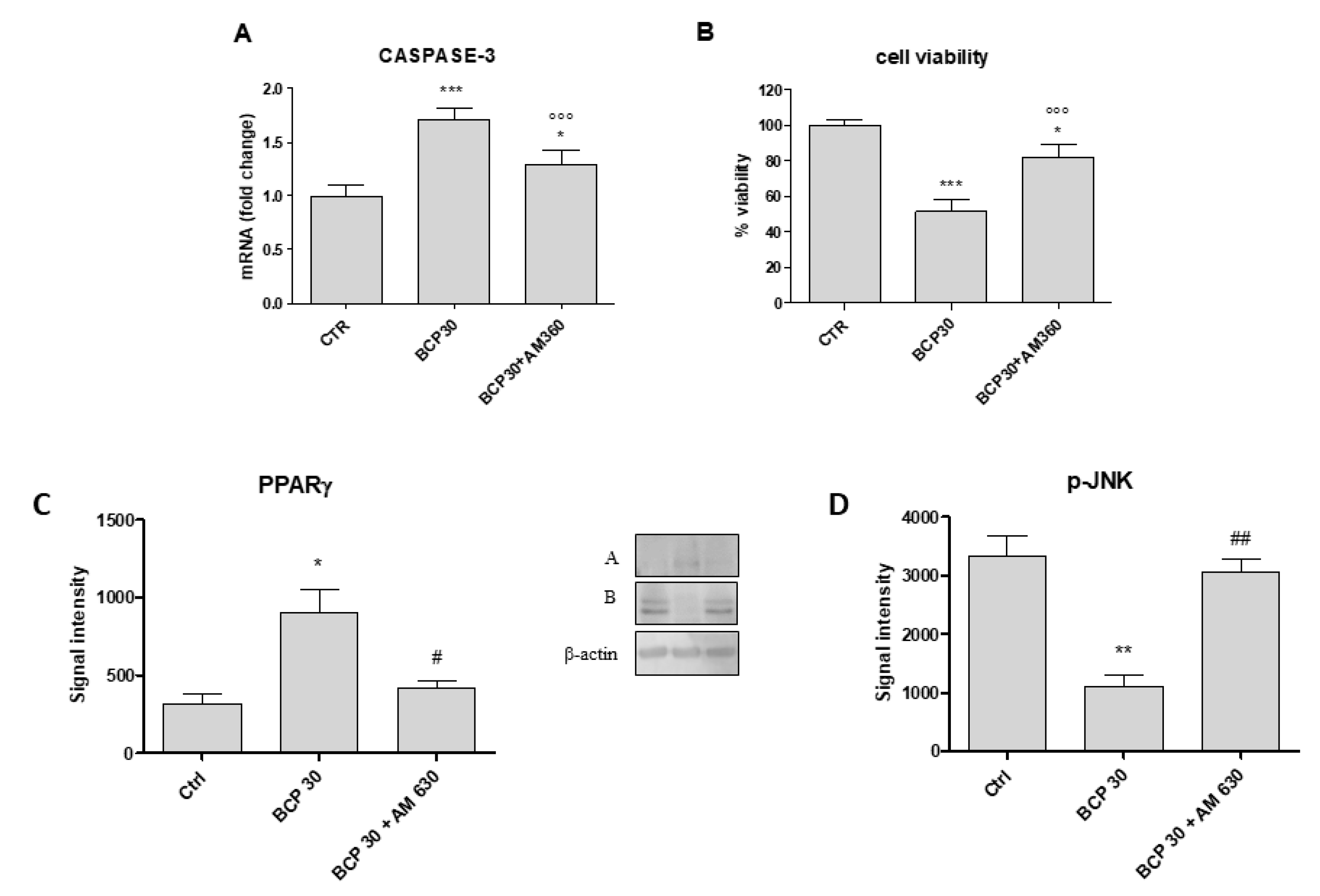
2.5. AM630, a CB2 Antagonist, Abrogates BCP Effects
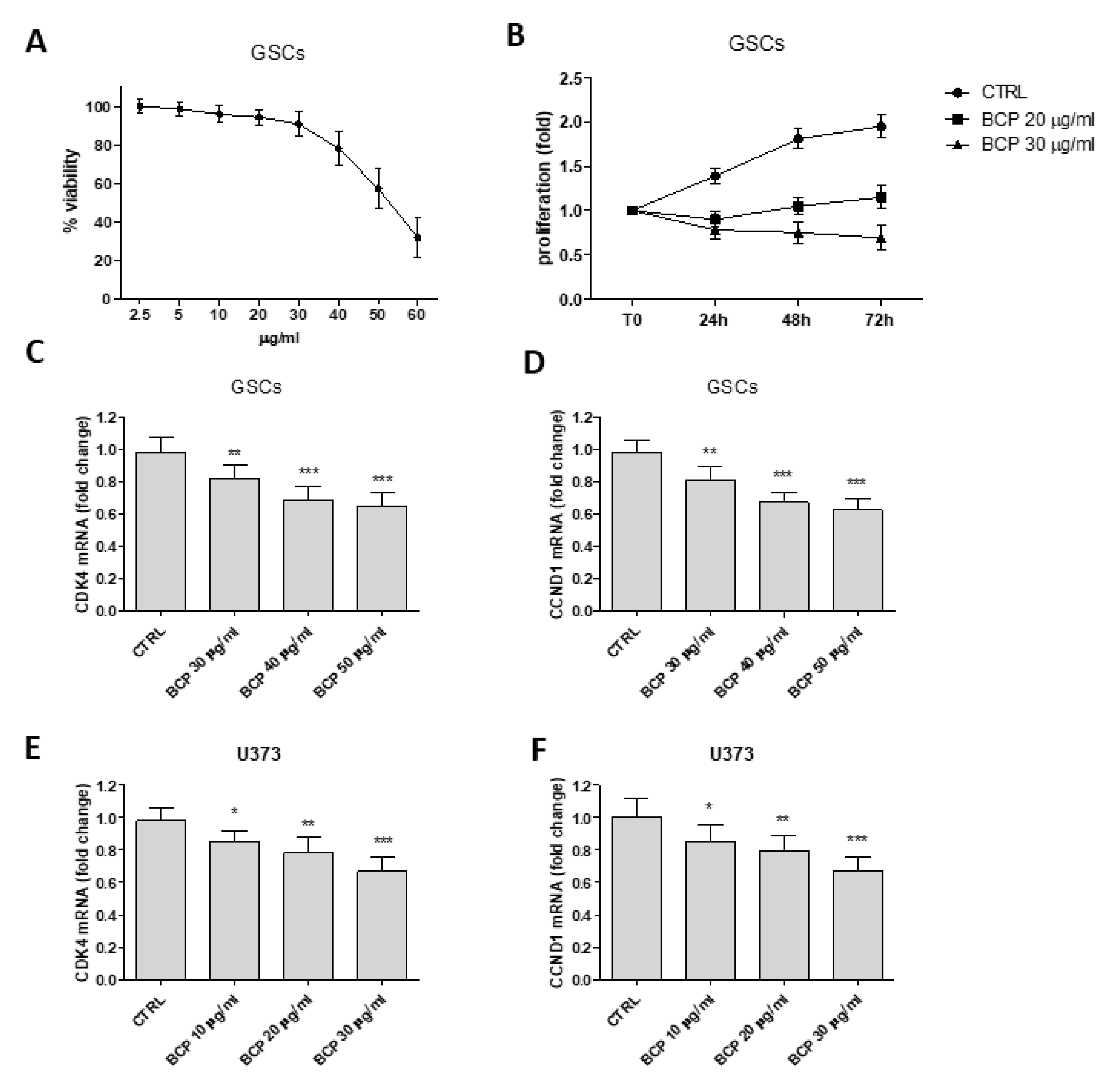
2.6. β-Caryophyllene Reduces the Proliferation of Glioma Stem Cells and Inhibits Cell Cycle
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Cell Treatment
4.3. MTT Assay
4.4. Tunel Assay
4.5. RNA Isolation, cDNA Synthesis, and Real-Time Quantitative PCR Amplification
4.6. Western Blot Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Adamson, C.; Kanu, O.O.; Mehta, A.I.; Di, C.; Lin, N.; Mattox, A.K.; Bigner, D.D. Glioblastoma multiforme: A review of where we have been and where we are going. Expert Opin. Investig. Drugs 2009, 18, 1061–1083. [Google Scholar] [CrossRef] [PubMed]
- Abbas, M.N.; Kausar, S.; Cui, H. Therapeutic potential of natural products in glioblastoma treatment: Targeting key glioblastoma signaling pathways and epigenetic alterations. Clin Transl. Oncol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Bhat, V.; Balasubramaniyan, B.; Vaillant, R.; Ezhilarasan, K.; Hummelink, F.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Aldape Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-κB Family of Transcription Factors and Its Regulation. Cold Spring Harb Perspect. Biol. 2009. [Google Scholar] [CrossRef]
- Yeung, Y.T.; McDonald, K.L.; Grewal, T.; Munoz, L. Interleukins in glioblastoma pathophysiology: Implications for therapy. Br. J. Pharmacol. 2013, 168, 591–606. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Ciechomska, I.; Kaminska, B. Cannabinoid signaling in glioma cells. Adv. Exp. Med. Biol. 2013, 986, 209–220. [Google Scholar] [PubMed]
- Mechoulam, R. Discovery of endocannabinoids and some random thoughts on their possible roles in neuroprotection and aggression. Prostaglandins Leukot Essent Fat. Acids 2002, 66, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Gertsch, J.; Leonti, M.; Raduner, S.; Racz, I.; Chen, J.Z.; Xie, X.Q.; Altmann, K.H.; Karsak, M.; Zimmer, A. Beta-caryophyllene is a dietary cannabinoid. Proc. Natl. Acad. Sci. USA 2008, 105, 9099–9104. [Google Scholar] [CrossRef] [PubMed]
