Can Hemp Help? Low-THC Cannabis and Non-THC Cannabinoids for the Treatment of Cancer

 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
Effects of Cannabis on Human Health
2. Cannabinoids
3. Role of Cannabinoids in Cancer
3.1. Cannabinoid Receptors (CBRs)
3.2. Non-THC Cannabinoids Are Ligands for Calcium Selective Ion Channels and Orphaned/De-Orphaned G-Protein-Coupled Receptors
3.2.1. Transient Receptor Potential Channels of the Vanilloid Subtype–TRPV1/2
3.2.2. De-Orphaned G-Protein-Coupled Receptor 55
3.2.3. Orphan G-Protein-Coupled Receptors GPR3, GPR6 and GPR12
3.3. Crosstalk between GPCRs and Non-GPCR Signalling Pathways
3.4. Cannabinoid Receptor-Dependent Cancer Effects
3.4.1. Cannabinoids Induce Apoptosis via Sustained Ceramide Accumulation
3.4.2. Downstream Targets of Ceramide
3.4.3. Cannabinoid Induced Anti-Cancer Effects
4. Clinical Evidence: Cannabis for Use by Patients with Cancer
5. Role of Non-THC Cannabinoids in Cancer-Associated Inflammation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zuardi, A.W. History of cannabis as a medicine: A review. Braz. J. Psychiatry 2006, 28, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Sanchez, C.; Guzman, M. Towards the use of cannabinoids as antitumour agents. Nat. Rev. Cancer 2012, 12, 436–444. [Google Scholar] [CrossRef]
- Cui, W.; Howells, E.; Devitt, B. Medicinal cannabis: The knowledge, attitudes, and practices of Australian patients with cancer. In Proceedings of the Medical Oncology Group of Australia Annual Scientific Meeting, Canberra, Australia, 14–16 August 2019; p. 59. [Google Scholar]
- Pacula, R.L.; Smart, R. Medical Marijuana and Marijuana Legalization. Annu. Rev. Clin. Psychol. 2017, 13, 397–419. [Google Scholar] [CrossRef]
- Russo, E.B. Taming THC: Potential cannabis synergy and phytocannabinoid-terpenoid entourage effects. Br. J. Pharmacol. 2011, 163, 1344–1364. [Google Scholar] [CrossRef]
- Carlini, E.A. The good and the bad effects of (-) trans-delta-9-tetrahydrocannabinol (Delta 9-THC) on humans. Toxicon 2004, 44, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Condie, R.; Herring, A.; Koh, W.S.; Lee, M.; Kaminski, N.E. Cannabinoid inhibition of adenylate cyclase-mediated signal transduction and interleukin 2 (IL-2) expression in the murine T-cell line, EL4.IL-2. J. Biol. Chem. 1996, 271, 13175–13183. [Google Scholar] [CrossRef] [PubMed]
- McKallip, R.J.; Nagarkatti, M.; Nagarkatti, P.S. Δ-9-tetrahydrocannabinol enhances breast cancer growth and metastasis by suppression of the antitumor immune response. J. Immunol. 2005, 174, 3281–3289. [Google Scholar] [CrossRef] [PubMed]
- Taha, T.; Meiri, D.; Talhamy, S.; Wollner, M.; Peer, A.; Bar-Sela, G. Cannabis Impacts Tumor Response Rate to Nivolumab in Patients with Advanced Malignancies. Oncologist 2019, 24, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Lile, J.A.; Kelly, T.H.; Charnigo, R.J.; Stinchcomb, A.L.; Hays, L.R. Pharmacokinetic and pharmacodynamic profile of supratherapeutic oral doses of Delta(9) -THC in cannabis users. J. Clin. Pharmacol. 2013, 53, 680–690. [Google Scholar] [CrossRef]
- Mechoulam, R.; Parker, L.A.; Gallily, R. Cannabidiol: An overview of some pharmacological aspects. J. Clin. Pharmacol. 2002, 42, 11S–19S. [Google Scholar] [CrossRef]
- Scott, K.A.; Dalgleish, A.G.; Liu, W.M. Anticancer effects of phytocannabinoids used with chemotherapy in leukaemia cells can be improved by altering the sequence of their administration. Int. J. Oncol. 2017, 51, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Ligresti, A.; Moriello, A.S.; Starowicz, K.; Matias, I.; Pisanti, S.; De Petrocellis, L.; Laezza, C.; Portella, G.; Bifulco, M.; Di Marzo, V. Antitumor activity of plant cannabinoids with emphasis on the effect of cannabidiol on human breast carcinoma. J. Pharmacol. Exp. Ther. 2006, 318, 1375–1387. [Google Scholar] [CrossRef] [PubMed]
- Sledzinski, P.; Zeyland, J.; Slomski, R.; Nowak, A. The current state and future perspectives of cannabinoids in cancer biology. Cancer Med. 2018, 7, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Parliament of NSW. Hemp Industry Act 2008 No 58; Parliamentary Counsel’s Office: Sydney, Australia, 2017.
- Parliament of Western Australia. Industrial Hemp Act 2004; Parliament of Western Australia: Perth, Australia, 2018.
