Adoptive Cell Therapy—Harnessing Antigen-Specific T Cells to Target Solid Tumours

 , , , , , and
, , , , , and 
Abstract
1. Introduction
2. TCR—Structure and Signalling
3. CAR—Structure and Signalling
4. Challenges for CAR T Cell Therapies of Solid Tumours
5. Antigens Used for T Cell-Based Therapy
6. Advantages of TCR vs. CAR T Cell Therapy
7. Strategies for Selection of T Cells for Cancer Therapy
7.1. Selected Ex Vivo Expanded Tumour-Reactive TILs vs. Unselected TILs After Shortened Expansion
7.2. Enrichment of T Cells Expressing Markers of Activation
7.3. Selection of TAA-Specific T cells
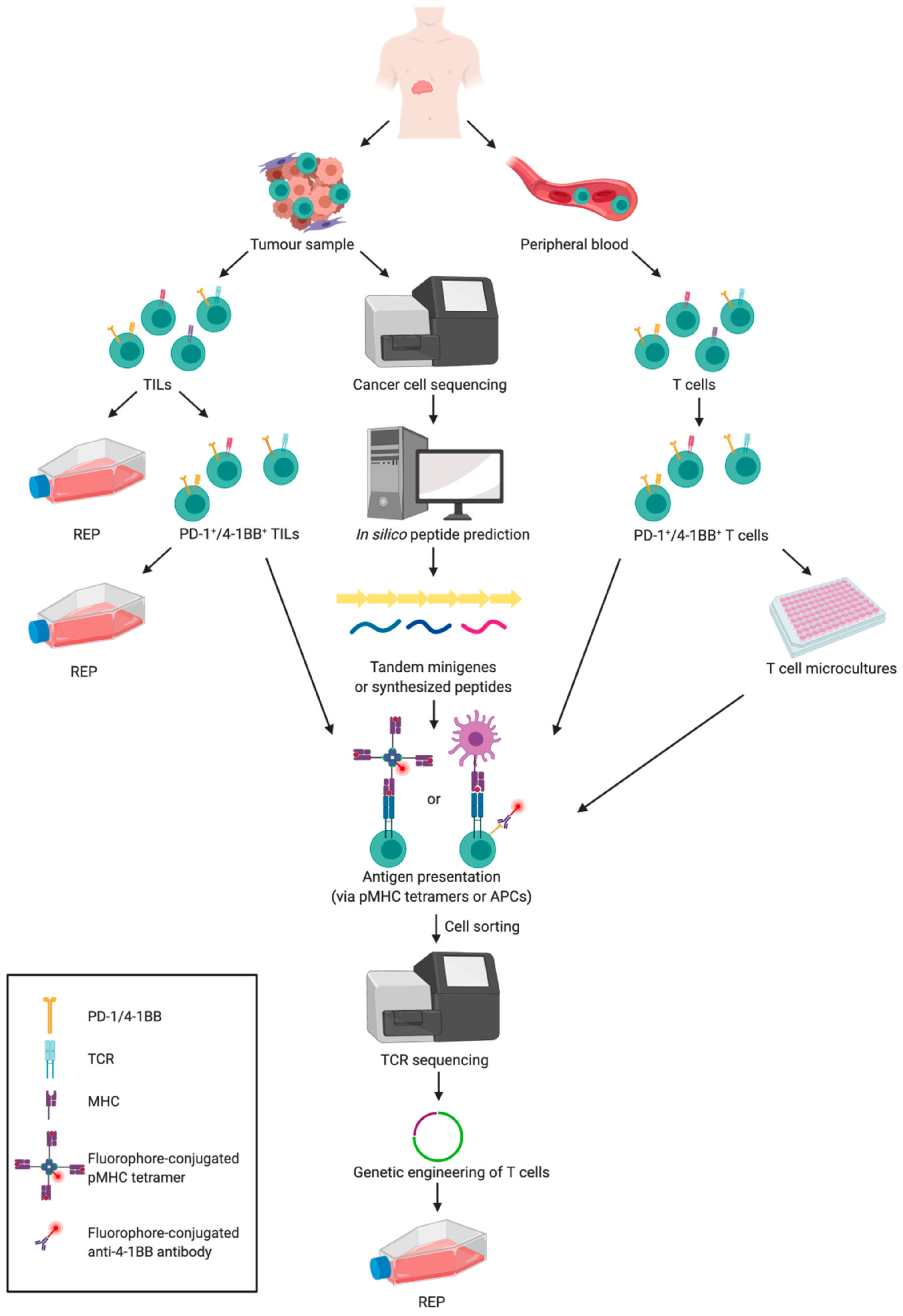
7.4. Selection of Neoantigen-Specific T Cells from the Tumour
7.5. Selection of Neoantigen-Specific T Cells from Peripheral Blood
7.6. Selection of T Cells Recognising Neoantigens with Driver Mutations
7.7. Genetic Engineering of T Cells to Express Specific TCR
8. Role of CD4+ Helper T Cells in an Anti-Tumour Response
9. MR1-Restricted T Cells—A Novel Population of T Cells with Pan-Cancer Therapeutic Potential
10. Choosing the Most Effective T Cell Differentiation State for ACT
11. Ex Vivo T Cells Expansion and Artificial Antigen-Presenting Systems
12. Improvements in Modulation of Anti-Tumour Potential of T Cells
12.1. Lymphodepletion
12.2. Stimulation of T Cells with Cytokines
12.3. Activating T Cell Co-Stimulatory Receptors
12.4. Targeting Immune Checkpoints
12.5. T Cell Therapies in Combination with Dendritic Cell-Based Vaccines
12.6. Enhancing Trafficking of T Cells to Solid Tumour Sites
12.7. Targeting T Cell Metabolism to Boost Their Anti-Tumour Potential
13. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Hochweller, K.; Wabnitz, G.H.; Samstag, Y.; Suffner, J.; Hämmerling, G.J.; Garbi, N. Dendritic cells control T cell tonic signaling required for responsiveness to foreign antigen. Proc. Natl. Acad. Sci. USA 2010, 107, 5931–5936. [Google Scholar] [CrossRef] [PubMed]
- Birnbaum, M.E.; Mendoza, J.L.; Sethi, D.K.; Dong, S.; Glanville, J.; Dobbins, J.; Ozkan, E.; Davis, M.M.; Wucherpfennig, K.W.; Garcia, K.C. Deconstructing the peptide-MHC specificity of T cell recognition. Cell 2014, 157, 1073–1087. [Google Scholar] [CrossRef] [PubMed]
- Gaud, G.; Lesourne, R.; Love, P.E. Regulatory mechanisms in T cell receptor signalling. Nat. Rev. Immunol. 2018, 18, 485–497. [Google Scholar] [CrossRef]
- Esin, S.; Shigematsu, M.; Nagai, S.; Eklund, A.; Wigzell, H.; Grunewald, J. Different percentages of peripheral blood gamma delta + T cells in healthy individuals from different areas of the world. Scand. J. Immunol. 1996, 43, 593–596. [Google Scholar] [CrossRef]
- Garcillán, B.; Marin, A.V.; Jiménez-Reinoso, A.; Briones, A.C.; Muñoz-Ruiz, M.; García-León, M.J.; Gil, J.; Allende, L.M.; Martínez-Naves, E.; Toribio, M.L.; et al. γδ T Lymphocytes in the Diagnosis of Human T Cell Receptor Immunodeficiencies. Front. Immunol. 2015, 6, 20. [Google Scholar] [CrossRef]
- Harris, D.T.; Kranz, D.M. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends Pharmacol. Sci. 2016, 37, 220–230. [Google Scholar] [CrossRef]
- Artyomov, M.N.; Lis, M.; Devadas, S.; Davis, M.M.; Chakraborty, A.K. CD4 and CD8 binding to MHC molecules primarily acts to enhance Lck delivery. Proc. Natl. Acad. Sci. USA 2010, 107, 16916–16921. [Google Scholar] [CrossRef]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef]
- Croft, M.; So, T.; Duan, W.; Soroosh, P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol. Rev. 2009, 229, 173–191. [Google Scholar] [CrossRef]
- Sanchez-Paulete, A.R.; Labiano, S.; Rodriguez-Ruiz, M.E.; Azpilikueta, A.; Etxeberria, I.; Bolaños, E.; Lang, V.; Rodriguez, M.; Aznar, M.A.; Jure-Kunkel, M.; et al. Deciphering CD137 (4-1BB) signaling in T-cell costimulation for translation into successful cancer immunotherapy. Eur. J. Immunol. 2016, 46, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Wikenheiser, D.J.; Stumhofer, J.S. ICOS Co-Stimulation: Friend or Foe? Front. Immunol. 2016, 7, 304. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Chandran, S.S.; Klebanoff, C.A. T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol. Rev. 2019, 290, 127–147. [Google Scholar] [CrossRef]
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T.; et al. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 2019, 25, 947–953. [Google Scholar] [CrossRef]
- Zheng, P.P.; Kros, J.M.; Li, J. Approved CAR T cell therapies: ice bucket challenges on glaring safety risks and long-term impacts. Drug Discov. Today 2018, 23, 1175–1182. [Google Scholar] [CrossRef]
- Friedman, K.M.; Garrett, T.E.; Evans, J.W.; Horton, H.M.; Latimer, H.J.; Seidel, S.L.; Horvath, C.J.; Morgan, R.A. Effective Targeting of Multiple B-Cell Maturation Antigen-Expressing Hematological Malignances by Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cells. Hum. Gene. Ther. 2018, 29, 585–601. [Google Scholar] [CrossRef]
- Smith, E.L.; Staehr, M.; Masakayan, R.; Tatake, I.J.; Purdon, T.J.; Wang, X.; Wang, P.; Liu, H.; Xu, Y.; Garrett-Thomson, S.C.; et al. Development and Evaluation of an Optimal Human Single-Chain Variable Fragment-Derived BCMA-Targeted CAR T Cell Vector. Mol. Ther. 2018, 26, 1447–1456. [Google Scholar] [CrossRef]
- Stamenkovic, I.; Seed, B. CD19, the earliest differentiation antigen of the B cell lineage, bears three extracellular immunoglobulin-like domains and an Epstein-Barr virus-related cytoplasmic tail. J. Exp. Med. 1988, 168, 1205–1210. [Google Scholar] [CrossRef]
- Wang, K.; Wei, G.; Liu, D. CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D.; June, C.H. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front. Immunol. 2018, 9, 2486. [Google Scholar] [CrossRef] [PubMed]
- DeRenzo, C.; Gottschalk, S. Genetic Modification Strategies to Enhance CAR T Cell Persistence for Patients With Solid Tumors. Front. Immunol. 2019, 10, 218. [Google Scholar] [CrossRef]
- Knochelmann, H.M.; Smith, A.S.; Dwyer, C.J.; Wyatt, M.M.; Mehrotra, S.; Paulos, C.M. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front. Immunol. 2018, 9, 1740. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, H.R.; Rodriguez, A.; Shepphird, J.; Brown, C.E.; Badie, B. Chimeric Antigen Receptors T Cell Therapy in Solid Tumor: Challenges and Clinical Applications. Front. Immunol. 2017, 8, 1850. [Google Scholar] [CrossRef] [PubMed]
- Craddock, J.A.; Lu, A.; Bear, A.; Pule, M.; Brenner, M.K.; Rooney, C.M.; Foster, A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010, 33, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Carpenito, C.; Sun, J.; Wang, L.C.; Kapoor, V.; Predina, J.; Powell, D.J.; Riley, J.L.; June, C.H.; Albelda, S.M. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011, 17, 4719–4730. [Google Scholar] [CrossRef] [PubMed]
