The Mitochondrial Protein VDAC1 at the Crossroads of Cancer Cell Metabolism: The Epigenetic Link


 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Xenograft Experiments
2.3. Gel Electrophoresis and Immunoblotting
2.4. Immunohistochemistry (IHC)
2.5. RNA Preparation, DNA Microarray Analysis, and Quantitative Real-Time PCR (q-RT-PCR)
2.6. Liquid Chromatography High-Resolution Mass Spectrometry (LC-HR-MS/MS) Analysis
2.7. Statistics and Data Analysis
3. Results
3.1. VDAC1 Depletion by si-RNA against Human (h)VDAC1 Inhibits Tumor Growth and Reprogramed Metabolism of U-87-MG Cell Line-Derived Tumors
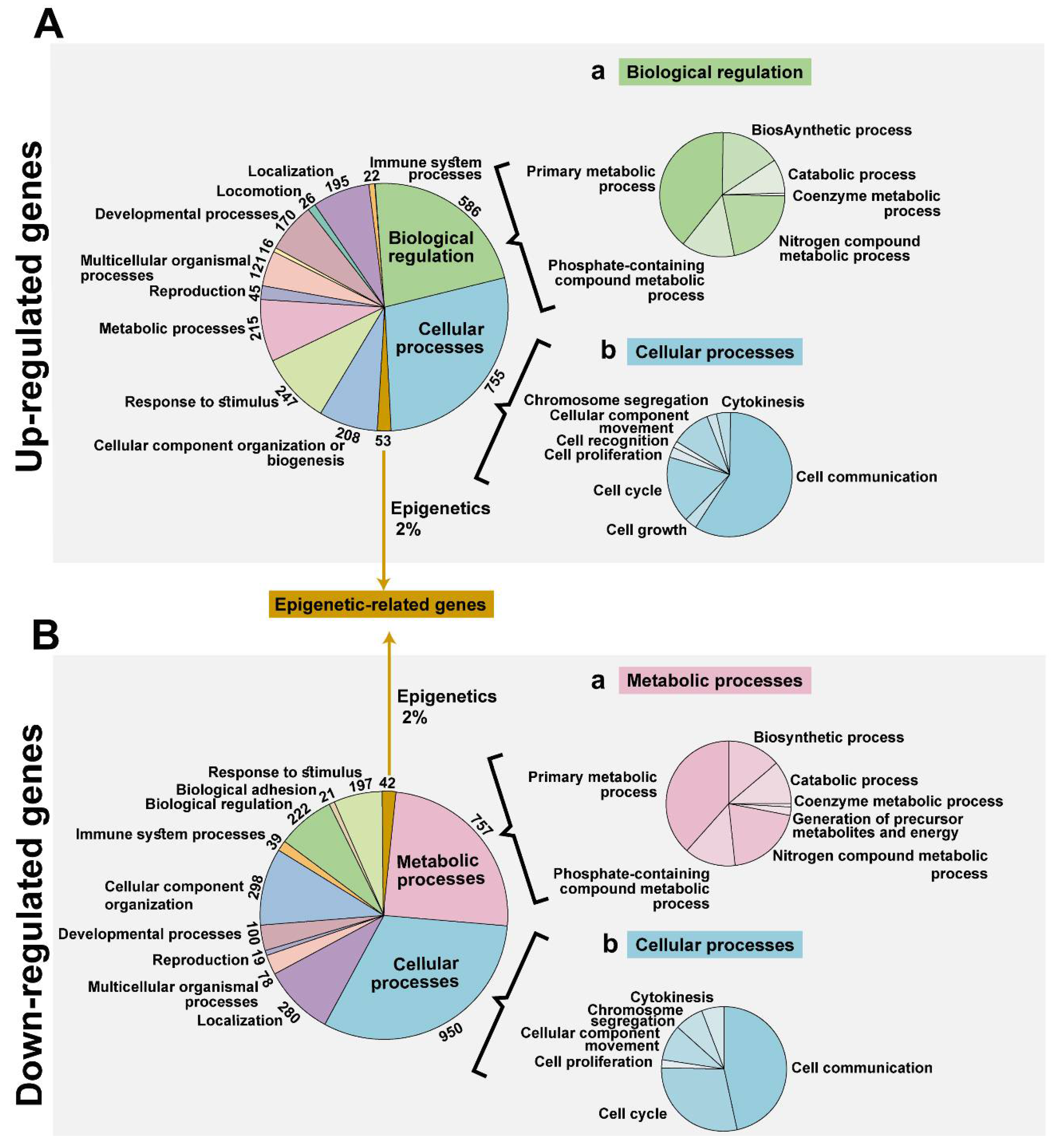
3.2. VDAC1 Depletion by si-hVDAC1-Induced Alteration of the Gene Expression Profile of si-hVDAC1-TTs
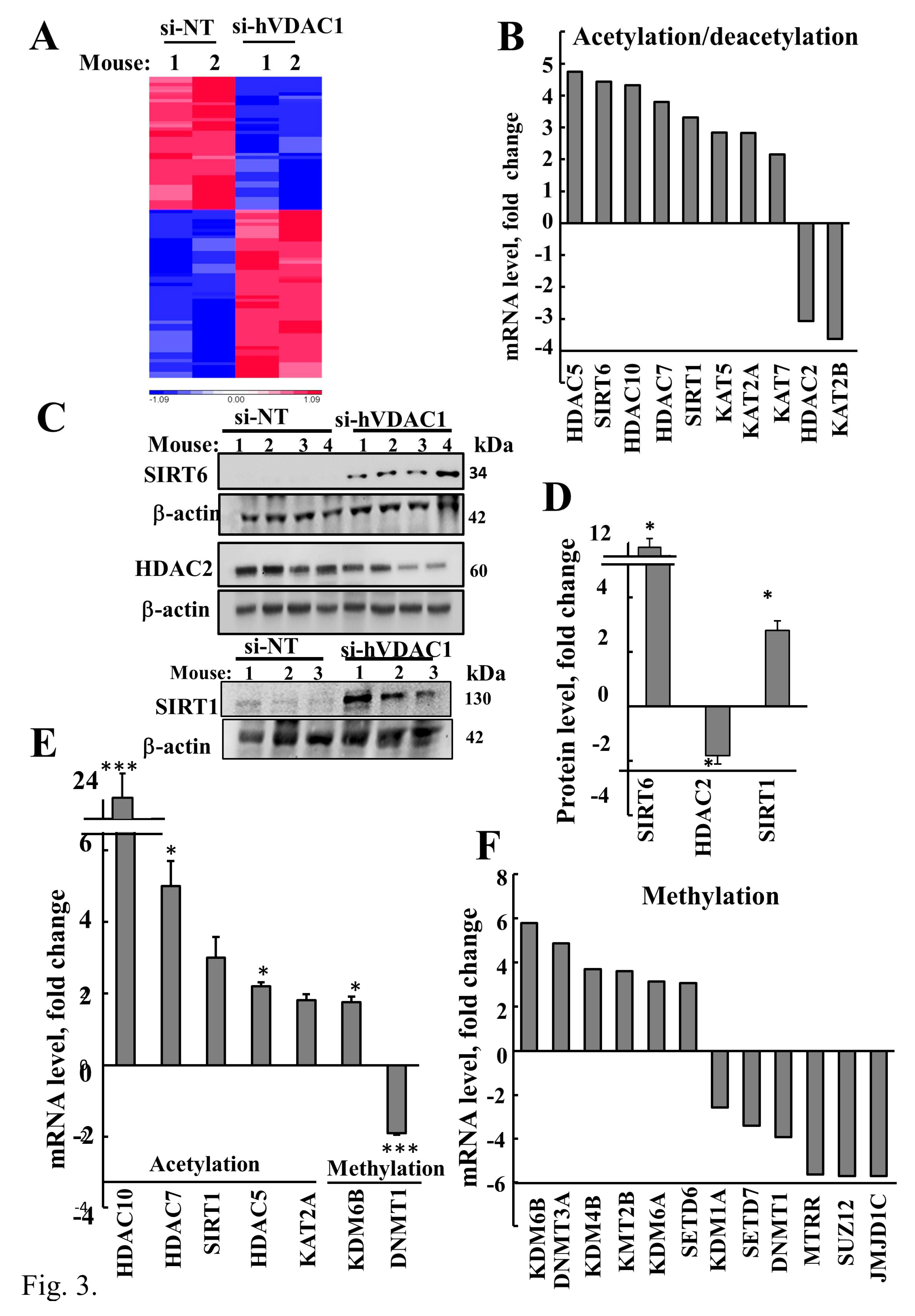
3.3. VDAC1 Depletion Altered Acetylation and Methylation of Histone 3 and Histone 4
4. Discussion
4.1. Epigenetics, Gene Transcription, Metabolism, and VDAC1
4.2. VDAC1 Depletion Altered Histones Acetylation and Methylation
4.3. SIRT1, SIRT6, and Metabolism Regulation
4.4. VDAC1 Depletion Altered Histones Modifications, Increasing Acetylation, and Decreasing Methylation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Schulze, A.; Harris, A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364. [Google Scholar] [CrossRef]
- Cai, Q.; Guo, Y.; Xiao, B.; Banerjee, S.; Saha, A.; Lu, J.; Glisovic, T.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C stabilizes Gemin3 to block p53-mediated apoptosis. PLoS Pathog. 2011, 7, e1002418. [Google Scholar] [CrossRef] [PubMed]
- Carrer, A.; Wellen, K.E. Metabolism and epigenetics: A link cancer cells exploit. Curr. Opin. Biotechnol. 2015, 34, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Etchegaray, J.-P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, R.; Gupta, S. Epigenetics and cancer. J. Appl. Physiol. 2010, 109, 598–605. [Google Scholar] [CrossRef]
- Dehan, P.; Kustermans, G.; Guenin, S.; Horion, J.; Boniver, J.; Delvenne, P. DNA methylation and cancer diagnosis: New methods and applications. Expert Rev. Mol. Diagn. 2009, 9, 651–657. [Google Scholar] [CrossRef]
- Hirst, M.; Marra, M.A. Epigenetics and human disease. Int. J. Biochem. Cell Biol. 2009, 41, 136–146. [Google Scholar] [CrossRef]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral Thinking: How Histone Modifications Regulate Gene Expression. Trends Genet. 2016, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Stefanska, B.; Karlic, H.; Varga, F.; Fabianowska-Majewska, K.; Haslberger, A.G. Epigenetic mechanisms in anti-cancer actions of bioactive food components—The implications in cancer prevention. Br. J. Pharmacol. 2012, 167, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wang, L.; Di, L.J. Compartmentation of metabolites in regulating epigenome of cancer. Mol. Med. 2016, 22, 349–360. [Google Scholar] [CrossRef][Green Version]