- Patel, B.; Wene, D.; Fan, Z.T. Qualitative and quantitative measurement of cannabinoids in cannabis using modified HPLC/DAD method. J. Pharm. Biomed. Anal. 2017, 146, 15–23. [Google Scholar] [CrossRef]
- National Academies of Sciences, Engineering, and Medicine. The Health Effects of Cannabis and Cannabinoids: The Current State of Evidence and Recommendations for Research; The National Academies Press: Washington, DC, USA, 2017; p. 486. [Google Scholar] [CrossRef]
- Shannon, S.; Lewis, N.; Lee, H.; Hughes, S. Cannabidiol in Anxiety and Sleep: A Large Case Series. Perm. J. 2019, 23, 18-041. [Google Scholar] [CrossRef]
- Thiele, E.; Marsh, E.; Mazurkiewicz-Beldzinska, M.; Halford, J.J.; Gunning, B.; Devinsky, O.; Checketts, D.; Roberts, C. Cannabidiol in patients with Lennox-Gastaut syndrome: Interim analysis of an open-label extension study. Epilepsia 2019, 60, 419–428. [Google Scholar] [CrossRef]
- Dall’Stella, P.B.; Docema, M.F.L.; Maldaun, M.V.C.; Feher, O.; Lancellotti, C.L.P. Case Report: Clinical Outcome and Image Response of Two Patients With Secondary High-Grade Glioma Treated With Chemoradiation, PCV, and Cannabidiol. Front. Oncol. 2018, 8, 643. [Google Scholar] [CrossRef]
- Imtiaz, S.; Shield, K.D.; Roerecke, M.; Cheng, J.; Popova, S.; Kurdyak, P.; Fischer, B.; Rehm, J. The burden of disease attributable to cannabis use in Canada in 2012. Addiction 2016, 111, 653–662. [Google Scholar] [CrossRef]
- Mandelbaum, D.E.; de la Monte, S.M. Adverse Structural and Functional Effects of Marijuana on the Brain: Evidence Reviewed. Pediatric Neurol. 2017, 66, 12–20. [Google Scholar] [CrossRef]
- Gruber, S.A.; Sagar, K.A.; Dahlgren, M.K.; Gonenc, A.; Smith, R.T.; Lambros, A.M.; Cabrera, K.B.; Lukas, S.E. The Grass Might Be Greener: Medical Marijuana Patients Exhibit Altered Brain Activity and Improved Executive Function after 3 Months of Treatment. Front. Pharmacol. 2017, 8, 983. [Google Scholar] [CrossRef]
- Gomez Del Pulgar, T.; De Ceballos, M.L.; Guzman, M.; Velasco, G. Cannabinoids protect astrocytes from ceramide-induced apoptosis through the phosphatidylinositol 3-kinase/protein kinase B pathway. J. Biol. Chem. 2002, 277, 36527–36533. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, M.; Cocchi, V.; Cavazza, L.; Bilel, S.; Hrelia, P.; Marti, M. Genotoxic Properties of Synthetic Cannabinoids on TK6 Human Cells by Flow Cytometry. Int. J. Mol. Sci. 2020, 21, 1150. [Google Scholar] [CrossRef] [PubMed]
- Koller, V.J.; Ferk, F.; Al-Serori, H.; Mišík, M.; Nersesyan, A.; Auwärter, V.; Grummt, T.; Knasmüller, S. Genotoxic properties of representatives of alkylindazoles and aminoalkyl-indoles which are consumed as synthetic cannabinoids. Food Chem. Toxicol. 2015, 80, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Barbosa-Leiker, C.; Burduli, E.; Smith, C.L.; Brooks, O.; Orr, M.; Gartstein, M. Daily Cannabis Use During Pregnancy and Postpartum in a State With Legalized Recreational Cannabis. J. Addict. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Howard, D.S.; Dhanraj, D.N.; Devaiah, C.G.; Lambers, D.S. Cannabis Use Based on Urine Drug Screens in Pregnancy and Its Association With Infant Birth Weight. J. Addict. Med. 2019, 13, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Roncero, C.; Valriberas-Herrero, I.; Mezzatesta-Gava, M.; Villegas, J.L.; Aguilar, L.; Grau-Lopez, L. Cannabis use during pregnancy and its relationship with fetal developmental outcomes and psychiatric disorders. A systematic review. Reprod. Health 2020, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Karila, L.; Roux, P.; Rolland, B.; Benyamina, A.; Reynaud, M.; Aubin, H.J.; Lancon, C. Acute and long-term effects of cannabis use: A review. Curr. Pharm Des. 2014, 20, 4112–4118. [Google Scholar] [CrossRef]
- Zuardi, A.W.; Crippa, J.A.; Hallak, J.E.; Bhattacharyya, S.; Atakan, Z.; Martin-Santos, R.; McGuire, P.K.; Guimaraes, F.S. A critical review of the antipsychotic effects of cannabidiol: 30 years of a translational investigation. Curr. Pharm Des. 2012, 18, 5131–5140. [Google Scholar] [CrossRef]
- Kis, B.; Ifrim, F.C.; Buda, V.; Avram, S.; Pavel, I.Z.; Antal, D.; Paunescu, V.; Dehelean, C.A.; Ardelean, F.; Diaconeasa, Z.; et al. Cannabidiol-from Plant to Human Body: A Promising Bioactive Molecule with Multi-Target Effects in Cancer. Int. J. Mol. Sci. 2019, 20, 5905. [Google Scholar] [CrossRef]
- Lucas, C.J.; Galettis, P.; Schneider, J. The pharmacokinetics and the pharmacodynamics of cannabinoids. Br. J. Clin. Pharmacol. 2018, 84, 2477–2482. [Google Scholar] [CrossRef]
- Ewing, L.E.; Skinner, C.M.; Quick, C.M.; Kennon-McGill, S.; McGill, M.R.; Walker, L.A.; ElSohly, M.A.; Gurley, B.J.; Koturbash, I. Hepatotoxicity of a Cannabidiol-Rich Cannabis Extract in the Mouse Model. Molecules 2019, 24, 1694. [Google Scholar] [CrossRef] [PubMed]
- Pisanti, S.; Malfitano, A.M.; Ciaglia, E.; Lamberti, A.; Ranieri, R.; Cuomo, G.; Abate, M.; Faggiana, G.; Proto, M.C.; Fiore, D.; et al. Cannabidiol: State of the art and new challenges for therapeutic applications. Pharmacol. Ther. 2017, 175, 133–150. [Google Scholar] [CrossRef]
- McPartland, J.M.; Russo, E.B. Cannabis and Cannabis Extracts Greater Than the Sum of Their Parts? J. Cannabis Ther. 2001, 1, 103–132. [Google Scholar] [CrossRef]
- Turner, C.E.; Elsohly, M.A.; Boeren, E.G. Constituents of Cannabis sativa L. XVII. A review of the natural constituents. J. Nat. Prod. 1980, 43, 169–234. [Google Scholar] [CrossRef] [PubMed]