- Schuberth, P.C.; Hagedorn, C.; Jensen, S.M.; Gulati, P.; van den Broek, M.; Mischo, A.; Soltermann, A.; Jüngel, A.; Marroquin Belaunzaran, O.; Stahel, R.; et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J. Transl. Med. 2013, 11, 187. [Google Scholar] [CrossRef]
- Tran, E.; Chinnasamy, D.; Yu, Z.; Morgan, R.A.; Lee, C.C.; Restifo, N.P.; Rosenberg, S.A. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J. Exp. Med. 2013, 210, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Priceman, S.J.; Tilakawardane, D.; Jeang, B.; Aguilar, B.; Murad, J.P.; Park, A.K.; Chang, W.C.; Ostberg, J.R.; Neman, J.; Jandial, R.; et al. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2. Clin. Cancer Res. 2018, 24, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Posey, A.D.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight. 2018, 3. [Google Scholar] [CrossRef]
- Zhao, Z.; Condomines, M.; van der Stegen, S.J.C.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Curran, K.J.; Seinstra, B.A.; Nikhamin, Y.; Yeh, R.; Usachenko, Y.; van Leeuwen, D.G.; Purdon, T.; Pegram, H.J.; Brentjens, R.J. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol. Ther. 2015, 23, 769–778. [Google Scholar] [CrossRef]
- Foster, A.E.; Mahendravada, A.; Shinners, N.P.; Chang, W.C.; Crisostomo, J.; Lu, A.; Khalil, M.; Morschl, E.; Shaw, J.L.; Saha, S.; et al. Regulated Expansion and Survival of Chimeric Antigen Receptor-Modified T Cells Using Small Molecule-Dependent Inducible MyD88/CD40. Mol. Ther. 2017, 25, 2176–2188. [Google Scholar] [CrossRef]
- Mata, M.; Gerken, C.; Nguyen, P.; Krenciute, G.; Spencer, D.M.; Gottschalk, S. Inducible Activation of MyD88 and CD40 in CAR T Cells Results in Controllable and Potent Antitumor Activity in Preclinical Solid Tumor Models. Cancer Discov. 2017, 7, 1306–1319. [Google Scholar] [CrossRef]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.A.; et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797. [Google Scholar] [CrossRef]
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.F.; Liu, H.; Dotti, G.; Balyasnikova, I.V.; Gottschalk, S. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Rα2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol. Res. 2017, 5, 571–581. [Google Scholar] [CrossRef]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Morsut, L.; Roybal, K.T.; Xiong, X.; Gordley, R.M.; Coyle, S.M.; Thomson, M.; Lim, W.A. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 2016, 164, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Roybal, K.T.; Williams, J.Z.; Morsut, L.; Rupp, L.J.; Kolinko, I.; Choe, J.H.; Walker, W.J.; McNally, K.A.; Lim, W.A. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 2016, 167, 419–432.e416. [Google Scholar] [CrossRef] [PubMed]
- Roybal, K.T.; Rupp, L.J.; Morsut, L.; Walker, W.J.; McNally, K.A.; Park, J.S.; Lim, W.A. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016, 164, 770–779. [Google Scholar] [CrossRef]
- Park, S.; Shevlin, E.; Vedvyas, Y.; Zaman, M.; Hsu, Y.S.; Min, I.M.; Jin, M.M. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci. Rep. 2017, 7, 14366. [Google Scholar] [CrossRef]
- Johnson, L.A.; Scholler, J.; Ohkuri, T.; Kosaka, A.; Patel, P.R.; McGettigan, S.E.; Nace, A.K.; Dentchev, T.; Thekkat, P.; Loew, A.; et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 2015, 7, 275ra222. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Posey, A.D.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454. [Google Scholar] [CrossRef]
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866. [Google Scholar] [CrossRef]
- Mohammed, S.; Sukumaran, S.; Bajgain, P.; Watanabe, N.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Fisher, W.E.; Leen, A.M.; Vera, J.F. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol. Ther. 2017, 25, 249–258. [Google Scholar] [CrossRef]
- Bajgain, P.; Tawinwung, S.; D’Elia, L.; Sukumaran, S.; Watanabe, N.; Hoyos, V.; Lulla, P.; Brenner, M.K.; Leen, A.M.; Vera, J.F. CAR T cell therapy for breast cancer: harnessing the tumor milieu to drive T cell activation. J. Immunother. Cancer 2018, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 2017, 7, 39833. [Google Scholar] [CrossRef] [PubMed]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Henderson, M.A.; Giuffrida, L.; Mills, J.K.; Sek, K.; Cross, R.S.; Davenport, A.J.; John, L.B.; Mardiana, S.; Slaney, C.Y.; et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J. Clin. Investig. 2017, 127, 929–941. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef]
- Bielamowicz, K.; Fousek, K.; Byrd, T.T.; Samaha, H.; Mukherjee, M.; Aware, N.; Wu, M.F.; Orange, J.S.; Sumazin, P.; Man, T.K.; et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018, 20, 506–518. [Google Scholar] [CrossRef]
- Caruso, H.; Heimberger, A.B. Comment on “Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma”. Neuro Oncol. 2018, 20, 1003–1004. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Richman, S.A.; Nunez-Cruz, S.; Moghimi, B.; Li, L.Z.; Gershenson, Z.T.; Mourelatos, Z.; Barrett, D.M.; Grupp, S.A.; Milone, M.C. High-Affinity GD2-Specific CAR T Cells Induce Fatal Encephalitis in a Preclinical Neuroblastoma Model. Cancer Immunol. Res. 2018, 6, 36–46. [Google Scholar] [CrossRef]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics. 2016, 3, 16011. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Mullins, C.S.; Linnebacher, M. Colorectal cancer vaccines: Tumor-associated antigens. World J. Gastroenterol. 2018, 24, 5418–5432. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: from basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Zamora, A.E.; Crawford, J.C.; Thomas, P.G. Hitting the Target: How T Cells Detect and Eliminate Tumors. J. Immunol. 2018, 200, 392–399. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L. The Emerging World of TCR-T Cell Trials Against Cancer: A Systematic Review. Technol. Cancer Res. Treat. 2019, 18, 1533033819831068. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Shen, L.; Zhang, J.; Lee, H.; Batista, M.T.; Johnston, S.A. RNA Transcription and Splicing Errors as a Source of Cancer Frameshift Neoantigens for Vaccines. Sci. Rep. 2019, 9, 14184. [Google Scholar] [CrossRef]