- Gronbaek, K.; Treppendahl, M.; Asmar, F.; Guldberg, P. Epigenetic changes in cancer as potential targets for prophylaxis and maintenance therapy. Basic Clin. Pharm. Toxicol. 2008, 103, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Urvalek, A.; Laursen, K.B.; Gudas, L.J. The roles of retinoic acid and retinoic acid receptors in inducing epigenetic changes. In The Biochemistry of Retinoic Acid Receptors I: Structure, Activation, and Function at the Molecular Level; Springer: Dordrecht, The Netherlands, 2014; pp. 129–149. [Google Scholar] [CrossRef]
- Chen, Z.X.; Riggs, A.D. DNA methylation and demethylation in mammals. J. Biol. Chem. 2011, 286, 18347–18353. [Google Scholar] [CrossRef]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr.; McKnight, S.L. Influence of Metabolism on Epigenetics and Disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef]
- Kaochar, S.; Tu, B.P. Gatekeepers of chromatin: Small metabolites elicit big changes in gene expression. Trends Biochem. Sci. 2012, 37, 477–483. [Google Scholar] [CrossRef]
- Shi, Y.; Shi, Y. Metabolic enzymes and coenzymes in transcription—A direct link between metabolism and transcription? Trends Genet. 2004, 20, 445–452. [Google Scholar] [CrossRef]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef]
- Sambucetti, L.C.; Fischer, D.D.; Zabludoff, S.; Kwon, P.O.; Chamberlin, H.; Trogani, N.; Xu, H.; Cohen, D. Histone deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading to specific chromatin acetylation and antiproliferative effects. J. Biol. Chem. 1999, 274, 34940–34947. [Google Scholar] [CrossRef] [PubMed]
- Sutendra, G.; Kinnaird, A.; Dromparis, P.; Paulin, R.; Stenson, T.H.; Haromy, A.; Hashimoto, K.; Zhang, N.; Flaim, E.; Michelakis, E.D. A nuclear pyruvate dehydrogenase complex is important for the generation of acetyl-CoA and histone acetylation. Cell 2014, 158, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 2016, 16, 694. [Google Scholar] [CrossRef]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef]
- Maddocks, O.D.; Labuschagne, C.F.; Adams, P.D.; Vousden, K.H. Serine metabolism supports the methionine cycle and DNA/RNA methylation through de novo ATP synthesis in cancer cells. Mol. Cell 2016, 61, 210–221. [Google Scholar] [CrossRef]
- Mentch, S.J.; Locasale, J.W. One-carbon metabolism and epigenetics: Understanding the specificity. Ann. N. Y. Acad. Sci. 2016, 1363, 91–98. [Google Scholar] [CrossRef]
- Mentch, S.J.; Mehrmohamadi, M.; Huang, L.; Liu, X.; Gupta, D.; Mattocks, D.; Padilla, P.G.; Ables, G.; Bamman, M.M.; Thalacker-Mercer, A.E. Histone methylation dynamics and gene regulation occur through the sensing of one-carbon metabolism. Cell Metab. 2015, 22, 861–873. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, Y.R.; Liu, K.; Yin, Z.; Liu, R.; Xia, Y.; Tan, L.; Yang, P.; Lee, J.H.; Li, X.J.; et al. KAT2A coupled with the alpha-KGDH complex acts as a histone H3 succinyltransferase. Nature 2017, 552, 273–277. [Google Scholar] [CrossRef]