- Chakravarti, B.; Ravi, J.; Ganju, R.K. Cannabinoids as therapeutic agents in cancer: Current status and future implications. Oncotarget 2014, 5, 5852–5872. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M. Metabolism of the Endocannabinoid Anandamide: Open Questions after 25 Years. Front. Mol. Neurosci. 2017, 10, 166. [Google Scholar] [CrossRef]
- Maccarrone, M.; Finazzi-Agro, A. The endocannabinoid system, anandamide and the regulation of mammalian cell apoptosis. Cell Death Differ. 2003, 10, 946–955. [Google Scholar] [CrossRef]
- Placzek, E.A.; Okamoto, Y.; Ueda, N.; Barker, E.L. Membrane microdomains and metabolic pathways that define anandamide and 2-arachidonyl glycerol biosynthesis and breakdown. Neuropharmacology 2008, 55, 1095–1104. [Google Scholar] [CrossRef]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [Google Scholar] [CrossRef]
- Galve-Roperh, I.; Chiurchiu, V.; Diaz-Alonso, J.; Bari, M.; Guzman, M.; Maccarrone, M. Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog. Lipid Res. 2013, 52, 633–650. [Google Scholar] [CrossRef]
- Sarfaraz, S.; Afaq, F.; Adhami, V.M.; Malik, A.; Mukhtar, H. Cannabinoid receptor agonist-induced apoptosis of human prostate cancer cells LNCaP proceeds through sustained activation of ERK1/2 leading to G1 cell cycle arrest. J. Biol. Chem. 2006, 281, 39480–39491. [Google Scholar] [CrossRef] [PubMed]
- Lukhele, S.T.; Motadi, L.R. Cannabidiol rather than Cannabis sativa extracts inhibit cell growth and induce apoptosis in cervical cancer cells. BMC Complement. Altern. Med. 2016, 16, 335. [Google Scholar] [CrossRef] [PubMed]
- Marcu, J.P.; Christian, R.T.; Lau, D.; Zielinski, A.J.; Horowitz, M.P.; Lee, J.; Pakdel, A.; Allison, J.; Limbad, C.; Moore, D.H.; et al. Cannabidiol enhances the inhibitory effects of Δ9-tetrahydrocannabinol on human glioblastoma cell proliferation and survival. Mol. Cancer Ther. 2010, 9, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Lorenzon, T.; Bari, M.; Melino, G.; Finazzi-Agro, A. Anandamide induces apoptosis in human cells via vanilloid receptors. Evidence for a protective role of cannabinoid receptors. J. Biol. Chem. 2000, 275, 31938–31945. [Google Scholar] [CrossRef] [PubMed]
- Cianchi, F.; Papucci, L.; Schiavone, N.; Lulli, M.; Magnelli, L.; Vinci, M.C.; Messerini, L.; Manera, C.; Ronconi, E.; Romagnani, P.; et al. Cannabinoid receptor activation induces apoptosis through tumor necrosis factor α-mediated ceramide de novo synthesis in colon cancer cells. Clin. Cancer Res. 2008, 14, 7691–7700. [Google Scholar] [CrossRef]
- McKallip, R.J.; Lombard, C.; Fisher, M.; Martin, B.R.; Ryu, S.; Grant, S.; Nagarkatti, P.S.; Nagarkatti, M. Targeting CB2 cannabinoid receptors as a novel therapy to treat malignant lymphoblastic disease. Blood 2002, 100, 627–634. [Google Scholar] [CrossRef] [PubMed]
- McKallip, R.J.; Jia, W.; Schlomer, J.; Warren, J.W.; Nagarkatti, P.S.; Nagarkatti, M. Cannabidiol-induced apoptosis in human leukemia cells: A novel role of cannabidiol in the regulation of p22phox and Nox4 expression. Mol. Pharmacol. 2006, 70, 897–908. [Google Scholar] [CrossRef]
- Ramer, R.; Heinemann, K.; Merkord, J.; Rohde, H.; Salamon, A.; Linnebacher, M.; Hinz, B. COX-2 and PPAR-gamma confer cannabidiol-induced apoptosis of human lung cancer cells. Mol. Cancer Ther. 2013, 12, 69–82. [Google Scholar] [CrossRef]
- Melck, D.; De Petrocellis, L.; Orlando, P.; Bisogno, T.; Laezza, C.; Bifulco, M.; Di Marzo, V. Suppression of nerve growth factor Trk receptors and prolactin receptors by endocannabinoids leads to inhibition of human breast and prostate cancer cell proliferation. Endocrinology 2000, 141, 118–126. [Google Scholar] [CrossRef]
- Sultan, A.S.; Marie, M.A.; Sheweita, S.A. Novel mechanism of cannabidiol-induced apoptosis in breast cancer cell lines. Breast 2018, 41, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Qamri, Z.; Preet, A.; Nasser, M.W.; Bass, C.E.; Leone, G.; Barsky, S.H.; Ganju, R.K. Synthetic cannabinoid receptor agonists inhibit tumor growth and metastasis of breast cancer. Mol. Cancer Ther. 2009, 8, 3117. [Google Scholar] [CrossRef]
- Borrelli, F.; Pagano, E.; Romano, B.; Panzera, S.; Maiello, F.; Coppola, D.; De Petrocellis, L.; Buono, L.; Orlando, P.; Izzo, A.A. Colon carcinogenesis is inhibited by the TRPM8 antagonist cannabigerol, a Cannabis-derived non-psychotropic cannabinoid. Carcinogenesis 2014, 35, 2787–2797. [Google Scholar] [CrossRef] [PubMed]
- Singer, E.; Judkins, J.; Salomonis, N.; Matlaf, L.; Soteropoulos, P.; McAllister, S.; Soroceanu, L. Reactive oxygen species-mediated therapeutic response and resistance in glioblastoma. Cell Death Dis. 2015, 6, e1601. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Santoni, M.; Santoni, G. Triggering of the TRPV2 channel by cannabidiol sensitizes glioblastoma cells to cytotoxic chemotherapeutic agents. Carcinogenesis 2013, 34, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Hernan Perez de la Ossa, D.; Lorente, M.; Gil-Alegre, M.E.; Torres, S.; Garcia-Taboada, E.; Aberturas Mdel, R.; Molpeceres, J.; Velasco, G.; Torres-Suarez, A.I. Local delivery of cannabinoid-loaded microparticles inhibits tumor growth in a murine xenograft model of glioblastoma multiforme. PLoS ONE 2013, 8, e54795. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.B.; Offidani, M.; Alesiani, F.; Discepoli, G.; Liberati, S.; Olivieri, A.; Santoni, M.; Santoni, G.; Leoni, P.; Nabissi, M. The effects of cannabidiol and its synergism with bortezomib in multiple myeloma cell lines. A role for transient receptor potential vanilloid type-2. Int. J. Cancer 2014, 134, 2534–2546. [Google Scholar] [CrossRef]