- Timmers, M.; Roex, G.; Wang, Y.; Campillo-Davo, D.; Van Tendeloo, V.F.I.; Chu, Y.; Berneman, Z.N.; Luo, F.; Van Acker, H.H.; Anguille, S. Chimeric Antigen Receptor-Modified T Cell Therapy in Multiple Myeloma: Beyond B Cell Maturation Antigen. Front. Immunol. 2019, 10, 1613. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.D.; Harris, D.T.; Soto, C.M.; Chervin, A.S.; Aggen, D.H.; Roy, E.J.; Kranz, D.M. A novel T cell receptor single-chain signaling complex mediates antigen-specific T cell activity and tumor control. Cancer Immunol. Immunother. 2014, 63, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yang, Z.; Horan, L.H.; Zhang, P.; Liu, L.; Zimdahl, B.; Green, S.; Lu, J.; Morales, J.F.; Barrett, D.M.; et al. A novel antibody-TCR (AbTCR) platform combines Fab-based antigen recognition with gamma/delta-TCR signaling to facilitate T-cell cytotoxicity with low cytokine release. Cell Discov. 2018, 4, 62. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J. Natl. Cancer. Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Shelton, T.E.; Even, J.; Rosenberg, S.A. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J. Immunother. 2003, 26, 332–342. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Restifo, N.P.; Royal, R.E.; Kammula, U.; White, D.E.; Mavroukakis, S.A.; et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 2005, 23, 2346–2357. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Hughes, M.S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; Wunderlich, J.R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 2011, 17, 4550–4557. [Google Scholar] [CrossRef]
- Tran, K.Q.; Zhou, J.; Durflinger, K.H.; Langhan, M.M.; Shelton, T.E.; Wunderlich, J.R.; Robbins, P.F.; Rosenberg, S.A.; Dudley, M.E. Minimally cultured tumor-infiltrating lymphocytes display optimal characteristics for adoptive cell therapy. J. Immunother. 2008, 31, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Gross, C.A.; Langhan, M.M.; Garcia, M.R.; Sherry, R.M.; Yang, J.C.; Phan, G.Q.; Kammula, U.S.; Hughes, M.S.; Citrin, D.E.; et al. CD8+ enriched "young" tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin. Cancer Res. 2010, 16, 6122–6131. [Google Scholar] [CrossRef] [PubMed]
- Prieto, P.A.; Durflinger, K.H.; Wunderlich, J.R.; Rosenberg, S.A.; Dudley, M.E. Enrichment of CD8+ cells from melanoma tumor-infiltrating lymphocyte cultures reveals tumor reactivity for use in adoptive cell therapy. J. Immunother. 2010, 33, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, J.; Yamazaki, S.; Sakaguchi, S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J. Immunol. 1999, 163, 5211–5218. [Google Scholar]
- Darrasse-Jèze, G.; Podsypanina, K. How numbers, nature, and immune status of foxp3(+) regulatory T-cells shape the early immunological events in tumor development. Front. Immunol. 2013, 4, 292. [Google Scholar] [CrossRef]
- Marek-Trzonkowska, N.; Piekarska, K.; Filipowicz, N.; Piotrowski, A.; Gucwa, M.; Vogt, K.; Sawitzki, B.; Siebert, J.; Trzonkowski, P. Mild hypothermia provides Treg stability. Sci. Rep. 2017, 7, 11915. [Google Scholar] [CrossRef]
- Marek, N.; Bieniaszewska, M.; Krzystyniak, A.; Juścińska, J.; Myśliwska, J.; Witkowski, P.; Hellmann, A.; Trzonkowski, P. The time is crucial for ex vivo expansion of T regulatory cells for therapy. Cell Transplant. 2011, 20, 1747–1758. [Google Scholar] [CrossRef]
- Itzhaki, O.; Hovav, E.; Ziporen, Y.; Levy, D.; Kubi, A.; Zikich, D.; Hershkovitz, L.; Treves, A.J.; Shalmon, B.; Zippel, D.; et al. Establishment and large-scale expansion of minimally cultured "young" tumor infiltrating lymphocytes for adoptive transfer therapy. J. Immunother. 2011, 34, 212–220. [Google Scholar] [CrossRef]
- Besser, M.J.; Shapira-Frommer, R.; Treves, A.J.; Zippel, D.; Itzhaki, O.; Schallmach, E.; Kubi, A.; Shalmon, B.; Hardan, I.; Catane, R.; et al. Minimally cultured or selected autologous tumor-infiltrating lymphocytes after a lympho-depleting chemotherapy regimen in metastatic melanoma patients. J. Immunother. 2009, 32, 415–423. [Google Scholar] [CrossRef]
- Pilon-Thomas, S.; Kuhn, L.; Ellwanger, S.; Janssen, W.; Royster, E.; Marzban, S.; Kudchadkar, R.; Zager, J.; Gibney, G.; Sondak, V.K.; et al. Efficacy of adoptive cell transfer of tumor-infiltrating lymphocytes after lymphopenia induction for metastatic melanoma. J. Immunother. 2012, 35, 615–620. [Google Scholar] [CrossRef]
- Radvanyi, L.G.; Bernatchez, C.; Zhang, M.; Fox, P.S.; Miller, P.; Chacon, J.; Wu, R.; Lizee, G.; Mahoney, S.; Alvarado, G.; et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin. Cancer Res. 2012, 18, 6758–6770. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.; Donia, M.; Ellebaek, E.; Borch, T.H.; Kongsted, P.; Iversen, T.Z.; Hölmich, L.R.; Hendel, H.W.; Met, Ö.; Andersen, M.H.; et al. Long-Lasting Complete Responses in Patients with Metastatic Melanoma after Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes and an Attenuated IL2 Regimen. Clin. Cancer Res. 2016, 22, 3734–3745. [Google Scholar] [CrossRef] [PubMed]
- Mullinax, J.E.; Hall, M.; Prabhakaran, S.; Weber, J.; Khushalani, N.; Eroglu, Z.; Brohl, A.S.; Markowitz, J.; Royster, E.; Richards, A.; et al. Combination of Ipilimumab and Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes for Patients with Metastatic Melanoma. Front. Oncol. 2018, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.; Westergaard, M.C.W.; Milne, K.; Nielsen, M.; Borch, T.H.; Poulsen, L.G.; Hendel, H.W.; Kennedy, M.; Briggs, G.; Ledoux, S.; et al. Adoptive cell therapy with tumor-infiltrating lymphocytes in patients with metastatic ovarian cancer: a pilot study. Oncoimmunology 2018, 7, e1502905. [Google Scholar] [CrossRef]
- Westergaard, M.C.W.; Andersen, R.; Chong, C.; Kjeldsen, J.W.; Pedersen, M.; Friese, C.; Hasselager, T.; Lajer, H.; Coukos, G.; Bassani-Sternberg, M.; et al. Tumour-reactive T cell subsets in the microenvironment of ovarian cancer. Br. J. Cancer 2019, 120, 424–434. [Google Scholar] [CrossRef]
- Andersen, R.; Westergaard, M.C.W.; Kjeldsen, J.W.; Müller, A.; Pedersen, N.W.; Hadrup, S.R.; Met, Ö.; Seliger, B.; Kromann-Andersen, B.; Hasselager, T.; et al. T-cell Responses in the Microenvironment of Primary Renal Cell Carcinoma-Implications for Adoptive Cell Therapy. Cancer Immunol. Res. 2018, 6, 222–235. [Google Scholar] [CrossRef]
- Turcotte, S.; Gros, A.; Hogan, K.; Tran, E.; Hinrichs, C.S.; Wunderlich, J.R.; Dudley, M.E.; Rosenberg, S.A. Phenotype and function of T cells infiltrating visceral metastases from gastrointestinal cancers and melanoma: Implications for adoptive cell transfer therapy. J. Immunol. 2013, 191, 2217–2225. [Google Scholar] [CrossRef]
- Wolfl, M.; Kuball, J.; Ho, W.Y.; Nguyen, H.; Manley, T.J.; Bleakley, M.; Greenberg, P.D. Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities. Blood 2007, 110, 201–210. [Google Scholar] [CrossRef]
- Ye, Q.; Song, D.G.; Poussin, M.; Yamamoto, T.; Best, A.; Li, C.; Coukos, G.; Powell, D.J. CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor. Clin. Cancer Res. 2014, 20, 44–55. [Google Scholar] [CrossRef]
