Model-Based Integration Analysis Revealed Presence of Novel Prognostic miRNA Targets and Important Cancer Driver Genes in Triple-Negative Breast Cancers



Abstract
1. Introduction
2. Results
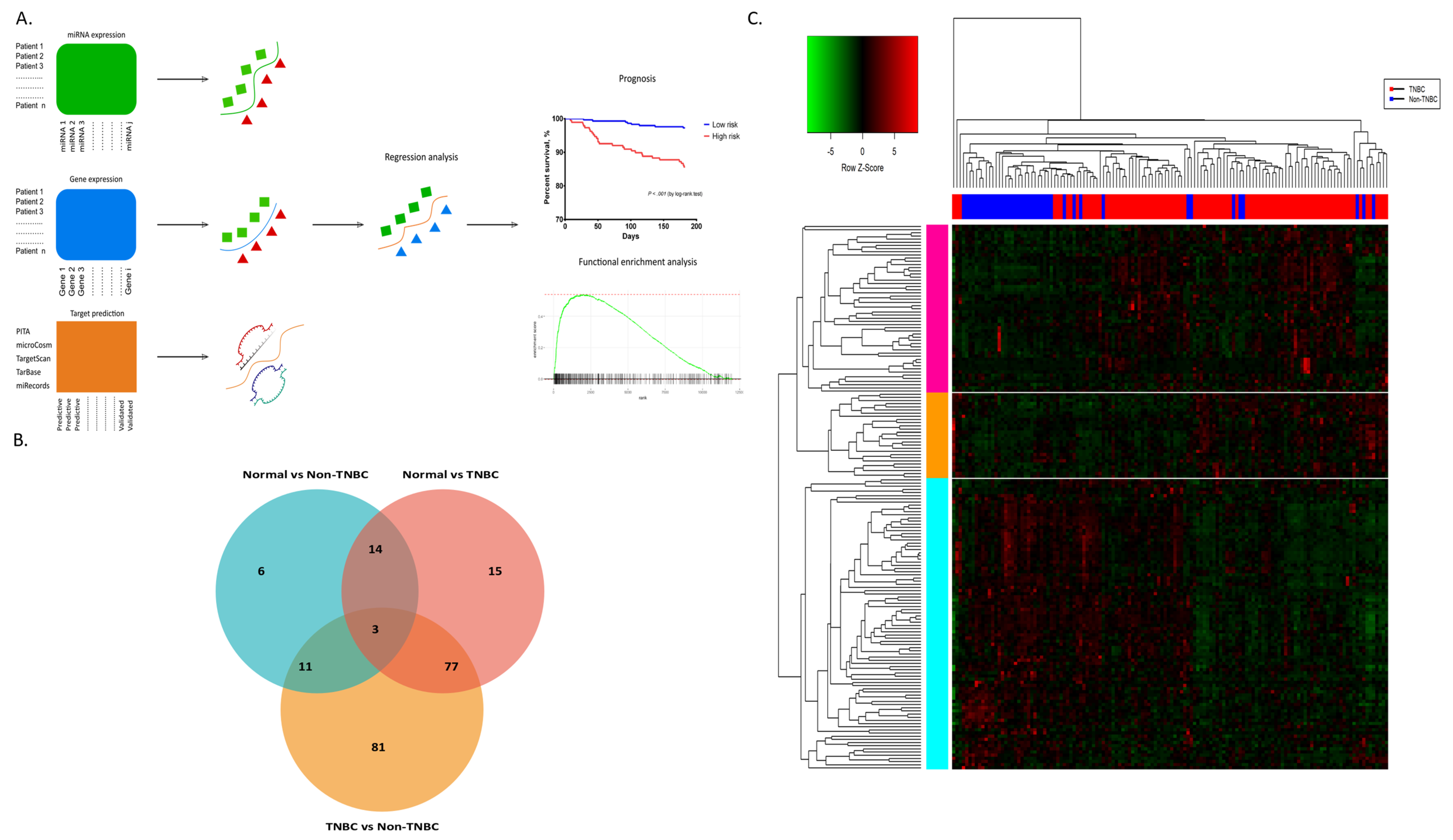
2.1. miRNAs Differentially Expressed among the Tumour Subtypes
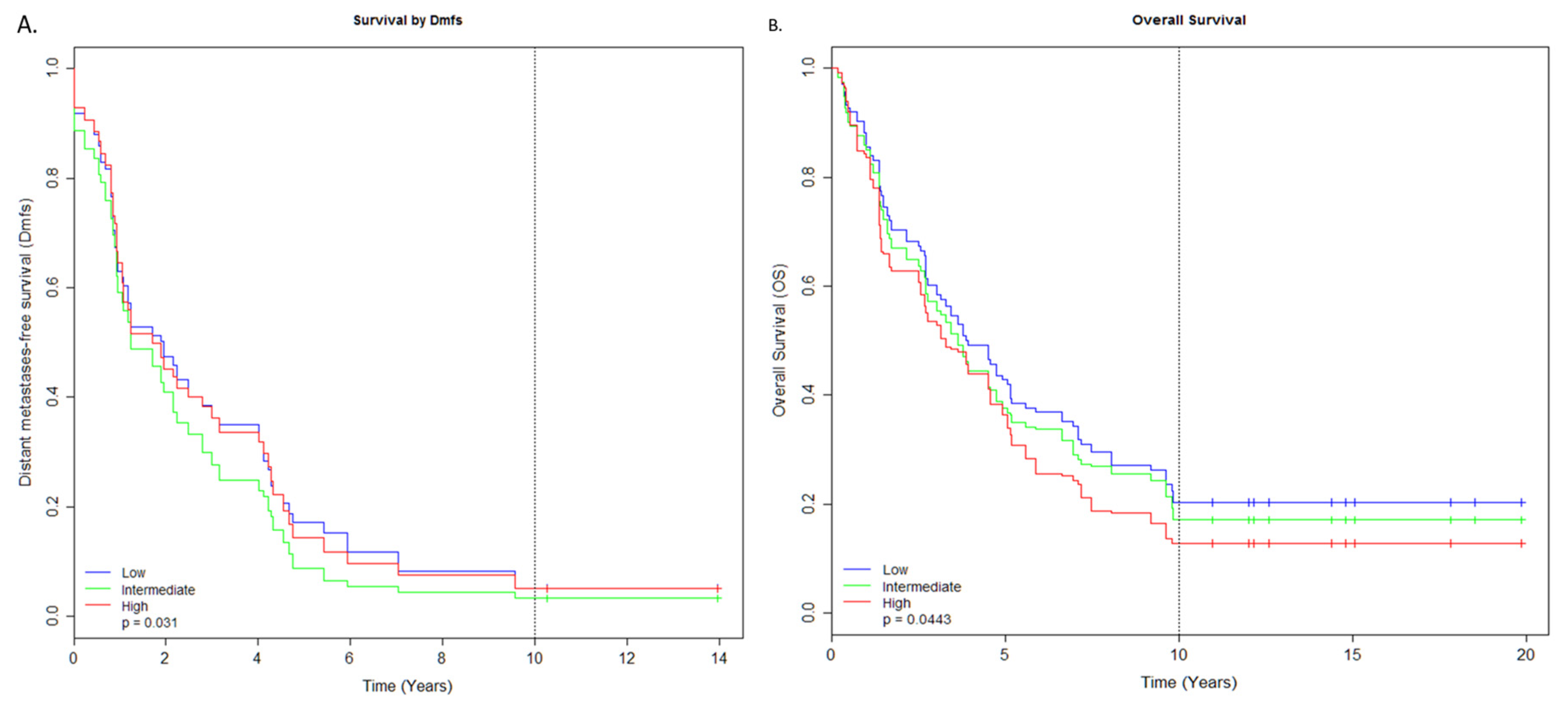
2.2. miRNAs Associated with Survival
2.3. In silico Validation
2.4. mRNA Differentially Expressed among the Tumour Subtype Classes
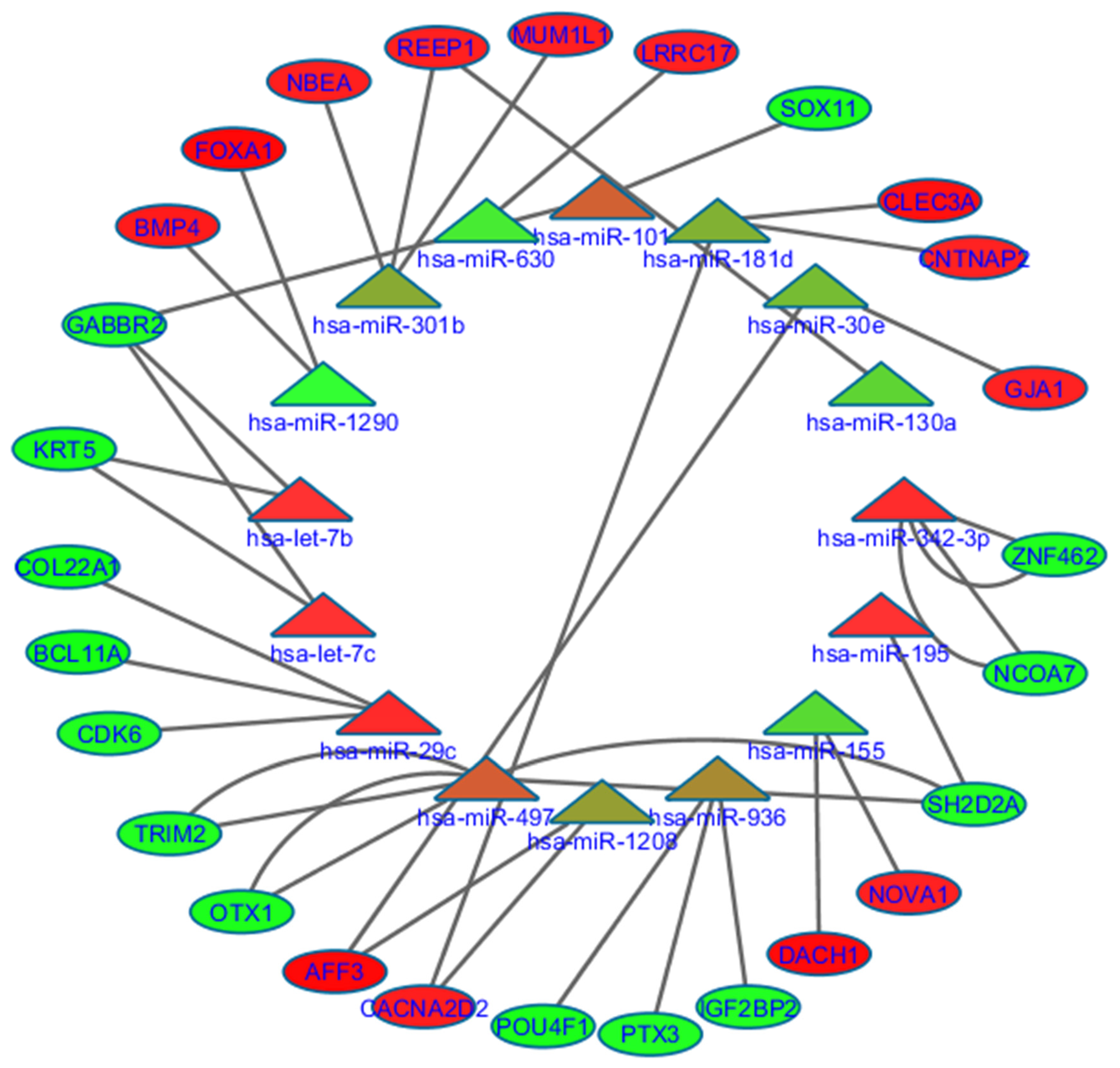
2.5. Prognostic miRNAs and Their Association with Predicted Targets and Enriched Functions
2.6. 18.-miRNAs Signature
2.7. 10.-miRNAs Signature