- Nabissi, M.; Morelli, M.B.; Offidani, M.; Amantini, C.; Gentili, S.; Soriani, A.; Cardinali, C.; Leoni, P.; Santoni, G. Cannabinoids synergize with carfilzomib, reducing multiple myeloma cells viability and migration. Oncotarget 2016, 7, 77543–77557. [Google Scholar] [CrossRef]
- Good, P.D.; Greer, R.M.; Huggett, G.E.; Hardy, J.R. An Open-Label Pilot Study Testing the Feasibility of Assessing Total Symptom Burden in Trials of Cannabinoid Medications in Palliative Care. J. Palliat. Med. 2019. [Google Scholar] [CrossRef]
- Brown, A.J. Novel cannabinoid receptors. Br. J. Pharmacol. 2007, 152, 567–575. [Google Scholar] [CrossRef]
- Berdyshev, E.V. Cannabinoid receptors and the regulation of immune response. Chem. Phys. Lipids 2000, 108, 169–190. [Google Scholar] [CrossRef]
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805. [Google Scholar] [CrossRef] [PubMed]
- Bénard, G.; Massa, F.; Puente, N.; Lourenço, J.; Bellocchio, L.; Soria-Gómez, E.; Matias, I.; Delamarre, A.; Metna-Laurent, M.; Cannich, A.; et al. Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012, 15, 558. Available online: https://www.nature.com/articles/nn.3053#supplementary-information (accessed on 12 February 2019). [CrossRef]
- Nagarkatti, P.; Pandey, R.; Rieder, S.A.; Hegde, V.L.; Nagarkatti, M. Cannabinoids as novel anti-inflammatory drugs. Future Med. Chem. 2009, 1, 1333–1349. [Google Scholar] [CrossRef] [PubMed]
- Roche, M.; Finn, D.P. Brain CB(2) Receptors: Implications for Neuropsychiatric Disorders. Pharmaceuticals 2010, 3, 2517–2553. [Google Scholar] [CrossRef]
- Galve-Roperh, I.; Sanchez, C.; Cortes, M.L.; Gomez del Pulgar, T.; Izquierdo, M.; Guzman, M. Anti-tumoral action of cannabinoids: Involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat. Med. 2000, 6, 313–319. [Google Scholar] [CrossRef]
- Sanchez, C.; de Ceballos, M.L.; Gomez del Pulgar, T.; Rueda, D.; Corbacho, C.; Velasco, G.; Galve-Roperh, I.; Huffman, J.W.; Ramon y Cajal, S.; Guzman, M. Inhibition of glioma growth in vivo by selective activation of the CB(2) cannabinoid receptor. Cancer Res. 2001, 61, 5784–5789. [Google Scholar]
- Rosenthaler, S.; Pohn, B.; Kolmanz, C.; Huu, C.N.; Krewenka, C.; Huber, A.; Kranner, B.; Rausch, W.D.; Moldzio, R. Differences in receptor binding affinity of several phytocannabinoids do not explain their effects on neural cell cultures. Neurotoxicol. Teratol. 2014, 46, 49–56. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB(1) and CB(2). Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef]
- Muller, C.; Morales, P.; Reggio, P.H. Cannabinoid Ligands Targeting TRP Channels. Front. Mol. Neurosci. 2018, 11, 487. [Google Scholar] [CrossRef]
- Im, J.H.; Kang, K.W.; Kim, S.Y.; Kim, Y.G.; An, Y.J.; Park, S.; Jeong, B.H.; Choi, S.Y.; Lee, J.S.; Kang, K.W. GPR119 agonist enhances gefitinib responsiveness through lactate-mediated inhibition of autophagy. J. Exp. Clin. Cancer Res. 2018, 37, 295. [Google Scholar] [CrossRef]
- Amantini, C.; Mosca, M.; Nabissi, M.; Lucciarini, R.; Caprodossi, S.; Arcella, A.; Giangaspero, F.; Santoni, G. Capsaicin-induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J. Neurochem. 2007, 102, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Cho, Y.Y.; Zheng, D.; Zhu, F.; Ericson, M.E.; Ma, W.Y.; Yao, K.; Dong, Z. Transient receptor potential type vanilloid 1 suppresses skin carcinogenesis. Cancer Res. 2009, 69, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Nabissi, M.; Morelli, M.B.; Amantini, C.; Farfariello, V.; Ricci-Vitiani, L.; Caprodossi, S.; Arcella, A.; Santoni, M.; Giangaspero, F.; De Maria, R.; et al. TRPV2 channel negatively controls glioma cell proliferation and resistance to Fas-induced apoptosis in ERK-dependent manner. Carcinogenesis 2010, 31, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.Y.; Lu, H.C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef]
- Liu, B.; Song, S.; Ruz-Maldonado, I.; Pingitore, A.; Huang, G.C.; Baker, D.; Jones, P.M.; Persaud, S.J. GPR55-dependent stimulation of insulin secretion from isolated mouse and human islets of Langerhans. Diabetes Obes. Metab. 2016, 18, 1263–1273. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjogren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Balenga, N.A.; Ford, L.A.; Ross, R.A.; Waldhoer, M.; Irving, A.J. The GPR55 ligand L-alpha-lysophosphatidylinositol promotes RhoA-dependent Ca2+ signaling and NFAT activation. FASEB J. 2009, 23, 183–193. [Google Scholar] [CrossRef]
- Falasca, M.; Corda, D. Elevated levels and mitogenic activity of lysophosphatidylinositol in k-ras-transformed epithelial cells. Eur. J. Biochem. 1994, 221, 383–389. [Google Scholar] [CrossRef]
- Falasca, M.; Iurisci, C.; Carvelli, A.; Sacchetti, A.; Corda, D. Release of the mitogen lysophosphatidylinositol from H-Ras-transformed fibroblasts; a possible mechanism of autocrine control of cell proliferation. Oncogene 1998, 16, 2357–2365. [Google Scholar] [CrossRef]
- Falasca, M.; Ferro, R. Role of the lysophosphatidylinositol/GPR55 axis in cancer. Adv. Biol. Regul. 2016, 60, 88–93. [Google Scholar] [CrossRef]
- Pineiro, R.; Maffucci, T.; Falasca, M. The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene 2011, 30, 142–152. [Google Scholar] [CrossRef]
