Tumor Size Matters—Understanding Concomitant Tumor Immunity in the Context of Hypofractionated Radiotherapy with Immunotherapy

 , and
, and 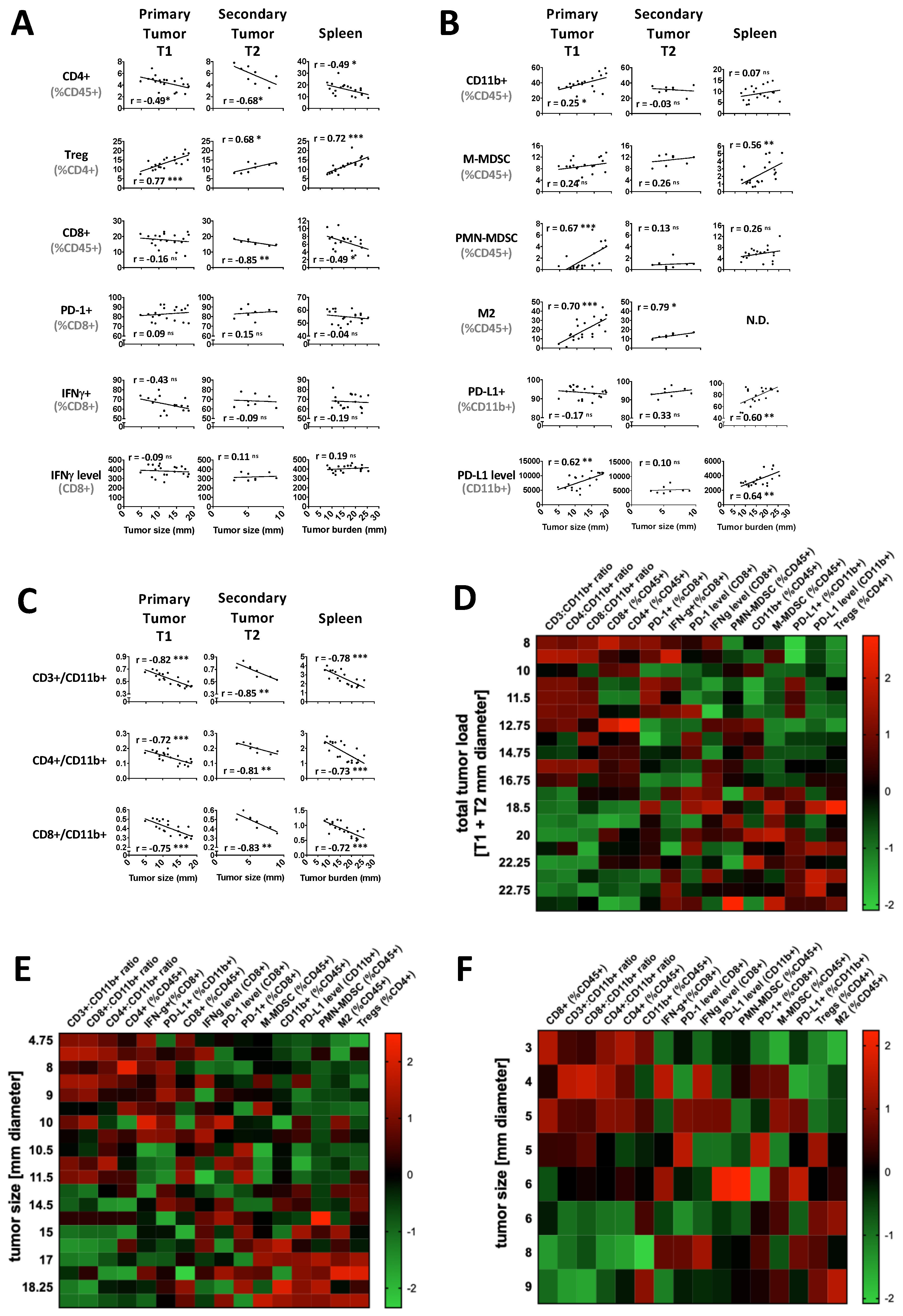{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
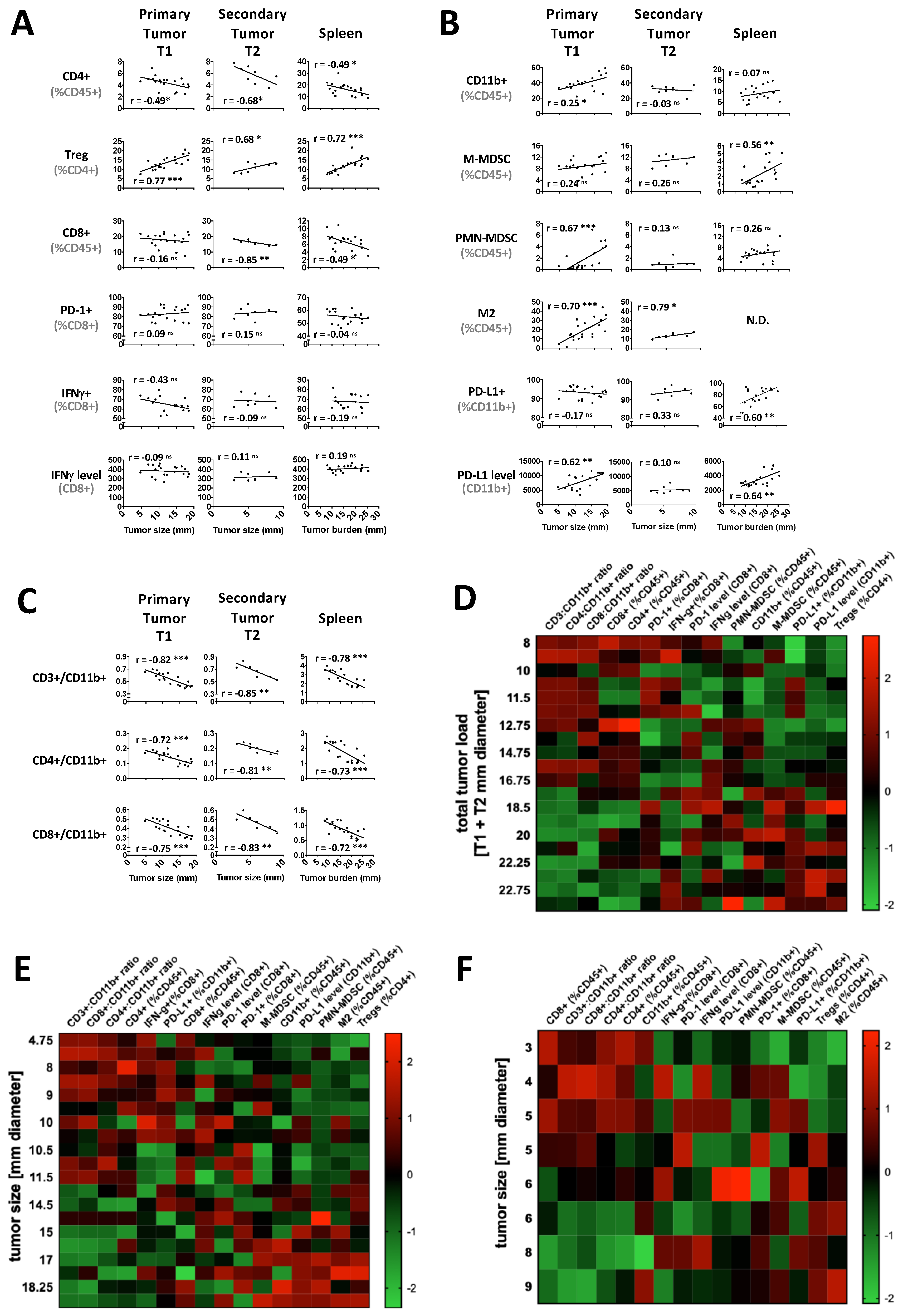
2.1. Tumor Load Determines the Immune Balance—Locally and Systemically
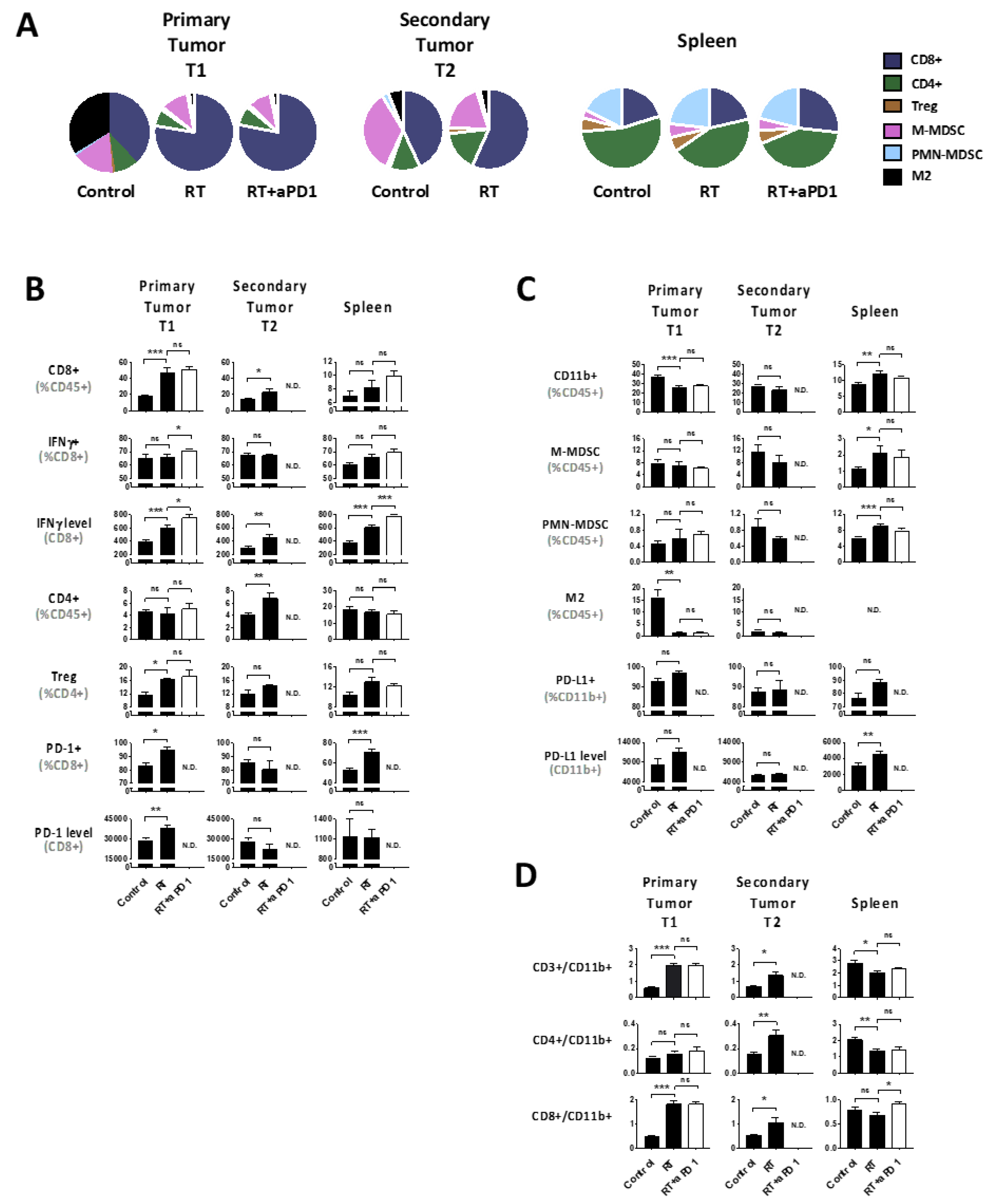
2.2. Radiation Plus Anti-PD-1 Monoclonal Antibody Can Drive Superior Systemic Tumor Control
2.3. hRT Alters the Immune Balance
2.4. Tumor hRT Plus Systemic PD-1 Blockade Increases the Functionality of T Cells—Locally and Systemically
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Cell Line and Reagents
4.3. Tumor Model and RT
4.4. Tissue Harvest and Immune Profiling
4.5. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Xia, B.; Zheng, T.; Lou, G. Immunoscore system combining CD8 and PD-1/PD-L1: A novel approach that predicts the clinical outcomes for cervical cancer. Int. J. Biol. Mark. 2019. [Google Scholar] [CrossRef] [Green Version]
- Schaue, D. A Century of Radiation Therapy and Adaptive Immunity. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Schaue, D.; Comin-Anduix, B.; Ribas, A.; Zhang, L.; Goodglick, L.; Sayre, J.W.; Debucquoy, A.; Haustermans, K.; McBride, W.H. T-cell responses to survivin in cancer patients undergoing radiation therapy. Clin. Cancer Res. 2008, 14, 4883–4890. [Google Scholar] [CrossRef]
- Formenti, S.C.; Lee, P.; Adams, S.; Goldberg, J.D.; Li, X.; Xie, M.W.; Ratikan, J.A.; Felix, C.; Hwang, L.; Faull, K.F.; et al. Focal Irradiation and Systemic TGFbeta Blockade in Metastatic Breast Cancer. Clin. Cancer Res. 2018, 24, 2493–2504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demaria, S.; Formenti, S. Role of T lymphocytes in tumor response to radiotherapy. Front. Oncol. 