- Seliktar-Ofir, S.; Merhavi-Shoham, E.; Itzhaki, O.; Yunger, S.; Markel, G.; Schachter, J.; Besser, M.J. Selection of Shared and Neoantigen-Reactive T Cells for Adoptive Cell Therapy Based on CD137 Separation. Front. Immunol. 2017, 8, 1211. [Google Scholar] [CrossRef]
- Inozume, T.; Hanada, K.; Wang, Q.J.; Ahmadzadeh, M.; Wunderlich, J.R.; Rosenberg, S.A.; Yang, J.C. Selection of CD8+PD-1+ lymphocytes in fresh human melanomas enriches for tumor-reactive T cells. J. Immunother. 2010, 33, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. PD-1 identifies the patient-specific CD8⁺ tumor-reactive repertoire infiltrating human tumors. J. Clin. Investig. 2014, 124, 2246–2259. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Poma, S.M.; Salas-Benito, D.; Lozano, T.; Casares, N.; Riezu-Boj, J.I.; Mancheño, U.; Elizalde, E.; Alignani, D.; Zubeldia, N.; Otano, I.; et al. Expansion of Tumor-Infiltrating CD8. Cancer Res. 2017, 77, 3672–3684. [Google Scholar] [CrossRef] [PubMed]
- Yee, C.; Thompson, J.A.; Byrd, D.; Riddell, S.R.; Roche, P.; Celis, E.; Greenberg, P.D. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc. Natl. Acad. Sci. USA 2002, 99, 16168–16173. [Google Scholar] [CrossRef] [PubMed]
- Mackensen, A.; Meidenbauer, N.; Vogl, S.; Laumer, M.; Berger, J.; Andreesen, R. Phase I study of adoptive T-cell therapy using antigen-specific CD8+ T cells for the treatment of patients with metastatic melanoma. J. Clin. Oncol. 2006, 24, 5060–5069. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; El-Gamil, M.; Dudley, M.E.; Li, Y.F.; Rosenberg, S.A.; Robbins, P.F. T cells associated with tumor regression recognize frameshifted products of the CDKN2A tumor suppressor gene locus and a mutated HLA class I gene product. J. Immunol. 2004, 172, 6057–6064. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Yao, X.; Li, Y.F.; El-Gamil, M.; Dudley, M.E.; Yang, J.C.; Almeida, J.R.; Douek, D.C.; Samuels, Y.; Rosenberg, S.A.; et al. Mutated PPP1R3B is recognized by T cells used to treat a melanoma patient who experienced a durable complete tumor regression. J. Immunol. 2013, 190, 6034–6042. [Google Scholar] [CrossRef]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef]
- Gfeller, D.; Bassani-Sternberg, M. Predicting Antigen Presentation-What Could We Learn From a Million Peptides? Front. Immunol. 2018, 9, 1716. [Google Scholar] [CrossRef]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Yao, X.; Crystal, J.S.; Li, Y.F.; El-Gamil, M.; Gross, C.; Davis, L.; Dudley, M.E.; Yang, J.C.; Samuels, Y.; et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res. 2014, 20, 3401–3410. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.D.; Moss, P.A.; Goulder, P.J.; Barouch, D.H.; McHeyzer-Williams, M.G.; Bell, J.I.; McMichael, A.J.; Davis, M.M. Phenotypic analysis of antigen-specific T lymphocytes. Science 1996, 274, 94–96. [Google Scholar] [CrossRef] [PubMed]
- Rodenko, B.; Toebes, M.; Hadrup, S.R.; van Esch, W.J.; Molenaar, A.M.; Schumacher, T.N.; Ovaa, H. Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat. Protoc. 2006, 1, 1120–1132. [Google Scholar] [CrossRef] [PubMed]
- Newell, E.W.; Klein, L.O.; Yu, W.; Davis, M.M. Simultaneous detection of many T-cell specificities using combinatorial tetramer staining. Nat. Methods 2009, 6, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Newell, E.W.; Sigal, N.; Nair, N.; Kidd, B.A.; Greenberg, H.B.; Davis, M.M. Combinatorial tetramer staining and mass cytometry analysis facilitate T-cell epitope mapping and characterization. Nat. Biotechnol. 2013, 31, 623–629. [Google Scholar] [CrossRef]
- Bentzen, A.K.; Marquard, A.M.; Lyngaa, R.; Saini, S.K.; Ramskov, S.; Donia, M.; Such, L.; Furness, A.J.; McGranahan, N.; Rosenthal, R.; et al. Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat. Biotechnol. 2016, 34, 1037–1045. [Google Scholar] [CrossRef]
- Zhang, S.Q.; Ma, K.Y.; Schonnesen, A.A.; Zhang, M.; He, C.; Sun, E.; Williams, C.M.; Jia, W.; Jiang, N. High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat. Biotechnol. 2018. [Google Scholar] [CrossRef]
- Fehlings, M.; Jhunjhunwala, S.; Kowanetz, M.; O’Gorman, W.E.; Hegde, P.S.; Sumatoh, H.; Lee, B.H.; Nardin, A.; Becht, E.; Flynn, S.; et al. Late-differentiated effector neoantigen-specific CD8+ T cells are enriched in peripheral blood of non-small cell lung carcinoma patients responding to atezolizumab treatment. J. Immunother. Cancer 2019, 7, 249. [Google Scholar] [CrossRef]
- Veatch, J.R.; Jesernig, B.L.; Kargl, J.; Fitzgibbon, M.; Lee, S.M.; Baik, C.; Martins, R.; Houghton, A.M.; Riddell, S.R. Endogenous CD4 + T Cells Recognize Neoantigens in Lung Cancer Patients, Including Recurrent Oncogenic KRAS and ERBB2 (Her2) Driver Mutations. Cancer Immunol. Res. 2019, 7, 910–922. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Robbins, P.F.; Lu, Y.C.; Prickett, T.D.; Gartner, J.J.; Jia, L.; Pasetto, A.; Zheng, Z.; Ray, S.; Groh, E.M.; et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 2016, 375, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Bobisse, S.; Genolet, R.; Roberti, A.; Tanyi, J.L.; Racle, J.; Stevenson, B.J.; Iseli, C.; Michel, A.; Le Bitoux, M.A.; Guillaume, P.; et al. Sensitive and frequent identification of high avidity neo-epitope specific CD8. Nat. Commun. 2018, 9, 1092. [Google Scholar] [CrossRef]
- Deniger, D.C.; Pasetto, A.; Robbins, P.F.; Gartner, J.J.; Prickett, T.D.; Paria, B.C.; Malekzadeh, P.; Jia, L.; Yossef, R.; Langhan, M.M.; et al. T-cell Responses to TP53 "Hotspot" Mutations and Unique Neoantigens Expressed by Human Ovarian Cancers. Clin. Cancer Res. 2018, 24, 5562–5573. [Google Scholar] [CrossRef]
- Liu, S.; Matsuzaki, J.; Wei, L.; Tsuji, T.; Battaglia, S.; Hu, Q.; Cortes, E.; Wong, L.; Yan, L.; Long, M.; et al. Efficient identification of neoantigen-specific T-cell responses in advanced human ovarian cancer. J. Immunother. Cancer 2019, 7, 156. [Google Scholar] [CrossRef]
- Zacharakis, N.; Chinnasamy, H.; Black, M.; Xu, H.; Lu, Y.C.; Zheng, Z.; Pasetto, A.; Langhan, M.; Shelton, T.; Prickett, T.; et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 2018, 24, 724–730. [Google Scholar] [CrossRef]
- Meng, Q.; Valentini, D.; Rao, M.; Moro, C.F.; Paraschoudi, G.; Jäger, E.; Dodoo, E.; Rangelova, E.; Del Chiaro, M.; Maeurer, M. Neoepitope targets of tumour-infiltrating lymphocytes from patients with pancreatic cancer. Br. J. Cancer 2019, 120, 97–108. [Google Scholar] [CrossRef]
- Scheper, W.; Kelderman, S.; Fanchi, L.F.; Linnemann, C.; Bendle, G.; de Rooij, M.A.J.; Hirt, C.; Mezzadra, R.; Slagter, M.; Dijkstra, K.; et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat. Med. 2019, 25, 89–94. [Google Scholar] [CrossRef]
- Cohen, C.J.; Gartner, J.J.; Horovitz-Fried, M.; Shamalov, K.; Trebska-McGowan, K.; Bliskovsky, V.V.; Parkhurst, M.R.; Ankri, C.; Prickett, T.D.; Crystal, J.S.; et al. Isolation of neoantigen-specific T cells from tumor and peripheral lymphocytes. J. Clin. Investig. 2015, 125, 3981–3991. [Google Scholar] [CrossRef]