- Hofmann, N.A.; Yang, J.; Trauger, S.A.; Nakayama, H.; Huang, L.; Strunk, D.; Moses, M.A.; Klagsbrun, M.; Bischoff, J.; Graier, W.F. The GPR 55 agonist, L-alpha-lysophosphatidylinositol, mediates ovarian carcinoma cell-induced angiogenesis. Br. J. Pharmacol. 2015, 172, 4107–4118. [Google Scholar] [CrossRef] [PubMed]
- Ford, L.A.; Roelofs, A.J.; Anavi-Goffer, S.; Mowat, L.; Simpson, D.G.; Irving, A.J.; Rogers, M.J.; Rajnicek, A.M.; Ross, R.A. A role for L-alpha-lysophosphatidylinositol and GPR55 in the modulation of migration, orientation and polarization of human breast cancer cells. Br. J. Pharmacol. 2010, 160, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Kargl, J.; Andersen, L.; Hasenohrl, C.; Feuersinger, D.; Stancic, A.; Fauland, A.; Magnes, C.; El-Heliebi, A.; Lax, S.; Uranitsch, S.; et al. GPR55 promotes migration and adhesion of colon cancer cells indicating a role in metastasis. Br. J. Pharmacol. 2016, 173, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Andradas, C.; Blasco-Benito, S.; Castillo-Lluva, S.; Dillenburg-Pilla, P.; Diez-Alarcia, R.; Juanes-Garcia, A.; Garcia-Taboada, E.; Hernando-Llorente, R.; Soriano, J.; Hamann, S.; et al. Activation of the orphan receptor GPR55 by lysophosphatidylinositol promotes metastasis in triple-negative breast cancer. Oncotarget 2016, 7, 47565–47575. [Google Scholar] [CrossRef]
- Yuan, Z.Y.; Dai, T.; Wang, S.S.; Peng, R.J.; Li, X.H.; Qin, T.; Song, L.B.; Wang, X. Overexpression of ETV4 protein in triple-negative breast cancer is associated with a higher risk of distant metastasis. Onco Targets Ther. 2014, 7, 1733–1742. [Google Scholar] [CrossRef]
- Ferro, R.; Adamska, A.; Lattanzio, R.; Mavrommati, I.; Edling, C.E.; Arifin, S.A.; Fyffe, C.A.; Sala, G.; Sacchetto, L.; Chiorino, G.; et al. GPR55 signalling promotes proliferation of pancreatic cancer cells and tumour growth in mice, and its inhibition increases effects of gemcitabine. Oncogene 2018. [Google Scholar] [CrossRef]
- Zheng, C.; Jiao, X.; Jiang, Y.; Sun, S. ERK1/2 activity contributes to gemcitabine resistance in pancreatic cancer cells. J. Int. Med. Res. 2013, 41, 300–306. [Google Scholar] [CrossRef]
- Laun, A.S.; Song, Z.H. GPR3 and GPR6, novel molecular targets for cannabidiol. Biochem. Biophys. Res. Commun. 2017, 490, 17–21. [Google Scholar] [CrossRef]
- Laun, A.S.; Shrader, S.H.; Brown, K.J.; Song, Z.H. GPR3, GPR6, and GPR12 as novel molecular targets: Their biological functions and interaction with cannabidiol. Acta Pharmacol. Sin. 2019, 40, 300–308. [Google Scholar] [CrossRef]
- Apostolou, P.; Toloudi, M.; Papasotiriou, I. Identification of genes involved in breast cancer and breast cancer stem cells. Breast Cancer 2015, 7, 183–191. [Google Scholar] [CrossRef]
- Breivogel, C.S.; McPartland, J.M.; Parekh, B. Investigation of non-CB1, non-CB2 WIN55212-2-sensitive G-protein-coupled receptors in the brains of mammals, birds, and amphibians. J. Recept. Signal Transduct. Res. 2018, 38, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Massi, P.; Vaccani, A.; Ceruti, S.; Colombo, A.; Abbracchio, M.P.; Parolaro, D. Antitumor effects of cannabidiol, a nonpsychoactive cannabinoid, on human glioma cell lines. J. Pharmacol. Exp. Ther. 2004, 308, 838–845. [Google Scholar] [CrossRef]
- Punzo, F.; Manzo, I.; Tortora, C.; Pota, E.; Angelo, V.; Bellini, G.; Di Paola, A.; Verace, F.; Casale, F.; Rossi, F. Effects of CB2 and TRPV1 receptors’ stimulation in pediatric acute T-lymphoblastic leukemia. Oncotarget 2018, 9, 21244–21258. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Casanova, M.L.; Blazquez, C.; Martinez-Palacio, J.; Villanueva, C.; Fernandez-Acenero, M.J.; Huffman, J.W.; Jorcano, J.L.; Guzman, M. Inhibition of skin tumor growth and angiogenesis in vivo by activation of cannabinoid receptors. J. Clin. Investig. 2003, 111, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, C.; Gonzalez-Feria, L.; Alvarez, L.; Haro, A.; Casanova, M.L.; Guzman, M. Cannabinoids inhibit the vascular endothelial growth factor pathway in gliomas. Cancer Res. 2004, 64, 5617–5623. [Google Scholar] [CrossRef]
- Elbaz, M.; Ahirwar, D.; Ravi, J.; Nasser, M.W.; Ganju, R.K. Novel role of cannabinoid receptor 2 in inhibiting EGF/EGFR and IGF-I/IGF-IR pathways in breast cancer. Oncotarget 2017, 8, 29668–29678. [Google Scholar] [CrossRef]
- Dun, M.D.; Chalkley, R.J.; Faulkner, S.; Keene, S.; Avery-Kiejda, K.A.; Scott, R.J.; Falkenby, L.G.; Cairns, M.J.; Larsen, M.R.; Bradshaw, R.A.; et al. Proteotranscriptomic Profiling of 231-BR Breast Cancer Cells: Identification of Potential Biomarkers and Therapeutic Targets for Brain Metastasis. Mol. Cell Proteom. 2015, 14, 2316–2330. [Google Scholar] [CrossRef]
- Degryse, S.; de Bock, C.E.; Demeyer, S.; Govaerts, I.; Bornschein, S.; Verbeke, D.; Jacobs, K.; Binos, S.; Skerrett-Byrne, D.A.; Murray, H.C.; et al. Mutant JAK3 phosphoproteomic profiling predicts synergism between JAK3 inhibitors and MEK/BCL2 inhibitors for the treatment of T-cell acute lymphoblastic leukemia. Leukemia 2018, 32, 788–800. [Google Scholar] [CrossRef]
- R Mokoena, D.; P George, B.; Abrahamse, H. Enhancing Breast Cancer Treatment Using a Combination of Cannabidiol and Gold Nanoparticles for Photodynamic Therapy. Int. J. Mol. Sci. 2019, 20, 4771. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Ciechomska, I.; Kaminska, B. Cannabinoid signaling in glioma cells. Adv. Exp. Med. Biol. 2013, 986, 209–220. [Google Scholar] [CrossRef]
- De Jesus, M.L.; Hostalot, C.; Garibi, J.M.; Salles, J.; Meana, J.J.; Callado, L.F. Opposite changes in cannabinoid CB1 and CB2 receptor expression in human gliomas. Neurochem. Int. 2010, 56, 829–833. [Google Scholar] [CrossRef]