- Shen, H.; Laird, P.W. Interplay between the cancer genome and epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Suvà, M.L.; Riggi, N.; Bernstein, B.E. Epigenetic Reprogramming in Cancer. Science 2013, 339, 1567–1570. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726. [Google Scholar] [CrossRef]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef]
- Moussaieff, A.; Kogan, N.M.; Aberdam, D. Concise Review: Energy Metabolites: Key Mediators of the Epigenetic State of Pluripotency. Stem Cells 2015, 33, 2374–2380. [Google Scholar] [CrossRef]
- Kim, H.K.; Noh, Y.H.; Nilius, B.; Ko, K.S.; Rhee, B.D.; Kim, N.; Han, J. Current and upcoming mitochondrial targets for cancer therapy. Semin. Cancer Biol. 2017, 47, 154–167. [Google Scholar] [CrossRef]
- Valcarcel-Jimenez, L.; Gaude, E.; Torrano, V.; Frezza, C.; Carracedo, A. Mitochondrial metabolism: Yin and Yang for tumor progression. Trends Endocrinol. Metab. 2017, 28, 748–757. [Google Scholar] [CrossRef]
- Maldonado, E.N.; Lemasters, J.J. Warburg revisited: Regulation of mitochondrial metabolism by voltage-dependent anion channels in cancer cells. J. Pharm. Exp. 2012, 342, 637–641. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Ben-Hail, D. VDAC, a multi-functional mitochondrial protein as a pharmacological target. Mitochondrion 2012, 12, 24–34. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Chandel, N.S.; Li, X.X.; Schumacker, P.T.; Colombini, M.; Thompson, C.B. Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc. Natl. Acad. Sci. USA 2000, 97, 4666–4671. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A.; Arif, T. Voltage-Dependent Anion Channel 1 as an Emerging Drug Target for Novel Anti-Cancer Therapeutics. Front. Oncol. 2017, 7, 154. [Google Scholar] [CrossRef]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef]
- Arif, T.; Vasilkovsky, L.; Refaely, Y.; Konson, A.; Shoshan-Barmatz, V. Silencing VDAC1 Expression by siRNA Inhibits Cancer Cell Proliferation and Tumor Growth in Vivo. Mol. Nucleic Acids 2014, 3, e159. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 2015, 1848, 2547–2575. [Google Scholar] [CrossRef]
- Abu-Hamad, S.; Sivan, S.; Shoshan-Barmatz, V. The expression level of the voltage-dependent anion channel controls life and death of the cell. Proc. Natl. Acad. Sci. USA 2006, 103, 5787–5792. [Google Scholar] [CrossRef]
- Koren, I.; Raviv, Z.; Shoshan-Barmatz, V. Downregulation of voltage-dependent anion channel-1 expression by RNA interference prevents cancer cell growth in vivo. Cancer Biol. 2010, 9, 1046–1052. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Golan, M. Mitochondrial VDAC1: Function in cell life and death and a target for cancer therapy. Curr. Med. Chem. 2012, 19, 714–735. [Google Scholar] [CrossRef]
- Arif, T.; Krelin, Y.; Nakdimon, I.; Benharroch, D.; Paul, A.; Dadon-Klein, D.; Shoshan-Barmatz, V. VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro Oncol. 2017, 19, 951–964. [Google Scholar] [CrossRef]