- Gros, A.; Parkhurst, M.R.; Tran, E.; Pasetto, A.; Robbins, P.F.; Ilyas, S.; Prickett, T.D.; Gartner, J.J.; Crystal, J.S.; Roberts, I.M.; et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 2016, 22, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.D.; Wick, D.A.; Nielsen, J.S.; Little, N.; Holt, R.A.; Nelson, B.H. A library-based screening method identifies neoantigen-reactive T cells in peripheral blood prior to relapse of ovarian cancer. Oncoimmunology 2017, 7, e1371895. [Google Scholar] [CrossRef] [PubMed]
- Gros, A.; Tran, E.; Parkhurst, M.R.; Ilyas, S.; Pasetto, A.; Groh, E.M.; Robbins, P.F.; Yossef, R.; Garcia-Garijo, A.; Fajardo, C.A.; et al. Recognition of human gastrointestinal cancer neoantigens by circulating PD-1+ lymphocytes. J. Clin. Investig. 2019, 129, 4992–5004. [Google Scholar] [CrossRef] [PubMed]
- Theaker, S.M.; Rius, C.; Greenshields-Watson, A.; Lloyd, A.; Trimby, A.; Fuller, A.; Miles, J.J.; Cole, D.K.; Peakman, M.; Sewell, A.K.; et al. T-cell libraries allow simple parallel generation of multiple peptide-specific human T-cell clones. J. Immunol. Methods 2016, 430, 43–50. [Google Scholar] [CrossRef]
- Yossef, R.; Tran, E.; Deniger, D.C.; Gros, A.; Pasetto, A.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Cafri, G.; Robbins, P.F.; et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight. 2018, 3. [Google Scholar] [CrossRef]
- Cafri, G.; Yossef, R.; Pasetto, A.; Deniger, D.C.; Lu, Y.C.; Parkhurst, M.; Gartner, J.J.; Jia, L.; Ray, S.; Ngo, L.T.; et al. Memory T cells targeting oncogenic mutations detected in peripheral blood of epithelial cancer patients. Nat. Commun. 2019, 10, 449. [Google Scholar] [CrossRef]
- Veatch, J.R.; Lee, S.M.; Fitzgibbon, M.; Chow, I.T.; Jesernig, B.; Schmitt, T.; Kong, Y.Y.; Kargl, J.; Houghton, A.M.; Thompson, J.A.; et al. Tumor-infiltrating BRAFV600E-specific CD4+ T cells correlated with complete clinical response in melanoma. J. Clin. Investig. 2018, 128, 1563–1568. [Google Scholar] [CrossRef]
- Chheda, Z.S.; Kohanbash, G.; Okada, K.; Jahan, N.; Sidney, J.; Pecoraro, M.; Yang, X.; Carrera, D.A.; Downey, K.M.; Shrivastav, S.; et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J. Exp. Med. 2018, 215, 141–157. [Google Scholar] [CrossRef]
- Lo, W.; Parkhurst, M.; Robbins, P.F.; Tran, E.; Lu, Y.C.; Jia, L.; Gartner, J.J.; Pasetto, A.; Deniger, D.; Malekzadeh, P.; et al. Immunologic Recognition of a Shared p53 Mutated Neoantigen in a Patient with Metastatic Colorectal Cancer. Cancer Immunol. Res. 2019, 7, 534–543. [Google Scholar] [CrossRef]
- Malekzadeh, P.; Pasetto, A.; Robbins, P.F.; Parkhurst, M.R.; Paria, B.C.; Jia, L.; Gartner, J.J.; Hill, V.; Yu, Z.; Restifo, N.P.; et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J. Clin. Investig. 2019, 129, 1109–1114. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Kuball, J.; Dossett, M.L.; Wolfl, M.; Ho, W.Y.; Voss, R.H.; Fowler, C.; Greenberg, P.D. Facilitating matched pairing and expression of TCR chains introduced into human T cells. Blood 2007, 109, 2331–2338. [Google Scholar] [CrossRef] [PubMed]
- Bendle, G.M.; Linnemann, C.; Hooijkaas, A.I.; Bies, L.; de Witte, M.A.; Jorritsma, A.; Kaiser, A.D.; Pouw, N.; Debets, R.; Kieback, E.; et al. Lethal graft-versus-host disease in mouse models of T cell receptor gene therapy. Nat. Med. 2010, 16, 565–570, 561p following 570. [Google Scholar] [CrossRef] [PubMed]
- van Loenen, M.M.; de Boer, R.; Amir, A.L.; Hagedoorn, R.S.; Volbeda, G.L.; Willemze, R.; van Rood, J.J.; Falkenburg, J.H.; Heemskerk, M.H. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc. Natl. Acad. Sci. USA 2010, 107, 10972–10977. [Google Scholar] [CrossRef] [PubMed]
- Leisegang, M.; Engels, B.; Meyerhuber, P.; Kieback, E.; Sommermeyer, D.; Xue, S.A.; Reuss, S.; Stauss, H.; Uckert, W. Enhanced functionality of T cell receptor-redirected T cells is defined by the transgene cassette. J. Mol. Med. (Berl.) 2008, 86, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Legut, M.; Dolton, G.; Mian, A.A.; Ottmann, O.G.; Sewell, A.K. CRISPR-mediated TCR replacement generates superior anticancer transgenic T cells. Blood 2018, 131, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409. [Google Scholar] [CrossRef]
- Albers, J.J.; Ammon, T.; Gosmann, D.; Audehm, S.; Thoene, S.; Winter, C.; Secci, R.; Wolf, A.; Stelzl, A.; Steiger, K.; et al. Gene editing enables T-cell engineering to redirect antigen specificity for potent tumor rejection. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Garber, K. Driving T-cell immunotherapy to solid tumors. Nat. Biotechnol. 2018, 36, 215–219. [Google Scholar] [CrossRef]
- Kennedy, R.; Celis, E. Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol. Rev. 2008, 222, 129–144. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.M.; Kastenmüller, W. CD4 + T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Linnemann, C.; van Buuren, M.M.; Bies, L.; Verdegaal, E.M.; Schotte, R.; Calis, J.J.; Behjati, S.; Velds, A.; Hilkmann, H.; Atmioui, D.E.; et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 2015, 21, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Alspach, E.; Lussier, D.M.; Miceli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, J.; Tsuji, T.; Luescher, I.F.; Shiku, H.; Mineno, J.; Okamoto, S.; Old, L.J.; Shrikant, P.; Gnjatic, S.; Odunsi, K. Direct tumor recognition by a human CD4(+) T-cell subset potently mediates tumor growth inhibition and orchestrates anti-tumor immune responses. Sci. Rep. 2015, 5, 14896. [Google Scholar] [CrossRef] [PubMed]
- Abelin, J.G.; Harjanto, D.; Malloy, M.; Suri, P.; Colson, T.; Goulding, S.P.; Creech, A.L.; Serrano, L.R.; Nasir, G.; Nasrullah, Y.; et al. Defining HLA-II Ligand Processing and Binding Rules with Mass Spectrometry Enhances Cancer Epitope Prediction. Immunity 2019, 51, 766–779.e717. [Google Scholar] [CrossRef] [PubMed]
- Moeller, M.; Haynes, N.M.; Kershaw, M.H.; Jackson, J.T.; Teng, M.W.; Street, S.E.; Cerutti, L.; Jane, S.M.; Trapani, J.A.; Smyth, M.J.; et al. Adoptive transfer of gene-engineered CD4+ helper T cells induces potent primary and secondary tumor rejection. Blood 2005, 106, 2995–3003. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.X.; Shu, S.; Disis, M.L.; Plautz, G.E. Adoptive transfer of tumor-primed, in vitro-activated, CD4+ T effector cells (TEs) combined with CD8+ TEs provides intratumoral TE proliferation and synergistic antitumor response. Blood 2007, 109, 4865–4876. [Google Scholar] [CrossRef]