- Rocha, F.C.; Dos Santos Júnior, J.G.; Stefano, S.C.; Da Silveira, D.X. Systematic review of the literature on clinical and experimental trials on the antitumor effects of cannabinoid in gliomas. J. Neurooncol. 2014, 116, 11–24. [Google Scholar] [CrossRef]
- Ramachandhiran, D.; Sankaranarayanan, C.; Murali, R.; Babukumar, S.; Vinothkumar, V. β-Caryophyllene promotes oxidative stress and apoptosis in KB cells through activation of mitochondrial-mediated pathway—An in-vitro and in-silico study. Arch. Physiol. Biochem. 2019, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Irrera, N.; D’Ascola, A.; Pallio, G.; Bitto, A.; Mazzon, E.; Mannino, F.; Squadrito, V.; Arcoraci, V.; Minutoli, L.; Campo, G.M.; et al. β-Caryophyllene Mitigates Collagen Antibody Induced Arthritis (CAIA) in Mice Through a Cross-Talk between CB2 and PPAR-γ Receptors. Biomolecules 2019, 31, 326. [Google Scholar] [CrossRef] [PubMed]
- Castillo, P.E.; Younts, T.J.; Chavez, A.E.; Hashimotodani, Y. Endocannabinoid signaling and synaptic function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, M.; Sawosz, E.; Strojny, B.; Jaworski, S.; Grodzik, M.; Chwalibog, A. NF-κB-related decrease of glioma angiogenic potentiall by graphite nanoparticles and graphene oxide nanoplatelets. Sci. Rep. 2018, 8, 14733. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Holland, E.C.; Celestino, J.; Dai, C.; Schaefer, L.; Sawaya, R.E.; Fuller, G.N. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat. Genet. 2000, 25, 55–57. [Google Scholar] [CrossRef]
- Singh, R.; Cuervo, A.M. Autophagy in the cellular energetic balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Youssef, D.A.; El-Fayoumi, H.M.; Mahmoud, M.F. Beta-caryophyllene alleviates diet-induced neurobehavioral changes in rats: The role of CB2 and PPAR-γ receptors. Biomed. Pharm. 2019, 110, 145–154. [Google Scholar] [CrossRef]