- Kim, Y.Z. Altered Histone Modifications in Gliomas. Brain Tumor Res. Treat. 2014, 2, 7–21. [Google Scholar] [CrossRef]
- Foltz, G.; Yoon, J.-G.; Lee, H.; Ma, L.; Tian, Q.; Hood, L.; Madan, A. Epigenetic Regulation of Wnt Pathway Antagonists in Human Glioblastoma Multiforme. Genes Cancer 2010, 1, 81–90. [Google Scholar] [CrossRef]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Nawarak, J.; Huang-Liu, R.; Kao, S.H.; Liao, H.H.; Sinchaikul, S.; Chen, S.T.; Cheng, S.L. Proteomics analysis of A375 human malignant melanoma cells in response to arbutin treatment. Biochim. Biophys. Acta 2009, 1794, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Brunet, J.; O’Loughlin, J.L.; Gunnell, K.E.; Sabiston, C.M. Physical activity and depressive symptoms after breast cancer: Cross-sectional and longitudinal relationships. Health Psychol. 2018, 37, 14–23. [Google Scholar] [CrossRef]
- Bojang, P., Jr.; Ramos, K.S. The promise and failures of epigenetic therapies for cancer treatment. Cancer Treat. Rev. 2014, 40, 153–169. [Google Scholar] [CrossRef]
- Jang, S.M.; Kim, J.W.; Kim, C.H.; An, J.H.; Johnson, A.; Song, P.I.; Rhee, S.; Choi, K.H. KAT5-mediated SOX4 acetylation orchestrates chromatin remodeling during myoblast differentiation. Cell Death Dis. 2015, 6, e1857. [Google Scholar] [CrossRef]
- Li, B.; Samanta, A.; Song, X.; Iacono, K.T.; Bembas, K.; Tao, R.; Basu, S.; Riley, J.L.; Hancock, W.W.; Shen, Y.; et al. FOXP3 interactions with histone acetyltransferase and class II histone deacetylases are required for repression. Proc. Natl. Acad. Sci. USA 2007, 104, 4571–4576. [Google Scholar] [CrossRef]
- Sapountzi, V.; Cote, J. MYST-family histone acetyltransferases: Beyond chromatin. Cell. Mol. Life Sci. 2011, 68, 1147–1156. [Google Scholar] [CrossRef]
- Weichert, W.; Roske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef]
- Urbich, C.; Rossig, L.; Kaluza, D.; Potente, M.; Boeckel, J.N.; Knau, A.; Diehl, F.; Geng, J.G.; Hofmann, W.K.; Zeiher, A.M.; et al. HDAC5 is a repressor of angiogenesis and determines the angiogenic gene expression pattern of endothelial cells. Blood 2009, 113, 5669–5679. [Google Scholar] [CrossRef]
- Czubryt, M.P.; McAnally, J.; Fishman, G.I.; Olson, E.N. Regulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1 alpha ) and mitochondrial function by MEF2 and HDAC5. Proc. Natl. Acad. Sci. USA 2003, 100, 1711–1716. [Google Scholar] [CrossRef]
- Bradley, E.W.; Carpio, L.R.; Olson, E.N.; Westendorf, J.J. Histone deacetylase 7 (Hdac7) suppresses chondrocyte proliferation and beta-catenin activity during endochondral ossification. J. Biol. Chem. 2015, 290, 118–126. [Google Scholar] [CrossRef]
- NCBI. HDAC7 Histone Deacetylase 7. Available online: https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=ShowDetailView&TermToSearch=51564 (accessed on 21 April 2020).