- Hunder, N.N.; Wallen, H.; Cao, J.; Hendricks, D.W.; Reilly, J.Z.; Rodmyre, R.; Jungbluth, A.; Gnjatic, S.; Thompson, J.A.; Yee, C. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N. Engl. J. Med. 2008, 358, 2698–2703. [Google Scholar] [CrossRef]
- Crowther, M.D.; Dolton, G.; Legut, M.; Caillaud, M.E.; Lloyd, A.; Attaf, M.; Galloway, S.A.E.; Rius, C.; Farrell, C.P.; Szomolay, B.; et al. Genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat. Immunol. 2020, 21, 178–185. [Google Scholar] [CrossRef]
- Gold, M.C.; Cerri, S.; Smyk-Pearson, S.; Cansler, M.E.; Vogt, T.M.; Delepine, J.; Winata, E.; Swarbrick, G.M.; Chua, W.J.; Yu, Y.Y.; et al. Human mucosal associated invariant T cells detect bacterially infected cells. PLoS Biol. 2010, 8, e1000407. [Google Scholar] [CrossRef]
- Le Bourhis, L.; Dusseaux, M.; Bohineust, A.; Bessoles, S.; Martin, E.; Premel, V.; Coré, M.; Sleurs, D.; Serriari, N.E.; Treiner, E.; et al. MAIT cells detect and efficiently lyse bacterially-infected epithelial cells. PLoS Pathog. 2013, 9, e1003681. [Google Scholar] [CrossRef] [PubMed]
- Lepore, M.; Kalinichenko, A.; Calogero, S.; Kumar, P.; Paleja, B.; Schmaler, M.; Narang, V.; Zolezzi, F.; Poidinger, M.; Mori, L.; et al. Functionally diverse human T cells recognize non-microbial antigens presented by MR1. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Kaech, S.M.; Cui, W. Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 2012, 12, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Gattinoni, L.; Restifo, N.P. Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy? J. Immunother. 2012, 35, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Marek, N.; Myśliwiec, M.; Raczyńska, K.; Zorena, K.; Myśliwska, J.; Trzonkowski, P. Increased spontaneous production of VEGF by CD4+ T cells in type 1 diabetes. Clin. Immunol. 2010, 137, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Jensen, M.C.; Lansdorp, P.M.; Gough, M.; Elliott, C.; Riddell, S.R. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Investig. 2008, 118, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Gattinoni, L.; Torabi-Parizi, P.; Kerstann, K.; Cardones, A.R.; Finkelstein, S.E.; Palmer, D.C.; Antony, P.A.; Hwang, S.T.; Rosenberg, S.A.; et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9571–9576. [Google Scholar] [CrossRef]
- Zhou, J.; Shen, X.; Huang, J.; Hodes, R.J.; Rosenberg, S.A.; Robbins, P.F. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J. Immunol. 2005, 175, 7046–7052. [Google Scholar] [CrossRef]
- Hinrichs, C.S.; Borman, Z.A.; Cassard, L.; Gattinoni, L.; Spolski, R.; Yu, Z.; Sanchez-Perez, L.; Muranski, P.; Kern, S.J.; Logun, C.; et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc. Natl. Acad. Sci. USA 2009, 106, 17469–17474. [Google Scholar] [CrossRef]
- Wu, F.; Zhang, W.; Shao, H.; Bo, H.; Shen, H.; Li, J.; Liu, Y.; Wang, T.; Ma, W.; Huang, S. Human effector T cells derived from central memory cells rather than CD8(+)T cells modified by tumor-specific TCR gene transfer possess superior traits for adoptive immunotherapy. Cancer Lett. 2013, 339, 195–207. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Scott, C.D.; Leonardi, A.J.; Yamamoto, T.N.; Cruz, A.C.; Ouyang, C.; Ramaswamy, M.; Roychoudhuri, R.; Ji, Y.; Eil, R.L.; et al. Memory T cell-driven differentiation of naive cells impairs adoptive immunotherapy. J. Clin. Investig. 2016, 126, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Klebanoff, C.A.; Restifo, N.P. Paths to stemness: building the ultimate antitumour T cell. Nat. Rev. Cancer 2012, 12, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Speiser, D.E.; Lichterfeld, M.; Bonini, C. T memory stem cells in health and disease. Nat. Med. 2017, 23, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Cieri, N.; Camisa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Provasi, E.; Bondanza, A.; Bordignon, C.; Peccatori, J.; Ciceri, F.; et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 2013, 121, 573–584. [Google Scholar] [CrossRef]
- Levine, B.L.; Bernstein, W.B.; Connors, M.; Craighead, N.; Lindsten, T.; Thompson, C.B.; June, C.H. Effects of CD28 costimulation on long-term proliferation of CD4+ T cells in the absence of exogenous feeder cells. J. Immunol. 1997, 159, 5921–5930. [Google Scholar] [PubMed]
- Maus, M.V.; Thomas, A.K.; Leonard, D.G.; Allman, D.; Addya, K.; Schlienger, K.; Riley, J.L.; June, C.H. Ex vivo expansion of polyclonal and antigen-specific cytotoxic T lymphocytes by artificial APCs expressing ligands for the T-cell receptor, CD28 and 4-1BB. Nat. Biotechnol. 2002, 20, 143–148. [Google Scholar] [CrossRef]
- Butler, M.O.; Hirano, N. Human cell-based artificial antigen-presenting cells for cancer immunotherapy. Immunol. Rev. 2014, 257, 191–209. [Google Scholar] [CrossRef]
- Suhoski, M.M.; Golovina, T.N.; Aqui, N.A.; Tai, V.C.; Varela-Rohena, A.; Milone, M.C.; Carroll, R.G.; Riley, J.L.; June, C.H. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol. Ther. 2007, 15, 981–988. [Google Scholar] [CrossRef]
- Marek-Trzonkowska, N.; Myśliwiec, M.; Iwaszkiewicz-Grześ, D.; Gliwiński, M.; Derkowska, I.; Żalińska, M.; Zieliński, M.; Grabowska, M.; Zielińska, H.; Piekarska, K.; et al. Factors affecting long-term efficacy of T regulatory cell-based therapy in type 1 diabetes. J. Transl. Med. 2016, 14, 332. [Google Scholar] [CrossRef]
- Li, Y.; Kurlander, R.J. Comparison of anti-CD3 and anti-CD28-coated beads with soluble anti-CD3 for expanding human T cells: differing impact on CD8 T cell phenotype and responsiveness to restimulation. J. Transl. Med. 2010, 8, 104. [Google Scholar] [CrossRef] [PubMed]
- Kagoya, Y.; Nakatsugawa, M.; Ochi, T.; Cen, Y.; Guo, T.; Anczurowski, M.; Saso, K.; Butler, M.O.; Hirano, N. Transient stimulation expands superior antitumor T cells for adoptive therapy. JCI Insight. 2017, 2, e89580. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Powell, D.J.; Rosenberg, S.A.; Restifo, N.P. Adoptive immunotherapy for cancer: building on success. Nat. Rev. Immunol. 2006, 6, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Petrozziello, E.; Sturmheit, T.; Mondino, A. Exploiting cytokines in adoptive T-cell therapy of cancer. Immunotherapy 2015, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, C.J.; Knochelmann, H.M.; Smith, A.S.; Wyatt, M.M.; Rangel Rivera, G.O.; Arhontoulis, D.C.; Bartee, E.; Li, Z.; Rubinstein, M.P.; Paulos, C.M. Fueling Cancer Immunotherapy With Common Gamma Chain Cytokines. Front. Immunol. 2019, 10, 263. [Google Scholar] [CrossRef]
- Gliwiński, M.; Piotrowska, M.; Iwaszkiewicz-Grześ, D.; Urban-Wójciuk, Z.; Trzonkowski, P. Therapy with CD4+CD25+ T regulatory cells – should we be afraid of cancer? Contemp. Oncol. (Pozn.) 2019, 23, 1–6. [Google Scholar] [CrossRef]