- Schley, M.; Stander, S.; Kerner, J.; Vajkoczy, P.; Schupfer, G.; Dusch, M.; Schmelz, M.; Konrad, C. Predominant CB2 receptor expression in endothelial cells of glioblastoma in humans. Brain Res. Bull. 2009, 79, 333–337. [Google Scholar] [CrossRef]
- Wu, X.; Han, L.; Zhang, X.; Li, L.; Jiang, C.; Qiu, Y.; Huang, R.; Xie, B.; Lin, Z.; Ren, J.; et al. Alteration of endocannabinoid system in human gliomas. J. Neurochem. 2012, 120, 842–849. [Google Scholar] [CrossRef]
- Ciaglia, E.; Torelli, G.; Pisanti, S.; Picardi, P.; D’Alessandro, A.; Laezza, C.; Malfitano, A.M.; Fiore, D.; Pagano Zottola, A.C.; Proto, M.C.; et al. Cannabinoid receptor CB1 regulates STAT3 activity and its expression dictates the responsiveness to SR141716 treatment in human glioma patients’ cells. Oncotarget 2015, 6, 15464–15481. [Google Scholar] [CrossRef]
- Dumitru, C.A.; Sandalcioglu, I.E.; Karsak, M. Cannabinoids in Glioblastoma Therapy: New Applications for Old Drugs. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef]
- Duchatel, R.J.; Jackson, E.R.; Alvaro, F.; Nixon, B.; Hondermarck, H.; Dun, M.D. Signal Transduction in Diffuse Intrinsic Pontine Glioma. Proteomics 2019, e1800479. [Google Scholar] [CrossRef]
- McAllister, S.D.; Soroceanu, L.; Desprez, P.-Y. The Antitumor Activity of Plant-Derived Non-Psychoactive Cannabinoids. J. Neuroimmune Pharmacol. 2015, 10, 255–267. [Google Scholar] [CrossRef]
- Herrera, B.; Carracedo, A.; Diez-Zaera, M.; Guzman, M.; Velasco, G. p38 MAPK is involved in CB2 receptor-induced apoptosis of human leukaemia cells. FEBS Lett. 2005, 579, 5084–5088. [Google Scholar] [CrossRef]
- Preet, A.; Qamri, Z.; Nasser, M.W.; Prasad, A.; Shilo, K.; Zou, X.; Groopman, J.E.; Ganju, R.K. Cannabinoid receptors, CB1 and CB2, as novel targets for inhibition of non-small cell lung cancer growth and metastasis. Cancer Prev. Res. 2011, 4, 65–75. [Google Scholar] [CrossRef]
- Caffarel, M.M.; Sarrio, D.; Palacios, J.; Guzman, M.; Sanchez, C. Delta9-tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation. Cancer Res. 2006, 66, 6615–6621. [Google Scholar] [CrossRef]
- Salazar, M.; Carracedo, A.; Salanueva, I.J.; Hernandez-Tiedra, S.; Lorente, M.; Egia, A.; Vazquez, P.; Blazquez, C.; Torres, S.; Garcia, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Investig. 2009, 119, 1359–1372. [Google Scholar] [CrossRef]
- Hart, S.; Fischer, O.M.; Ullrich, A. Cannabinoids induce cancer cell proliferation via tumor necrosis factor alpha-converting enzyme (TACE/ADAM17)-mediated transactivation of the epidermal growth factor receptor. Cancer Res. 2004, 64, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- Mimeault, M.; Pommery, N.; Wattez, N.; Bailly, C.; Henichart, J.P. Anti-proliferative and apoptotic effects of anandamide in human prostatic cancer cell lines: Implication of epidermal growth factor receptor down-regulation and ceramide production. Prostate 2003, 56, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Gironella, M.; Lorente, M.; Garcia, S.; Guzman, M.; Velasco, G.; Iovanna, J.L. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res. 2006, 66, 6748–6755. [Google Scholar] [CrossRef] [PubMed]
- Herrera, B.; Carracedo, A.; Diez-Zaera, M.; Gomez del Pulgar, T.; Guzman, M.; Velasco, G. The CB2 cannabinoid receptor signals apoptosis via ceramide-dependent activation of the mitochondrial intrinsic pathway. Exp. Cell Res. 2006, 312, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.; Carracedo, A.; Salanueva, I.J.; Hernandez-Tiedra, S.; Egia, A.; Lorente, M.; Vazquez, P.; Torres, S.; Iovanna, J.L.; Guzman, M.; et al. TRB3 links ER stress to autophagy in cannabinoid anti-tumoral action. Autophagy 2009, 5, 1048–1049. [Google Scholar] [CrossRef]
- Massi, P.; Solinas, M.; Cinquina, V.; Parolaro, D. Cannabidiol as potential anticancer drug. Br. J. Clin. Pharmacol. 2013, 75, 303–312. [Google Scholar] [CrossRef]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the cross-talk between apoptosis and autophagy. Mol. Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef]
- Chen, Q.; Kang, J.; Fu, C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct. Target. Ther. 2018, 3, 18. [Google Scholar] [CrossRef]
- Jacobsson, S.O.; Wallin, T.; Fowler, C.J. Inhibition of rat C6 glioma cell proliferation by endogenous and synthetic cannabinoids. Relative involvement of cannabinoid and vanilloid receptors. J. Pharmacol. Exp. Ther. 2001, 299, 951–959. [Google Scholar]
- Goncharov, I.; Weiner, L.; Vogel, Z. Δ9-tetrahydrocannabinol increases C6 glioma cell death produced by oxidative stress. Neuroscience 2005, 134, 567–574. [Google Scholar] [CrossRef]
- Massi, P.; Vaccani, A.; Bianchessi, S.; Costa, B.; Macchi, P.; Parolaro, D. The non-psychoactive cannabidiol triggers caspase activation and oxidative stress in human glioma cells. Cell. Mol. Life Sci. 2006, 63, 2057–2066. [Google Scholar] [CrossRef]
- Sillar, J.R.; Germon, Z.P.; DeIuliis, G.N.; Dun, M.D. The Role of Reactive Oxygen Species in Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 6003. [Google Scholar] [CrossRef]
- Chandra, L.C.; Kumar, V.; Torben, W.; Vande Stouwe, C.; Winsauer, P.; Amedee, A.; Molina, P.E.; Mohan, M. Chronic administration of Delta9-tetrahydrocannabinol induces intestinal anti-inflammatory microRNA expression during acute simian immunodeficiency virus infection of rhesus macaques. J. Virol. 2015, 89, 1168–1181. [Google Scholar] [CrossRef]