2012, 2, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, W.H.; Howie, S.E. Induction of tolerance to a murine fibrosarcoma in two zones of dosage--the involvement of suppressor cells. Br. J. Cancer 1986, 53, 707–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milas, L.; Hunter, N.; Withers, H.R. Concomitant immunity to pulmonary metastases of a murine fibrosarcoma: Influence of removal of primary tumor by radiation or surgery, of active specific immunization and treatment with Corynebacterium granulosum. Int. J. Radiat. Oncol. Biol. Phys. 1976, 1, 1171–1178. [Google Scholar] [CrossRef]
- Stone, H.B.; Peters, L.J.; Milas, L. Effect of host immune capability on radiocurability and subsequent transplantability of a murine fibrosarcoma. J. Natl. Cancer Inst. 1979, 63, 1229–1235. [Google Scholar] [PubMed]
- Yang, J.C.; Chang, A.E.; Baker, A.R.; Sindelar, W.F.; Danforth, D.N.; Topalian, S.L.; DeLaney, T.; Glatstein, E.; Steinberg, S.M.; Merino, M.J.; et al. Randomized prospective study of the benefit of adjuvant radiation therapy in the treatment of soft tissue sarcomas of the extremity. J. Clin. Oncol. 1998, 16, 197–203. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, B.; Davis, A.M.; Turcotte, R.; Bell, R.; Catton, C.; Chabot, P.; Wunder, J.; Kandel, R.; Goddard, K.; Sadura, A.; et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: A randomised trial. Lancet 2002, 359, 2235–2241. [Google Scholar] [CrossRef]
- Italiano, A.; Le Cesne, A.; Mendiboure, J.; Blay, J.-Y.; Piperno-Neumann, S.; Chevreau, C.; Delcambre, C.; Penel, N.; Terrier, P.; Ranchere-Vince, D.; et al. Prognostic factors and impact of adjuvant treatments on local and metastatic relapse of soft-tissue sarcoma patients in the competing risks setting. Cancer 2014, 120, 3361–3369. [Google Scholar] [CrossRef] [Green Version]