- Liao, W.; Lin, J.X.; Leonard, W.J. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity 2013, 38, 13–25. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Saibil, S.D.; Sotov, V.; Le, M.X.; Khoja, L.; Ghazarian, D.; Bonilla, L.; Majeed, H.; Hogg, D.; Joshua, A.M.; et al. Phase II clinical trial of adoptive cell therapy for patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and low-dose interleukin-2. Cancer Immunol. Immunother. 2019, 68, 773–785. [Google Scholar] [CrossRef]
- Rosenzwajg, M.; Lorenzon, R.; Cacoub, P.; Pham, H.P.; Pitoiset, F.; El Soufi, K.; RIbet, C.; Bernard, C.; Aractingi, S.; Banneville, B.; et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann. Rheum. Dis. 2019, 78, 209–217. [Google Scholar] [CrossRef]
- Saadoun, D.; Rosenzwajg, M.; Joly, F.; Six, A.; Carrat, F.; Thibault, V.; Sene, D.; Cacoub, P.; Klatzmann, D. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N. Engl. J. Med. 2011, 365, 2067–2077. [Google Scholar] [CrossRef]
- Klatzmann, D.; Abbas, A.K. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat. Rev. Immunol. 2015, 15, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, B.; Liu, K.; Dudley, M.E.; Johnson, L.A.; Kaiser, A.; Downey, S.; Zheng, Z.; Shelton, T.E.; Matsuda, K.; Robbins, P.F.; et al. Adoptive cell therapy for patients with melanoma, using tumor-infiltrating lymphocytes genetically engineered to secrete interleukin-2. Hum. Gene. Ther. 2008, 19, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Sockolosky, J.T.; Trotta, E.; Parisi, G.; Picton, L.; Su, L.L.; Le, A.C.; Chhabra, A.; Silveria, S.L.; George, B.M.; King, I.C.; et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science 2018, 359, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Ren, Z.; Yang, K.; Liu, Z.; Cao, S.; Deng, S.; Xu, L.; Liang, Y.; Guo, J.; Bian, Y.; et al. A next-generation tumor-targeting IL-2 preferentially promotes tumor-infiltrating CD8. Nat. Commun. 2019, 10, 3874. [Google Scholar] [CrossRef]
- Caserta, S.; Alessi, P.; Basso, V.; Mondino, A. IL-7 is superior to IL-2 for ex vivo expansion of tumour-specific CD4(+) T cells. Eur. J. Immunol. 2010, 40, 470–479. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Sportès, C.; Ahmadzadeh, M.; Fry, T.J.; Ngo, L.T.; Schwarz, S.L.; Stetler-Stevenson, M.; Morton, K.E.; Mavroukakis, S.A.; Morre, M.; et al. IL-7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T-regulatory cells. J. Immunother. 2006, 29, 313–319. [Google Scholar] [CrossRef]
- Sportès, C.; Hakim, F.T.; Memon, S.A.; Zhang, H.; Chua, K.S.; Brown, M.R.; Fleisher, T.A.; Krumlauf, M.C.; Babb, R.R.; Chow, C.K.; et al. Administration of rhIL-7 in humans increases in vivo TCR repertoire diversity by preferential expansion of naive T cell subsets. J. Exp. Med. 2008, 205, 1701–1714. [Google Scholar] [CrossRef]
- Ding, Z.C.; Habtetsion, T.; Cao, Y.; Li, T.; Liu, C.; Kuczma, M.; Chen, T.; Hao, Z.; Bryan, L.; Munn, D.H.; et al. Adjuvant IL-7 potentiates adoptive T cell therapy by amplifying and sustaining polyfunctional antitumor CD4+ T cells. Sci. Rep. 2017, 7, 12168. [Google Scholar] [CrossRef]
- Robinson, T.O.; Schluns, K.S. The potential and promise of IL-15 in immuno-oncogenic therapies. Immunol. Lett. 2017, 190, 159–168. [Google Scholar] [CrossRef]
- Tang, L.; Zheng, Y.; Melo, M.B.; Mabardi, L.; Castaño, A.P.; Xie, Y.Q.; Li, N.; Kudchodkar, S.B.; Wong, H.C.; Jeng, E.K.; et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat. Biotechnol. 2018, 36, 707–716. [Google Scholar] [CrossRef]
- Kunert, A.; Chmielewski, M.; Wijers, R.; Berrevoets, C.; Abken, H.; Debets, R. Intra-tumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. Oncoimmunology 2017, 7, e1378842. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, F.; Jiang, Y.; Chen, J.; Wu, K.; Chen, X.; Lin, Y.; Zhang, H.; Li, L.; Zhang, Y. Adoptive Transfer of Interleukin-21-stimulated Human CD8+ T Memory Stem Cells Efficiently Inhibits Tumor Growth. J. Immunother. 2018, 41, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Santegoets, S.J.; Turksma, A.W.; Suhoski, M.M.; Stam, A.G.; Albelda, S.M.; Hooijberg, E.; Scheper, R.J.; van den Eertwegh, A.J.; Gerritsen, W.R.; Powell, D.J.; et al. IL-21 promotes the expansion of CD27+ CD28+ tumor infiltrating lymphocytes with high cytotoxic potential and low collateral expansion of regulatory T cells. J. Transl. Med. 2013, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Snyder, K.M.; Suhoski, M.M.; Maus, M.V.; Kapoor, V.; June, C.H.; Mackall, C.L. 4-1BB is superior to CD28 costimulation for generating CD8+ cytotoxic lymphocytes for adoptive immunotherapy. J. Immunol. 2007, 179, 4910–4918. [Google Scholar] [CrossRef] [PubMed]
- Chacon, J.A.; Wu, R.C.; Sukhumalchandra, P.; Molldrem, J.J.; Sarnaik, A.; Pilon-Thomas, S.; Weber, J.; Hwu, P.; Radvanyi, L. Co-stimulation through 4-1BB/CD137 improves the expansion and function of CD8(+) melanoma tumor-infiltrating lymphocytes for adoptive T-cell therapy. PLoS One 2013, 8, e60031. [Google Scholar] [CrossRef]
- Weigelin, B.; Bolaños, E.; Teijeira, A.; Martinez-Forero, I.; Labiano, S.; Azpilikueta, A.; Morales-Kastresana, A.; Quetglas, J.I.; Wagena, E.; Sánchez-Paulete, A.R.; et al. Focusing and sustaining the antitumor CTL effector killer response by agonist anti-CD137 mAb. Proc. Natl. Acad. Sci. USA 2015, 112, 7551–7556. [Google Scholar] [CrossRef]
- Chester, C.; Sanmamed, M.F.; Wang, J.; Melero, I. Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood 2018, 131, 49–57. [Google Scholar] [CrossRef]
- Song, A.; Song, J.; Tang, X.; Croft, M. Cooperation between CD4 and CD8 T cells for anti-tumor activity is enhanced by OX40 signals. Eur. J. Immunol. 2007, 37, 1224–1232. [Google Scholar] [CrossRef]
- Wu, X.; Gu, Z.; Chen, Y.; Chen, B.; Chen, W.; Weng, L.; Liu, X. Application of PD-1 Blockade in Cancer Immunotherapy. Comput. Struct. Biotechnol. J. 2019, 17, 661–674. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Peng, W.; Liu, C.; Xu, C.; Lou, Y.; Chen, J.; Yang, Y.; Yagita, H.; Overwijk, W.W.; Lizée, G.; Radvanyi, L.; et al. PD-1 blockade enhances T-cell migration to tumors by elevating IFN-γ inducible chemokines. Cancer Res. 2012, 72, 5209–5218. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, A.G.; Roberts, I.M.; Thompson, J.A.; Margolin, K.A.; Bhatia, S.; Lee, S.M.; Sloan, H.L.; Lai, I.P.; Farrar, E.A.; Wagener, F.; et al. T-Cell Therapy Using Interleukin-21-Primed Cytotoxic T-Cell Lymphocytes Combined With Cytotoxic T-Cell Lymphocyte Antigen-4 Blockade Results in Long-Term Cell Persistence and Durable Tumor Regression. J. Clin. Oncol. 2016, 34, 3787–3795. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.Z.; Goswami, S.; Fu, T.; Guan, B.; Chen, J.; Xiong, L.; Zhang, J.; Ng Tang, D.; Zhang, X.; Vence, L.; et al. Blockade of CTLA-4 and PD-1 Enhances Adoptive T-cell Therapy Efficacy in an ICOS-Mediated Manner. Cancer Immunol. Res. 2019, 7, 1803–1812. [Google Scholar] [CrossRef] [PubMed]