- Olivas-Aguirre, M.; Torres-López, L.; Valle-Reyes, J.S.; Hernández-Cruz, A.; Pottosin, I.; Dobrovinskaya, O. Cannabidiol directly targets mitochondria and disturbs calcium homeostasis in acute lymphoblastic leukemia. Cell Death Dis. 2019, 10, 779. [Google Scholar] [CrossRef]
- O’Sullivan, S.E.; Sun, Y.; Bennett, A.J.; Randall, M.D.; Kendall, D.A. Time-dependent vascular actions of cannabidiol in the rat aorta. Eur. J. Pharmacol. 2009, 612, 61–68. [Google Scholar] [CrossRef]
- Kenyon, J.; Liu, W.; Dalgleish, A. Report of Objective Clinical Responses of Cancer Patients to Pharmaceutical-grade Synthetic Cannabidiol. Anticancer Res. 2018, 38, 5831–5835. [Google Scholar] [CrossRef]
- Likar, R.; Koestenberger, M.; Stultschnig, M.; Nahler, G. Concomitant Treatment of Malignant Brain Tumours with CBD—A Case Series and Review of the Literature. Anticancer Res. 2019, 39, 5797–5801. [Google Scholar] [CrossRef]
- Pellati, F.; Borgonetti, V.; Brighenti, V.; Biagi, M.; Benvenuti, S.; Corsi, L. Cannabis sativa L. and Nonpsychoactive Cannabinoids: Their Chemistry and Role against Oxidative Stress, Inflammation, and Cancer. Biomed. Res. Int. 2018, 2018, 1691428. [Google Scholar] [CrossRef]
- Herring, A.C.; Koh, W.S.; Kaminski, N.E. Inhibition of the cyclic AMP signaling cascade and nuclear factor binding to CRE and kappaB elements by cannabinol, a minimally CNS-active cannabinoid. Biochem. Pharmacol. 1998, 55, 1013–1023. [Google Scholar] [CrossRef]
- Elbaz, M.; Nasser, M.W.; Ravi, J.; Wani, N.A.; Ahirwar, D.K.; Zhao, H.; Oghumu, S.; Satoskar, A.R.; Shilo, K.; Carson, W.E.; et al. Modulation of the tumor microenvironment and inhibition of EGF/EGFR pathway: Novel anti-tumor mechanisms of Cannabidiol in breast cancer. Mol. Oncol. 2015, 9, 906–919. [Google Scholar] [CrossRef]
- Izzo, A.A.; Camilleri, M. Cannabinoids in intestinal inflammation and cancer. Pharmacol. Res. 2009, 60, 117–125. [Google Scholar] [CrossRef]
- Staudt, D.; Murray, H.C.; McLachlan, T.; Alvaro, F.; Enjeti, A.K.; Verrills, N.M.; Dun, M.D. Targeting Oncogenic Signaling in Mutant FLT3 Acute Myeloid Leukemia: The Path to Least Resistance. Int. J. Mol. Sci. 2018, 19, 3198. [Google Scholar] [CrossRef]
- Likar, R.; Kostenberger, M.; Nahler, G. Cannabidiol in cancer treatment. Schmerz 2020. [Google Scholar] [CrossRef]
- Faulkner, S.; Dun, M.D.; Hondermarck, H. Proteogenomics: Emergence and promise. Cell. Mol. Life Sci. 2015, 72, 953–957. [Google Scholar] [CrossRef]
- Murray, H.C.; Dun, M.D.; Verrills, N.M. Harnessing the power of proteomics for identification of oncogenic, druggable signalling pathways in cancer. Expert Opin. Drug Discov. 2017, 12, 431–447. [Google Scholar] [CrossRef]




{kind=link}
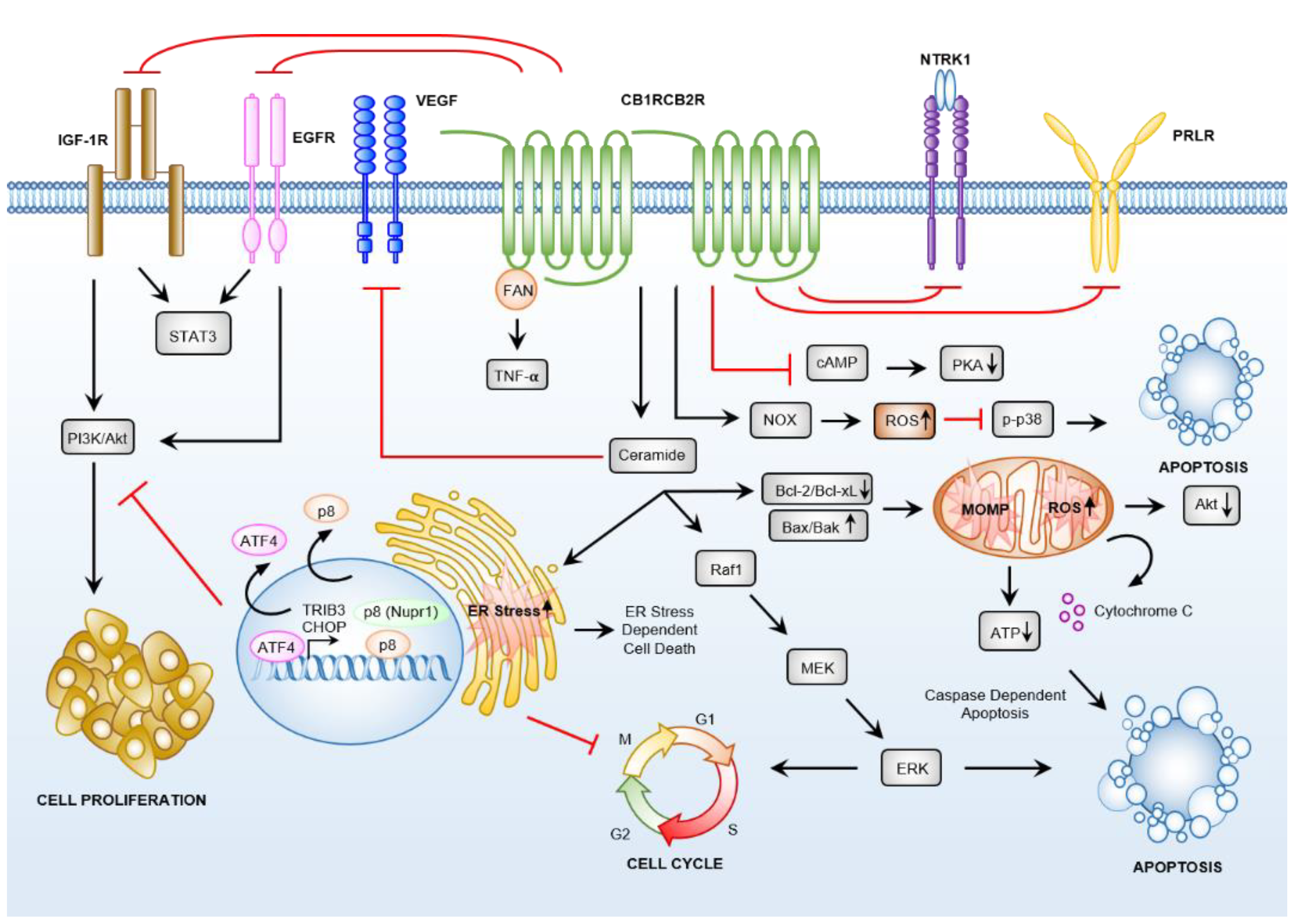{kind=link}
{kind=link}
| Cancer Type | Model/Cell Line | Compound | Effective Dose | Effects | References |
|---|---|---|---|---|---|
| Breast | MCF-7 | Cannabidiol (CBD) | 8.2 µM/2.6 mg/L | Inhibition of cancer cell growth and proliferation | [13] |
| CBD-rich extract (~70% CBD) | 6.0 µM/2.7 mg/L | ||||
| ∆9-tetrahydrocannabinol (∆9-THC) | 14.2 µM/4.5 mg/L | ||||
| AEA | 1.4 µM/0.5 mg/L | [53] | |||
| MDA-MB-231 | CBD | 2.2 µM/0.7 mg/L | Inhibition of cancer cell growth, induction of apoptosis | [54] | |
| WIN-55,212-2, JWH-133 | 10 µM/4.3 mg/L, 10 µM/3.1 mg/L | Inhibition of proliferation | [55] | ||
| Xenograft-MBA-MD-231 cells | CBD | 5 mg/kg (i.p.) | Reduced tumour size and volume | [13] | |
| CBD-rich extract (~70% CBD) | 6.5 mg/kg (i.p.) | ||||
| WIN-55,212-2, JWH-133 | 5 mg/kg (i.p.) | Reduced tumour growth, angiogenesis and metastasis | [55] | ||
| Cervical | SiHa | CBD | 3.2 µg/mL/3.2 mg/L | Inhibition of cancer cell growth, induction of apoptosis | [46] |
| HeLa | 3.2 µg/mL/3.2 mg/L | ||||
| ME-180 | 1.5 µg/mL/1.5 mg/L | ||||
| Colon | HCT8 | CB-13 | >50 nmol/L/0.02 mg/L | Inhibition of cancer cell growth | [49] |
| SW480 | |||||
| HCA7 | |||||
| HCT15 | |||||
| HCT-116 | CBG | ≥3.0 µM/0.9 mg/L | Reduced viability of cancer cells | [56] | |
| Xenograft-HCT-116 | CBG | 3 and 10 mg/kg (i.p.) | Inhibition of tumour growth | ||
| Glioma | U251 | CBD | 0.6 µM/0.2 mg/L | Inhibition of cancer cell growth | [47] |
| ∆9-THC | 3.3 µM/1 mg/L | ||||
| U87 | CBD | 0.6 µM/0.2 mg/L | |||
| ∆9-THC | 3.3 µM/1 mg/L | ||||
| GSC3832 | CBD | 3.5 µM/1.1 mg/L | Inhibition of viability | [57] | |
| GSC387 | CBD | 2.6 µM/0.8 mg/L | |||
| U87MG | CBD | >25 µM/>7.9 mg/L | Reduced viability and induce cancer cell death | [58] | |
| Xenograft-U87 | CBD | 6.7 mg with 75 mg micro-particles | Inhibition of tumour growth | [59] | |
| Multiple Myeloma | U266 | CBD | 32.2 µM/10.1 mg/L | Reduced cancer cell viability, increased cytotoxicity, inhibition of cancer cell migration | [60,61] |
| ∆9-THC | 39.5 μM/12.4 mg/L | ||||
| CBD + ∆9-THC + carfilzomib | (0–50 μM/0–15.7 mg/L) CBD + (12.5–50 μM/3.9–15.7 mg/L) ∆9-THC + (12.5–100 nM/0.009–0.072 mg/L) carfilzomib | ||||
| U266TRPV2 | CBD | 19.8 μM/6.2 mg/L | |||
| RPMI | CBD | 22.4 μM/7.0 mg/L | |||
| ∆9-THC | 30.8 μM/9.7 mg/L | ||||
| CBD + ∆9-THC + carfilzomib | (0–50 μM/0–15.7 mg/L) CBD + (12.5–50 μM/3.9–15.7 mg/L) ∆9-THC + (0.9–7.5 nM/0.0006–0.005 mg/L) carfilzomib | ||||
| RPMITRPV2 | CBD | 13.5 μM/4.2 mg/L |
| Trial No. | Cancer Type/s | Study Type/Phase | Treatments | Dose of Cannabinoids or Cannabis Products | Delivery | Outcome ƛ |
|---|---|---|---|---|---|---|
| NCT01812603; NCT01812616 | Glioblastoma multiforme (GBM) | Interventional (Clinical Trial)/Phase 1 & Phase 2 | Combination of Temozolomide (TMZ) and Sativex (1:1 ∆9-THC:CBD) | Dose-intense TMZ with a maximum of 32.4 mg THC and 30 mg CBD per day | Oral spray | Increased 39% of 1-year survival rate |