- Song, C.; Zhu, S.; Wu, C.; Kang, J. Histone deacetylase (HDAC) 10 suppresses cervical cancer metastasis through inhibition of matrix metalloproteinase (MMP) 2 and 9 expression. J. Biol. Chem. 2013, 288, 28021–28033. [Google Scholar] [CrossRef]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef]
- Stunkel, W.; Peh, B.K.; Tan, Y.C.; Nayagam, V.M.; Wang, X.; Salto-Tellez, M.; Ni, B.; Entzeroth, M.; Wood, J. Function of the SIRT1 protein deacetylase in cancer. Biotechnol. J. 2007, 2, 1360–1368. [Google Scholar] [CrossRef]
- Michishita, E.; McCord, R.A.; Berber, E.; Kioi, M.; Padilla-Nash, H.; Damian, M.; Cheung, P.; Kusumoto, R.; Kawahara, T.L.; Barrett, J.C.; et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 2008, 452, 492–496. [Google Scholar] [CrossRef]
- Schwer, B.; Schumacher, B.; Lombard, D.B.; Xiao, C.; Kurtev, M.V.; Gao, J.; Schneider, J.I.; Chai, H.; Bronson, R.T.; Tsai, L.H.; et al. Neural sirtuin 6 (Sirt6) ablation attenuates somatic growth and causes obesity. Proc. Natl. Acad. Sci. USA 2010, 107, 21790–21794. [Google Scholar] [CrossRef]
- Ladopoulos, V.; Hofemeister, H.; Hoogenkamp, M.; Riggs, A.D.; Stewart, A.F.; Bonifer, C. The histone methyltransferase KMT2B is required for RNA polymerase II association and protection from DNA methylation at the MagohB CpG island promoter. Mol. Cell. Biol. 2013, 33, 1383–1393. [Google Scholar] [CrossRef]
- Levy, D.; Kuo, A.J.; Chang, Y.; Schaefer, U.; Kitson, C.; Cheung, P.; Espejo, A.; Zee, B.M.; Liu, C.L.; Tangsombatvisit, S.; et al. Lysine methylation of the NF-kappaB subunit RelA by SETD6 couples activity of the histone methyltransferase GLP at chromatin to tonic repression of NF-kappaB signaling. Nat. Immunol. 2011, 12, 29–36. [Google Scholar] [CrossRef]
- Barsyte-Lovejoy, D.; Li, F.; Oudhoff, M.J.; Tatlock, J.H.; Dong, A.; Zeng, H.; Wu, H.; Freeman, S.A.; Schapira, M.; Senisterra, G.A.; et al. (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc. Natl. Acad. Sci. USA 2014, 111, 12853–12858. [Google Scholar] [CrossRef]
- De la Cruz, C.C.; Kirmizis, A.; Simon, M.D.; Isono, K.; Koseki, H.; Panning, B. The polycomb group protein SUZ12 regulates histone H3 lysine 9 methylation and HP1 alpha distribution. Chromosome Res. 2007, 15, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Pasini, D.; Bracken, A.P.; Hansen, J.B.; Capillo, M.; Helin, K. The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol. Cell. Biol. 2007, 27, 3769–3779. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, S.; Akiyoshi, M.; Tokura, M.; Miyachi, H.; Nakai, Y.; Kimura, H.; Shinkai, Y.; Tachibana, M. JMJD1C, a JmjC domain-containing protein, is required for long-term maintenance of male germ cells in mice. Biol. Reprod. 2013, 89, 93. [Google Scholar] [CrossRef] [PubMed]
- Dhayalan, A.; Rajavelu, A.; Rathert, P.; Tamas, R.; Jurkowska, R.Z.; Ragozin, S.; Jeltsch, A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 2010, 285, 26114–26120. [Google Scholar] [CrossRef]
- Yang, L.; Rau, R.; Goodell, M.A. DNMT3A in haematological malignancies. Nat. Rev. Cancer 2015, 15, 152–165. [Google Scholar] [CrossRef]
- Datta, J.; Ghoshal, K.; Sharma, S.M.; Tajima, S.; Jacob, S.T. Biochemical fractionation reveals association of DNA methyltransferase (Dnmt) 3b with Dnmt1 and that of Dnmt 3a with a histone H3 methyltransferase and Hdac1. J. Cell. Biochem. 2003, 88, 855–864. [Google Scholar] [CrossRef]
- Young, L.C.; McDonald, D.W.; Hendzel, M.J. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following gamma-irradiation. J. Biol. Chem. 2013, 288, 21376–21388. [Google Scholar] [CrossRef]
- Fiskus, W.; Sharma, S.; Shah, B.; Portier, B.P.; Devaraj, S.G.; Liu, K.; Iyer, S.P.; Bearss, D.; Bhalla, K.N. Highly effective combination of LSD1 (KDM1A) antagonist and pan-histone deacetylase inhibitor against human AML cells. Leukemia 2014, 28, 2155–2164. [Google Scholar] [CrossRef]
- Jiang, W.; Wang, J.; Zhang, Y. Histone H3K27me3 demethylases KDM6A and KDM6B modulate definitive endoderm differentiation from human ESCs by regulating WNT signaling pathway. Cell Res. 2013, 23, 122–130. [Google Scholar] [CrossRef]
- Rothbart, S.B.; Strahl, B.D. Interpreting the language of histone and DNA modifications. Biochim. Biophys. Acta 2014, 1839, 627–643. [Google Scholar] [CrossRef]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily conserved and nonconserved cellular localizations and functions of human SIRT proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Rando, O.J. Metabolic Inputs into the Epigenome. Cell Metab. 2017, 25, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Ramassone, A.; Pagotto, S.; Veronese, A.; Visone, R. Epigenetics and MicroRNAs in Cancer. Int. J. Mol. Sci. 2018, 19, 459. [Google Scholar] [CrossRef]
- Schvartzman, J.M.; Thompson, C.B.; Finley, L.W.S. Metabolic regulation of chromatin modifications and gene expression. J. Cell. Biol. 2018, 217, 2247–2259. [Google Scholar] [CrossRef]
- Mack, S.C.; Hubert, C.G.; Miller, T.E.; Taylor, M.D.; Rich, J.N. An epigenetic gateway to brain tumor cell identity. Nat. Neurosci. 2016, 19, 10–19. [Google Scholar] [CrossRef]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003, 4, 117. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef]
- Shahbazian, M.D.; Grunstein, M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100. [Google Scholar] [CrossRef]