- Pistis, M.; O’Sullivan, S.E. The Role of Nuclear Hormone Receptors in Cannabinoid Function. Adv. Pharm. 2017, 80, 291–328. [Google Scholar]
- Puffenbarger, R.A.; Boothe, A.C.; Cabral, G.A. Cannabinoids inhibit LPS-inducible cytokine mRNA expression in rat microglial cells. Glia 2000, 29, 58–69. [Google Scholar] [CrossRef]
- Gertsch, J.; Schoop, R.; Kuenzle, U.; Suter, A. Echinacea alkylamides modulate TNF alpha gene expression via cannabinoid receptor CB2 and multiple signal transduction pathways. FEBS Lett. 2004, 577, 563–569. [Google Scholar] [CrossRef]
- Swantek, J.L.; Cobb, M.H.; Geppert, T.D. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor alpha (TNF alpha) translation: Glucocorticoids inhibit TNF alpha translation by blocking JNK/SAPK. Mol. Cell Biol. 1997, 17, 6274–6282. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yao, S. JNK-Bcl-2/Bcl-xL-Bax/Bak pathway mediates the crosstalk between Matrine-induced autophagy and apoptosis via interplay with Beclin 1. Int. J. Mol. Sci. 2015, 16, 25744–25758. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Wu, C.L.; Wang, X.; Ban, Q.; Quan, C.; Liu, M.; Dong, H.; Li, J.; Kim, G.Y.; Choi, Y.H.; et al. SP600125 enhances C-2-induced cell death by the switch from autophagy to apoptosis in bladder cancer cells. J. Exp. Clin. Cancer Res. 2019, 38, 448. [Google Scholar] [CrossRef]
- Murata, K.; Matsumura, S.; Yoshioka, Y.; Ueno, Y.; Matsuda, H. Screening of beta-secretase and acetylcholinesterase inhibitors from plant resources. J. Nat. Med. 2015, 69, 123–129. [Google Scholar] [CrossRef]
- Tian, X.; Liu, H.; Xiang, F.; Xu, L.; Dong, Z. β-Caryophyllene protects against ischemic stroke by promoting polarization of microglia toward M2 phenotype via the TLR4 pathway. Life Sci. 2019, 237, 116915. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer 5′-3′ | Reverse Primer 5′-3′ |
|---|---|---|
| ACTB | TTGTTACAGGAAGTCCCTTGCC | ATGCTATCACCTCCCCTGTGT |
| CASPASE-3 | CTGAGGCATGGTGAAGAAGGA | GTCCAGTTCTGTACCACGGCA |