- Prehn, R.T.; Main, J.M. Immunity to methylcholanthrene-induced sarcomas. J. Natl. Cancer Inst. 1957, 18, 769–778. [Google Scholar]
- Howie, S.; McBride, W.H. Tumor-specific T helper activity can be abrogated by two distinct suppressor cell mechanisms. Eur. J. Immunol. 1982, 12, 671–675. [Google Scholar] [CrossRef] [PubMed]
- McBride, W.H.; Howie, S. Paradoxical presence of T cell anergy during successful T cell-dependent tumour immunotherapy: Characterization of a state of T cell ‘amnaesia’ following systemic administration of C. parvum. Clin. Exp. Immunol. 1984, 57, 139–148. [Google Scholar] [PubMed]
- Peters, L.J.; McBride, W.H.; Mason, K.A.; Milas, L. A role for T lymphocytes in tumour inhibition and enhancement caused by systemic administration of Corynebacterium parvum. J. Reticuloendothel. Soc. 1978, 24, 9–18. [Google Scholar] [PubMed]
- Ben-Ami, E.; Barysauskas, C.M.; Solomon, S.; Tahlil, K.; Malley, R.; Hohos, M.; Polson, K.; Loucks, M.; Severgnini, M.; Patel, T.; et al. Immunotherapy with single agent nivolumab for advanced leiomyosarcoma of the uterus: Results of a phase 2 study. Cancer 2017, 123, 3285–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- Sharma, A.; Bode, B.; Studer, G.; Moch, H.; Okoniewski, M.; Knuth, A.; von Boehmer, L.; van den Broek, M. Radiotherapy of human sarcoma promotes an intratumoral immune effector signature. Clin. Cancer Res. 2013, 19, 4843–4853. [Google Scholar] [CrossRef] [Green Version]
- Luke, J.J.; Lemons, J.M.; Karrison, T.G.; Pitroda, S.P.; Melotek, J.M.; Zha, Y.; Al-Hallaq, H.A.; Arina, A.; Khodarev, N.N.; Janisch, L.; et al. Safety and Clinical Activity of Pembrolizumab and Multisite Stereotactic Body Radiotherapy in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 1611–1618. [Google Scholar] [CrossRef]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef]