| NCT02255292 | Solid tumour | Interventional (Clinical Trial)/Phase 2 | CBD | Unknown | Unknown | Not yet recruiting |
| NCT01489826 | Solid tumour | Interventional (Clinical Trial)/Phase 1 | Dexanabinol (HU-211; a synthetic cannabinoid) | 2–36 mg/kg once weekly–3 doses in 21-day cycle | Intravenous infusion | Progression-free survival increased |
| NCT01654497 | Brain cancer | Interventional (Clinical Trial)/Phase 1 | Dexanabinol (HU-211) | 2–44 mg/kg once weekly–4 doses in 28-day cycle | Intravenous infusion | No relevant results available |
| NCT03431363 | Head and neck cancer | Observational | Medically certified cannabis with adjuvant chemoradiation | Dosing options to be stratified into 3 groups viz. standard, frail/elderly (age > 65 or ECOG 2), and cannabis-experienced | Smoke | Recruiting |
| NCT02423239 | Hepatocellular carcinoma; pancreatic cancer | Interventional (Clinical Trial)/Phase 1 | Dexanabinol (HU-211) monotherapy and in combination with chemotherapy | MTD ** once a week | Intravenous infusion | Ongoing |
| NCT03245658 | Pancreatic cancer | Interventional (Clinical Trial)/Phase 2 | 1:2 ∆9-THC:CBD | Individually titrated doses on daily basis; for 4 weeks | Oral drops | Not yet recruiting |
| NCT03529448 | GBM | Interventional (Clinical Trial)/ Phase 1 & Phase 2 | TN-TC11G (1:1 ∆9-THC:CBD) combination with temozolomide and radiotherapy | Total daily dose of 10–160 mg, after meal | Unknown | Not yet recruiting |
| NCT03617692 | Non-small-cell lung carcinoma (NSCLC) metastasis | Observational | Cannabis products | Products, dose and administration frequency decided by study participants | Oral administration | Recruiting |
| NCT03052738 | Paediatric CNS tumour | Observational | Medical marijuana-derived products | Method of delivery, strain used, dosing and frequency decided by study participants | Recruiting | |
| NCT03687034 | Glioblastoma | Interventional (Clinical Trial)/Phase 1 | CBD with standard of care | Escalating doses of CBD | Oral sublingual formulation | Not yet recruiting |
| NCT03607643 | GI malignancies (pancreas, liver rectum, colon, or gall bladder), multiple myeloma, or GBM | Interventional (Clinical Trial)/Phase 1 & Phase 2 | CBD with standard of care chemotherapy | 100 mg twice daily before meal | Oral sublingual formulation | Not yet recruiting |
| ACTRN12617001287325 | GBM | Interventional (Clinical Trial)/Phase 2 | 1:1 ∆9-THC:CBD (6 mg/mL:6 mg/mL) or 1:4 CBD:∆9-THC (3.8 mg/mL:15 mg/mL) and standard treatment *** | Starts at 0.25 mL at night and each night titrated up or downwards by 0.05 mL based on participant’s response | Oral oily liquids | No relevant results available |
| ACTRN12619000265178 | Any cancer | Interventional (Clinical Trial)/phase 4 | ∆9-THC or 1:1 ∆9-THC:CBD. Combined with standard treatment for advanced cancer and symptoms | Starts at 2.5 mg THC three times a day in cannabis naive patients, and 5 mg THC three times a day in previous users. Dosage adjusted based on patient’s response up to a maximum of 30 mg THC per day. | Oral oily liquids | Recruiting |
| ACTRN12619000037101 | Any cancer | Interventional (Clinical Trial)/Phase 2 | 1:1 ∆9-THC:CBD | Total daily dose of 2.5 mg:2.5 mg–30 mg:30 mg | Oral oily liquid | Recruiting |
| ACTRN12618001220257 | Any cancer | Interventional (Clinical Trial)/Phase 2 | CBD | Total daily dose of 50 mg–600 mg | Oral oily liquid | Recruiting |
| ACTRN12618001205224 | Any cancer | Interventional (Clinical Trial)/Phase 1 | CBD or ∆9-THC with palliative care | Total daily doses of 50 mg–600 mg/day for CBD or 2.5 mg–30 mg for THC | Oral oily liquid | Doses of THC and CBD used in the study were generally well tolerated and up to 50% of the participants had an overall improvement in their condition since starting cannabis but results need to be replicated in placebo controlled trial [62] |
| ACTRN12616001036404 | Any cancer | Interventional (Clinical Trial)/Phase 2 & Phase 3 | 1:1 ∆9-THC/CBD with chemotherapy | 2.5 mg THC and 2.5 mg CBD, once the day before chemotherapy and three times daily for 5 days for the first 5 days of participants’ chemotherapy cycle and for three consecutive cycles. | Oral Capsule | Recruiting |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Afrin, F.; Chi, M.; Eamens, A.L.; Duchatel, R.J.; Douglas, A.M.; Schneider, J.; Gedye, C.; Woldu, A.S.; Dun, M.D. Can Hemp Help? Low-THC Cannabis and Non-THC Cannabinoids for the Treatment of Cancer. Cancers 2020, 12, 1033. https://doi.org/10.3390/cancers12041033
Afrin F, Chi M, Eamens AL, Duchatel RJ, Douglas AM, Schneider J, Gedye C, Woldu AS, Dun MD. Can Hemp Help? Low-THC Cannabis and Non-THC Cannabinoids for the Treatment of Cancer. Cancers. 2020; 12(4):1033. https://doi.org/10.3390/cancers12041033
Chicago/Turabian StyleAfrin, Farjana, Mengna Chi, Andrew L. Eamens, Ryan J. Duchatel, Alicia M. Douglas, Jennifer Schneider, Craig Gedye, Ameha S. Woldu, and Matthew D. Dun. 2020. "Can Hemp Help? Low-THC Cannabis and Non-THC Cannabinoids for the Treatment of Cancer" Cancers 12, no. 4: 1033. https://doi.org/10.3390/cancers12041033
APA StyleAfrin, F., Chi, M., Eamens, A. L., Duchatel, R. J., Douglas, A. M., Schneider, J., Gedye, C., Woldu, A. S., & Dun, M. D. (2020). Can Hemp Help? Low-THC Cannabis and Non-THC Cannabinoids for the Treatment of Cancer. Cancers, 12(4), 1033. https://doi.org/10.3390/cancers12041033