- Delcuve, G.P.; Khan, D.H.; Davie, J.R. Roles of histone deacetylases in epigenetic regulation: Emerging paradigms from studies with inhibitors. Clin. Epigenetics 2012, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Segre, C.V.; Chiocca, S. Regulating the regulators: The post-translational code of class I HDAC1 and HDAC2. J. Biomed. Biotechnol. 2011, 2011, 690848. [Google Scholar] [CrossRef] [PubMed]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed]
- Martens, J.H.; O’Sullivan, R.J.; Braunschweig, U.; Opravil, S.; Radolf, M.; Steinlein, P.; Jenuwein, T. The profile of repeat-associated histone lysine methylation states in the mouse epigenome. EMBO J. 2005, 24, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Carafa, V.; Altucci, L.; Nebbioso, A. Dual Tumor Suppressor and Tumor Promoter Action of Sirtuins in Determining Malignant Phenotype. Front. Pharm. 2019, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Tennen, R.I.; Chua, K.F. Chromatin regulation and genome maintenance by mammalian SIRT6. Trends Biochem. Sci. 2011, 36, 39–46. [Google Scholar] [CrossRef]
- Zhang, P.; Tu, B.; Wang, H.; Cao, Z.; Tang, M.; Zhang, C.; Gu, B.; Li, Z.; Wang, L.; Yang, Y.; et al. Tumor suppressor p53 cooperates with SIRT6 to regulate gluconeogenesis by promoting FoxO1 nuclear exclusion. Proc. Natl. Acad. Sci. USA 2014, 111, 10684–10689. [Google Scholar] [CrossRef]
- Kim, H.S.; Xiao, C.; Wang, R.H.; Lahusen, T.; Xu, X.; Vassilopoulos, A.; Vazquez-Ortiz, G.; Jeong, W.I.; Park, O.; Ki, S.H.; et al. Hepatic-specific disruption of SIRT6 in mice results in fatty liver formation due to enhanced glycolysis and triglyceride synthesis. Cell Metab. 2010, 12, 224–236. [Google Scholar] [CrossRef]
- Min, L.; Ji, Y.; Bakiri, L.; Qiu, Z.; Cen, J.; Chen, X.; Chen, L.; Scheuch, H.; Zheng, H.; Qin, L.; et al. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat. Cell. Biol. 2012, 14, 1203–1211. [Google Scholar] [CrossRef]
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [CrossRef]
- Kawahara, T.L.; Michishita, E.; Adler, A.S.; Damian, M.; Berber, E.; Lin, M.; McCord, R.A.; Ongaigui, K.C.; Boxer, L.D.; Chang, H.Y.; et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 2009, 136, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.L.; Rapicavoli, N.A.; Wu, A.R.; Qu, K.; Quake, S.R.; Chang, H.Y. Dynamic chromatin localization of Sirt6 shapes stress- and aging-related transcriptional networks. PLoS Genet. 2011, 7, e1002153. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.R.; Milner, J. SIRT1, metabolism and cancer. Curr. Opin. Oncol. 2012, 24, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; D’Urso, A.; Toiber, D.; Sebastian, C.; Henry, R.E.; Vadysirisack, D.D.; Guimaraes, A.; Marinelli, B.; Wikstrom, J.D.; Nir, T.; et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 2010, 140, 280–293. [Google Scholar] [CrossRef]
- Dominy, J.E., Jr.; Lee, Y.; Jedrychowski, M.P.; Chim, H.; Jurczak, M.J.; Camporez, J.P.; Ruan, H.B.; Feldman, J.; Pierce, K.; Mostoslavsky, R.; et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol. Cell 2012, 48, 900–913. [Google Scholar] [CrossRef]
- Castonguay, Z.; Auger, C.; Thomas, S.C.; Chahma, M.; Appanna, V.D. Nuclear lactate dehydrogenase modulates histone modification in human hepatocytes. Biochem. Biophys. Res. Commun. 2014, 454, 172–177. [Google Scholar] [CrossRef]
- Marsoni, S.; Damia, G.; Camboni, G. A work in progress: The clinical development of histone deacetylase inhibitors. Epigenetics 2008, 3, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Sanders, B.D.; Jackson, B.; Brent, M.; Taylor, A.M.; Dang, W.; Berger, S.L.; Schreiber, S.L.; Howitz, K.; Marmorstein, R. Identification and characterization of novel sirtuin inhibitor scaffolds. Bioorg. Med. Chem. 2009, 17, 7031–7041. [Google Scholar] [CrossRef][Green Version]
- Wozniak, G.G.; Strahl, B.D. Hitting the ‘mark’: Interpreting lysine methylation in the context of active transcription. Biochim. Biophys. Acta 2014, 1839, 1353–1361. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Arif, T.; Amsalem, Z.; Shoshan-Barmatz, V. Metabolic Reprograming Via Silencing of Mitochondrial VDAC1 Expression Encourages Differentiation of Cancer Cells. Mol. Nucleic Acids 2019, 17, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Arif, T.; Stern, O.; Pittala, S.; Chalifa-Caspi, V.; Shoshan-Barmatz, V. Rewiring of Cancer Cell Metabolism by Mitochondrial VDAC1 Depletion Results in Time-Dependent Tumor Reprogramming: Glioblastoma as a Proof of Concept. Cells 2019, 8, 1330. [Google Scholar] [CrossRef]
- Richly, H.; Lange, M.; Simboeck, E.; Di Croce, L. Setting and resetting of epigenetic marks in malignant transformation and development. BioEssays 2010, 32, 669–679. [Google Scholar] [CrossRef]
- Ohm, J.E.; McGarvey, K.M.; Yu, X.; Cheng, L.; Schuebel, K.E.; Cope, L.; Mohammad, H.P.; Chen, W.; Daniel, V.C.; Yu, W.; et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007, 39, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, C.T.; Sandstrom, R.S.; Huang, S.A.; Wang, Y.; Schotta, G.; Berger, M.S.; Lin, C.A. Cross-species Analyses Unravel the Complexity of H3K27me3 and H4K20me3 in the Context of Neural Stem Progenitor Cells. Neuroepigenetics 2016, 6, 10–25. [Google Scholar] [CrossRef] [PubMed]
- Evertts, A.G.; Manning, A.L.; Wang, X.; Dyson, N.J.; Garcia, B.A.; Coller, H.A. H4K20 methylation regulates quiescence and chromatin compaction. Mol. Biol. Cell 2013, 24, 3025–3037. [Google Scholar] [CrossRef]