- Mardiana, S.; Solomon, B.J.; Darcy, P.K.; Beavis, P.A. Supercharging adoptive T cell therapy to overcome solid tumor-induced immunosuppression. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Poschke, I.; Lövgren, T.; Adamson, L.; Nyström, M.; Andersson, E.; Hansson, J.; Tell, R.; Masucci, G.V.; Kiessling, R. A phase I clinical trial combining dendritic cell vaccination with adoptive T cell transfer in patients with stage IV melanoma. Cancer Immunol. Immunother. 2014, 63, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Chodon, T.; Comin-Anduix, B.; Chmielowski, B.; Koya, R.C.; Wu, Z.; Auerbach, M.; Ng, C.; Avramis, E.; Seja, E.; Villanueva, A.; et al. Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin. Cancer Res. 2014, 20, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, T.S.; Berent-Maoz, B.; Cheung-Lau, G.; Huang, R.R.; Wang, X.; Tsoi, J.; Kaplan-Lefko, P.; Cabrera, P.; Tran, J.; Pang, J.; et al. A Pilot Trial of the Combination of Transgenic NY-ESO-1-reactive Adoptive Cellular Therapy with Dendritic Cell Vaccination with or without Ipilimumab. Clin. Cancer Res. 2019, 25, 2096–2108. [Google Scholar] [CrossRef]
- Idorn, M.; Skadborg, S.K.; Kellermann, L.; Halldórsdóttir, H.R.; Holmen Olofsson, G.; Met, Ö.; Thor Straten, P. Chemokine receptor engineering of T cells with CXCR2 improves homing towards subcutaneous human melanomas in xenograft mouse model. Oncoimmunology 2018, 7, e1450715. [Google Scholar] [CrossRef]
- Peng, W.; Ye, Y.; Rabinovich, B.A.; Liu, C.; Lou, Y.; Zhang, M.; Whittington, M.; Yang, Y.; Overwijk, W.W.; Lizée, G.; et al. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin. Cancer Res. 2010, 16, 5458–5468. [Google Scholar] [CrossRef]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 2016, 45, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Kishton, R.J.; Sukumar, M.; Restifo, N.P. Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metab. 2017, 26, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Kouidhi, S.; Ben Ayed, F.; Benammar Elgaaied, A. Targeting Tumor Metabolism: A New Challenge to Improve Immunotherapy. Front. Immunol. 2018, 9, 353. [Google Scholar] [CrossRef] [PubMed]
- Addou-Klouche, L. NK Cells in Cancer Immunotherapy. In Natural Killer Cells; Aribi, M., Ed.; IntechOpen: London, UK, 2017. [Google Scholar]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Challenge | Possible Solution | References |
|---|---|---|
| chimeric antigen receptor (CAR) T cell migration to the tumour site | Introduction of C-C chemokine receptor type 2 (CCR2) into CAR T cell—chemotaxis toward CC motif chemokine ligand 2 (CCL2) secreted by cancer cells | [27,28] |
| Development of CAR targeting fibroblast activation protein (FAP) expressed on immunosuppressive stromal cells often associated with epithelial tumours | [29,30] | |
| Local CAR T cell administration (i.e., intratumoural injections) | [31,32] | |
| Limited in vivo persistence and proliferation of CAR T cells | Using distinct co-stimulatory molecules for CD4+ and CD8+ T cell subsets (i.e., CD4.ICOS-CAR T cells and CD8.41BB-CAR T cells) | [33] |
| Addition of a second, independent co-stimulatory molecule, i.e., 4-1BB ligand, CD40L | [34,35] | |
| Addition of inducible MyD88/CD40 (iMC), to activate downstream toll-like receptor (TLR) and CD40 signalling pathways using a small molecule ligand, rimiducid | [36,37] | |
| Constitutive expression of a cytokine that is bound to cell membrane of CAR T cell or secreted by the cell, i.e., interleukin (IL) 15, IL-12 | [38,39,40] | |
| Constitutive activation of intracellular IL-7 cytokine receptor triggering IL-7 axis without stimulating bystander lymphocytes | [41] | |
| Limited tumour specificity and off-target effects | The use of combinatorial antigen sensing circuits, i.e., synthetic Notch, where engagement of a tissue-specific antigen by a surface receptor induces transcription of a CAR recognising a tumour-associated antigen | [42,43,44] |
| Reduction of CAR T cell affinity | [45] | |
| Designing CAR T cells targeting antigens that contain tumour-specific modifications/mutations (i.e., mutated variant III of epidermal growth factor receptor; EGFRvIII) | [46,47,48] | |
| Overcoming immunosuppressive tumour microenvironment (TME) | CAR T cell transduction with dominant-negative transforming growth factor β receptor II (dnTGF-βRII)—a decoy receptor for immunosuppressive TGFβ produced by tumour | [49] |
| Introducing switch receptors transforming inhibitory cytokine signals into a stimulus (i.e., fusing immunosuppressive IL-4 receptor exodomain to the immunostimulatory IL-7 receptor endodomain) | [50,51] | |
| Introduction of hypoxia-inducible factor 1-alpha (HIF-1α), a transcription factor stabilised in response to hypoxia, to CAR T cells resulting in increased CAR expression specifically in hypoxic TME | [52] | |
| Co-expression of catalase to protect CAR T cells, as well as bystander T cells, from reactive oxygen species (ROS) in TME | [53] | |
| Inhibition of adenosine receptors with their antagonists or shRNA to prevent immunosuppressive effects exerted by tumour derived adenosine | [54] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chruściel, E.; Urban-Wójciuk, Z.; Arcimowicz, Ł.; Kurkowiak, M.; Kowalski, J.; Gliwiński, M.; Marjański, T.; Rzyman, W.; Biernat, W.; Dziadziuszko, R.; et al. Adoptive Cell Therapy—Harnessing Antigen-Specific T Cells to Target Solid Tumours. Cancers 2020, 12, 683. https://doi.org/10.3390/cancers12030683
Chruściel E, Urban-Wójciuk Z, Arcimowicz Ł, Kurkowiak M, Kowalski J, Gliwiński M, Marjański T, Rzyman W, Biernat W, Dziadziuszko R, et al. Adoptive Cell Therapy—Harnessing Antigen-Specific T Cells to Target Solid Tumours. Cancers. 2020; 12(3):683. https://doi.org/10.3390/cancers12030683
Chicago/Turabian StyleChruściel, Elżbieta, Zuzanna Urban-Wójciuk, Łukasz Arcimowicz, Małgorzata Kurkowiak, Jacek Kowalski, Mateusz Gliwiński, Tomasz Marjański, Witold Rzyman, Wojciech Biernat, Rafał Dziadziuszko, and et al. 2020. "Adoptive Cell Therapy—Harnessing Antigen-Specific T Cells to Target Solid Tumours" Cancers 12, no. 3: 683. https://doi.org/10.3390/cancers12030683
APA StyleChruściel, E., Urban-Wójciuk, Z., Arcimowicz, Ł., Kurkowiak, M., Kowalski, J., Gliwiński, M., Marjański, T., Rzyman, W., Biernat, W., Dziadziuszko, R., Montesano, C., Bernardini, R., & Marek-Trzonkowska, N. (2020). Adoptive Cell Therapy—Harnessing Antigen-Specific T Cells to Target Solid Tumours. Cancers, 12(3), 683. https://doi.org/10.3390/cancers12030683