- McBride, W.H.; Chiang, C.S.; Olson, J.L.; Wang, C.C.; Hong, J.H.; Pajonk, F.; Dougherty, G.J.; Iwamoto, K.S.; Pervan, M.; Liao, Y.P. A sense of danger from radiation. Radiat. Res. 2004, 162, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Schoenhals, J.E.; Li, A.; Valdecanas, D.R.; Ye, H.; Zang, F.; Tang, C.; Tang, M.; Liu, C.-G.; Liu, X.; et al. Suppression of Type I IFN Signaling in Tumors Mediates Resistance to Anti-PD-1 Treatment That Can Be Overcome by Radiotherapy. Cancer Res. 2017, 77, 839–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, S.; Wang, B.; Kawashima, N.; Braunstein, S.; Badura, M.; Cameron, T.O.; Babb, J.S.; Schneider, R.J.; Formenti, S.C.; Dustin, M.L.; et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J. Immunol. 2008, 181, 3099–3107. [Google Scholar] [CrossRef]
- Chakraborty, M.; Abrams, S.I.; Camphausen, K.; Liu, K.; Scott, T.; Coleman, C.N.; Hodge, J.W. Irradiation of tumor cells up-regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J. Immunol. 2003, 170, 6338–6347. [Google Scholar] [CrossRef] [Green Version]
- Kalbasi, A.; June, C.H.; Haas, N.; Vapiwala, N. Radiation and immunotherapy: A synergistic combination. J. Clin. Investig. 2013, 123, 2756–2763. [Google Scholar] [CrossRef]
- Demaria, S.; Kawashima, N.; Yang, A.M.; Devitt, M.L.; Babb, J.S.; Allison, J.P.; Formenti, S.C. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin. Cancer Res. 2005, 11, 728–734. [Google Scholar]
- Schaue, D.; Ratikan, J.A.; Iwamoto, K.S.; McBride, W.H. Maximizing tumor immunity with fractionated radiation. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, 1306–1310. [Google Scholar] [CrossRef] [Green Version]