- Abroun, S.; Saki, N.; Ahmadvand, M.; Asghari, F.; Salari, F.; Rahim, F. STATs: An Old Story, Yet Mesmerizing. Cell J. 2015, 17, 395–411. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Fold Change (p-Value) | Function |
|---|---|---|
| KAT2A (Lysine acetyltransferase 2A) | 2.83 (0.04) | Histone acetyltransferase (HAT) functions in the regulation of gene expression, and promotes transcriptional activation [54]. Can act both as an acetyltransferase and a succinyltransferase [30]. |
| KAT2B (Lysine acetyltransferase 2B) | −3.63 (0.04) | HAT has significant histone acetyltransferase activity and is associated with cell proliferation and transcriptional activation promotion [55]. |
| KAT5 (Lysine acetyltransferase 5) | 2.84 (0.03) | Catalytic subunit of histone acetyltransferase complex which is involved in transcriptional activation of select genes, principally by acetylation of nucleosomal histones H4 and H2A [56]. Regulates cell growth and proliferation [57]. |
| KAT7 (Lysine acetyltransferase 7) | 2.15 (0.04) | Part of the acetyltransferase complex. Involved in transcription and DNA replication via acetylation of histones H3 and H4 [58] |
| HDAC2 (Histone deacetylase 2) | −3.063 (0.04) | Highly expressed in cancer, associated with tumor de-differentiation and invasion [59]. |
| HDAC5 (Histone deacetylase 5) | 4.747 (0.05) | Possesses deacetylase activity for histone lysine residues and other proteins (p53?) [60]. Associated with cell differentiation and is a negative regulator of cell migration and angiogenesis [61]. Represses transcription when tethered to a promoter. It also interacts with myocyte enhancer factor-2 (MEF2) protein, resulting in repression of MEF2-dependent genes [62]. |
| HDAC7 (Histone deacetylase 7) | 3.793 (0.03) | Has little intrinsic deacetylase activity but may serve various other functions related to suppression of development, proliferation, and inflammation [63]. Promotes repression mediated via the transcriptional co-repressors SMRT and MEF2(A) [64]. |
| HDAC10 (Histone deacetylase 10) | 4.323 (0.04) | Acts as a suppressor of cancer metastasis and cell cycle regulation [65]. Low expression of HDAC10 associated with poor prognosis of cancer [66]. |
| SIRT1 (NAD+-dependent protein deacetylase sirtuin-1) | 3.307 (0.03) | NAD+-dependent protein linked to cellular survival pathways by virtue of maintaining the tumor suppressor gene p53 [67]. |
| SIRT6 (NAD+-dependent protein deacetylase sirtuin-6) | 4.433 (0.03) | NAD+-dependent protein deacetylase of histone H3K9 and H3K56. Plays various roles in metabolism, stress resistance and lifespan [68,69]. |
| Enzyme | Fold Change (p-Value) | Function |
|---|---|---|
| KMT2B (Lysine methyltransferase 2B) | 3.6 (0.03) | Histone methyltransferase. Methylates histone H3K4, involved in transcriptional activation [70]. |
| SETD6 (SET-domain-containing 6) | 3.06 (0.03) | Methyltransferase that adds a methyl group to histone H2A, which is involved in nuclear receptor-dependent transcription [64], leading to down-regulation of NF-κB transcription factor activity [71]. |
| SETD7 (SET-domain-containing Lysine methyltransferase 7) | −3.4 (0.03) | Histone methyltransferase that specifically mono-methylates H3K4. Implicated in multiple signaling and disease-related pathways, with a broad diversity of reported substrates [72]. |
| SUZ12 (Polycomb repressive complex 2 subunit) | −5.7 (0.02) | Component of the polycomb complex. Methylates H3K27, leading to transcriptional repression of the affected target gene [73,74]. |
| JMJD1C (Jumonji-domain-containing 1C) | −5.7 (0.03) | A candidate histone demethylase (H3K9) thought to be a co-activator of key transcription factors [75]. |
| DNMT3A (DNA methyltransferase 3A) | 4.854 (0.03) | Mediates genome-wide de novo methylation and establishment of DNA methylation patterns during development. Recruited to tri-methylated H3K36 [76]. Critically important new tumor suppressor [77]. |
| DNMT1 (DNA methyltransferase 1) | −3.918 (0.08) | Methylates CpG residues. Mediates transcriptional repression by direct binding to HDAC2 in association with DNMT3B and dimethylation of promoter histone H3 at H3K4 and H3K9 [78]. |
| KDM4B (Lysine demethylase 4B) | 3.685 (0.03) | Histone demethylase that specifically demethylates H3K9. Contributes to the regulation of cellular differentiation and proliferation [79]. |
| KDM1A (Lysine demethylase 1A) | −2.559 (0.04) | Histone demethylase demethylates both H3K4me and H3K9me/me2 of histone H3. Thereby, acting as a coactivator or a corepressor, depending on the context [80]. |
| KDM6A (Lysine demethylase 6A) | 3.127 (0.03) | Histone demethylase that specifically demethylates H3K27me2/me3. Plays a role in cell differentiation [81]. |
| KDM6B (Lysine demethylase 6B) | 5.775 (0.03) | Histone demethylase that specifically demethylates H3K27me2/me3. Plays a role in cell differentiation [81]. |
| MTRR (Methionine synthase reductase) | −5.629 (0.03) | Involved in the reductive regeneration of co-factor vitamin B12 required for the maintenance of methionine synthase in a functional state. Necessary for utilization of methyl groups for DNA methylation [81]. |
| Proteomics: Histones and Histone-Associated Proteins | ||
|---|---|---|
| Protein | Fold Change (p-value) | Function |