| CASPASE-9 | TGCGAACTAACAGGCAAGCA | GTCTGAGAACCTCTGGTTTGC |
| BCL-2 | GAGGATTGTGGCCTTCTTTGAG | AGCCTCCGTTATCCTGGATC |
| BAX | GGACGAACTGGACAGTAACATG | GCAAAGTAGAAAAGGGCGACA |
| TNF-α | GATAGATGGGCTCATACCAGGG | TCTTCAAGGGCCAAGGCT |
| Beclin-1 | TGAGAGACTGGATCAGGAGG | CGCATCTGGTTTTCAACACTC |
| LC3 | AAGGCGCTTACAGCTCAATG | CTGGGAGGCATAGACCATGT |
| CDK4 | AGCCGAAACGATCAAGGAT | GCTTGACTGTTCCACCACTTG |
| CCND1 | CACCTTATTCATGGCTGAAGTC | ACAAACCTCCACTGGATGGT |
| CD133 | CTTGGCTCAGACTGGTAAATCC | CCACTTTCTCACTGATAGAG |
| OCT4 | AGAAGGATGTGGTCCGAGTG | GCACCTCAGTTTGAATGCATG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irrera, N.; D’Ascola, A.; Pallio, G.; Bitto, A.; Mannino, F.; Arcoraci, V.; Rottura, M.; Ieni, A.; Minutoli, L.; Metro, D.; et al. β-Caryophyllene Inhibits Cell Proliferation through a Direct Modulation of CB2 Receptors in Glioblastoma Cells. Cancers 2020, 12, 1038. https://doi.org/10.3390/cancers12041038
Irrera N, D’Ascola A, Pallio G, Bitto A, Mannino F, Arcoraci V, Rottura M, Ieni A, Minutoli L, Metro D, et al. β-Caryophyllene Inhibits Cell Proliferation through a Direct Modulation of CB2 Receptors in Glioblastoma Cells. Cancers. 2020; 12(4):1038. https://doi.org/10.3390/cancers12041038
Chicago/Turabian StyleIrrera, Natasha, Angela D’Ascola, Giovanni Pallio, Alessandra Bitto, Federica Mannino, Vincenzo Arcoraci, Michelangelo Rottura, Antonio Ieni, Letteria Minutoli, Daniela Metro, and et al. 2020. "β-Caryophyllene Inhibits Cell Proliferation through a Direct Modulation of CB2 Receptors in Glioblastoma Cells" Cancers 12, no. 4: 1038. https://doi.org/10.3390/cancers12041038
APA StyleIrrera, N., D’Ascola, A., Pallio, G., Bitto, A., Mannino, F., Arcoraci, V., Rottura, M., Ieni, A., Minutoli, L., Metro, D., Vaccaro, M., Altavilla, D., & Squadrito, F. (2020). β-Caryophyllene Inhibits Cell Proliferation through a Direct Modulation of CB2 Receptors in Glioblastoma Cells. Cancers, 12(4), 1038. https://doi.org/10.3390/cancers12041038