- Vanpouille-Box, C.; Formenti, S.C.; Demaria, S. TREX1 dictates the immune fate of irradiated cancer cells. Oncoimmunology 2017, 6, e1339857. [Google Scholar] [CrossRef] [Green Version]
- Dovedi, S.J.; Adlard, A.L.; Lipowska-Bhalla, G.; McKenna, C.; Jones, S.; Cheadle, E.J.; Stratford, I.J.; Poon, E.; Morrow, M.; Stewart, R.; et al. Acquired resistance to fractionated radiotherapy can be overcome by concurrent PD-L1 blockade. Cancer Res. 2014, 74, 5458–5468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koseła-Paterczyk, H.; Szacht, M.; Morysiński, T.; Ługowska, I.; Dziewirski, W.; Falkowski, S.; Zdzienicki, M.; Pieńkowski, A.; Szamotulska, K.; Switaj, T.; et al. Preoperative hypofractionated radiotherapy in the treatment of localized soft tissue sarcomas. Eur. J. Surg. Oncol. 2014, 40, 1641–1647. [Google Scholar] [CrossRef] [PubMed]
- Kalbasi, A.; Kamrava, M.; Nelson, S.D.; Dry, S.M.; Hernandez, J.; Chmielowski, B.; Singh, A.S.; Federman, N.C.; Bukata, S.V.; Bernthal, N.M.; et al. 5-Day Hypofractionated Preoperative Radiation Therapy in Soft Tissue Sarcoma: Preliminary Toxicity and Pathologic Outcomes from a Prospective Phase 2 Study. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, E753–E754. [Google Scholar] [CrossRef] [Green Version]
- Klein, G. Mechanisms of escape from immune surveillance. Natl. Cancer Inst. Monogr. 1976, 44, 135–136. [Google Scholar]
- Kolsch, E.; Mengersen, R. Low numbers of tumor cells suppress the host immune system. Adv. Exp. Med. Biol. 1976, 66, 431–436. [Google Scholar]
- Milas, L.; Hunter, N.; Mason, K.; Withers, H.R. Immunological resistance to pulmonary metastases in C3Hf-Bu mice bearing syngeneic fibrosarcoma of different sizes. Cancer Res. 1974, 34, 61–71. [Google Scholar]