| HPF1 (Histone PARylation factor 1) | −14.886 (0.03) | Promotes histone serine ADP-ribosylation in response to DNA damage, limiting DNA damage-induced PARP1 hyper-auto-modification, thus ensuring genome stability. |
| BZW1 (Basic leucine zipper and W2 domain-containing protein 1) | −2.109 (0.02) | Enhances histone H4 gene transcription. |
| CBX5 (Chromobox protein homolog 5) | −9.244 (0.04) | Component of heterochromatin that recognizes and binds histone H3 tails methylated at K9 (H3K9me), leading to epigenetic repression. |
| HAT1 (Histone acetyltransferase type B catalytic subunit) | −3.833 (0.01) | Acetylates soluble but not nucleosomal histone H4 at K5 and K12 and, to a lesser extent, acetylates H2AK5. |
| HDAC3 (Histone deacetylase 3) | −2.284 (<0.01) | Responsible for the deacetylation of lysine residues in the N-terminal region of the core histones. Participates in BCL6 transcriptional repressor activity by deacetylating H3K27 on enhancer elements. |
| H2AFZ (H2A histone family member Z) | −2.7 (0.02) | Variant histone H2A which replaces conventional H2A in a subset of nucleosomes. |
| HP1BP3 (Heterochromatin protein 1-binding protein 3) | −2.103 (0.03) | Component of heterochromatin that maintains heterochromatin integrity during G1/S progression and regulates the duration of G1 phase to critically influence cell proliferative capacity. Mediates chromatin condensation during hypoxia, leading to increased tumor cell viability, radio-resistance, chemo-resistance, and self-renewal. |
| H1F0 (Histone H1.0) | 2.548 (<0.01) | Histones H1 are necessary for the condensation of nucleosome chains into higher-order structures. H1F0 histones are found in cells in terminal stages of differentiation or those that have low rates of cell division. |
| Proteomics: Transcription Factors | ||
|---|---|---|
| Protein | Fold Change (p-Value) | Function |
| LARP7 (La-related protein 7) | −127.546 (0.04) | Negative transcriptional regulator of polymerase II genes. |
| MATR3 (Matrin-3) | −4.372 (<0.01) | May play a role in transcription or interact with other nuclear matrix proteins to form the internal fibrogranular network. |
| TOP1 (DNA topoisomerase 1 alpha) | −4.154 (0.04) | Releases the supercoiling and torsional tension of DNA introduced during replication and transcription by transiently cleaving and rejoining one strand of the DNA duplex. |
| SMARCD2 (SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily D member 2) | −3.63 (0.04) | Involved in transcriptional activation and repression of select genes by chromatin remodeling (alteration of DNA–nucleosome topology). |
| SUB1 (Activated RNA polymerase II transcriptional coactivator p15) | −3.481 (0.03) | General co-activator that cooperatively functions with tumor associated fibroblasts (TAFs) and mediates functional interactions between upstream activators and the general transcriptional machinery. |
| RALY (RNA-binding protein Raly) | −2.3 (0.02) | RNA-binding protein that acts as a transcriptional co-factor for cholesterol biosynthetic genes in the liver. |
| CNOT9 (CCR4-NOT transcription complex subunit 9) | −1.9 (<0.01) | Component of the CCR4-NOT complex, which is one of the major cellular mRNA deadenylases and is linked to cellular processes, including bulk mRNA degradation, miRNA-mediated repression, translational repression during translational initiation, and general transcription regulation. |
| STAT1 (Signal transducer and activator of transcription 1-alpha/beta) | −1.7 (0.03) | Signal transducer and transcription activator that mediates cellular responses to interferons (IFNs), other cytokines, and other growth factors. |
| CSRP1 (Cysteine- and glycine-rich protein 1) | 4.8766 (<0.01) | May be involved in regulatory processes important for development and cellular differentiation. |
| ETS1 (ETS proto-oncogene 1) | 5 (0.02) | Transcription factor. Directly controls the expression of cytokine and chemokine genes in a wide variety of different cellular contexts. May control the differentiation, survival, and proliferation of lymphoid cells. May also regulate angiogenesis by regulating the expression of genes controlling endothelial cell migration and invasion. |
| STAT6 (Signal transducer and activator of transcription 6) | 11.5 (0.01) | Carries out dual functions, affecting signal transduction and the activation of transcription. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amsalem, Z.; Arif, T.; Shteinfer-Kuzmine, A.; Chalifa-Caspi, V.; Shoshan-Barmatz, V. The Mitochondrial Protein VDAC1 at the Crossroads of Cancer Cell Metabolism: The Epigenetic Link. Cancers 2020, 12, 1031. https://doi.org/10.3390/cancers12041031
Amsalem Z, Arif T, Shteinfer-Kuzmine A, Chalifa-Caspi V, Shoshan-Barmatz V. The Mitochondrial Protein VDAC1 at the Crossroads of Cancer Cell Metabolism: The Epigenetic Link. Cancers. 2020; 12(4):1031. https://doi.org/10.3390/cancers12041031
Chicago/Turabian StyleAmsalem, Zohar, Tasleem Arif, Anna Shteinfer-Kuzmine, Vered Chalifa-Caspi, and Varda Shoshan-Barmatz. 2020. "The Mitochondrial Protein VDAC1 at the Crossroads of Cancer Cell Metabolism: The Epigenetic Link" Cancers 12, no. 4: 1031. https://doi.org/10.3390/cancers12041031
APA StyleAmsalem, Z., Arif, T., Shteinfer-Kuzmine, A., Chalifa-Caspi, V., & Shoshan-Barmatz, V. (2020). The Mitochondrial Protein VDAC1 at the Crossroads of Cancer Cell Metabolism: The Epigenetic Link. Cancers, 12(4), 1031. https://doi.org/10.3390/cancers12041031