- Suit, H.D.; Suchato, C. Hyperbaric oxygen and radiotherapy of a fibrosarcoma and of a squamous-cell carcinoma of C3H mice. Radiology 1967, 89, 713–719. [Google Scholar] [CrossRef]
- Allen, B.M.; Hiam, K.J.; Burnett, C.E.; Venida, A.; DeBarge, R.; Carmi, Y.; Spitzer, M.H. The Development, Function, and Plasticity of the Immune Macroenvironment in Cancer. bioRxiv 2019, 805473. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nesseler, J.P.; Lee, M.-H.; Nguyen, C.; Kalbasi, A.; Sayre, J.W.; Romero, T.; Nickers, P.; McBride, W.H.; Schaue, D. Tumor Size Matters—Understanding Concomitant Tumor Immunity in the Context of Hypofractionated Radiotherapy with Immunotherapy. Cancers 2020, 12, 714. https://doi.org/10.3390/cancers12030714
Nesseler JP, Lee M-H, Nguyen C, Kalbasi A, Sayre JW, Romero T, Nickers P, McBride WH, Schaue D. Tumor Size Matters—Understanding Concomitant Tumor Immunity in the Context of Hypofractionated Radiotherapy with Immunotherapy. Cancers. 2020; 12(3):714. https://doi.org/10.3390/cancers12030714
Chicago/Turabian StyleNesseler, Jean Philippe, Mi-Heon Lee, Christine Nguyen, Anusha Kalbasi, James W. Sayre, Tahmineh Romero, Philippe Nickers, William H. McBride, and Dörthe Schaue. 2020. "Tumor Size Matters—Understanding Concomitant Tumor Immunity in the Context of Hypofractionated Radiotherapy with Immunotherapy" Cancers 12, no. 3: 714. https://doi.org/10.3390/cancers12030714
APA StyleNesseler, J. P., Lee, M.-H., Nguyen, C., Kalbasi, A., Sayre, J. W., Romero, T., Nickers, P., McBride, W. H., & Schaue, D. (2020). Tumor Size Matters—Understanding Concomitant Tumor Immunity in the Context of Hypofractionated Radiotherapy with Immunotherapy. Cancers, 12(3), 714. https://doi.org/10.3390/cancers12030714