3. Discussion
4. Material and Methods
4.1. Data and Pre-Processing
4.2. Collection of Predicted, Validated Targets and Calculation of Correlation Index
4.3. Statistical Analysis
4.4. Gene Set Enrichment Analysis of Predicted Targets
4.5. Independent Cohorts for Validation for miRNA Prognostic Signature
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Volinia, S.; Croce, C.M. Prognostic microRNA/mRNA signature from the integrated analysis of patients with invasive breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 7413–7417. [Google Scholar] [CrossRef]
- Karn, T.; Pusztai, L.; Holtrich, U.; Iwamoto, T.; Shiang, C.Y.; Schmidt, M.; Müller, V.; Solbach, C.; Gaetje, R.; Hanker, L.; et al. Homogeneous Datasets of Triple Negative Breast Cancers Enable the Identification of Novel Prognostic and Predictive Signatures. PLoS ONE 2011, 6, e28403. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674. [Google Scholar] [CrossRef] [PubMed]
- Piva, R.; Spandidos, D.A.; Gambari, R. From microRNA functions to microRNA therapeutics: Novel targets and novel drugs in breast cancer research and treatment (Review). Int. J. Oncol. 2013, 43, 985. [Google Scholar] [CrossRef] [PubMed]
- Cascione, L.; Gasparini, P.; Lovat, F.; Carasi, S.; Pulvirenti, A.; Ferro, A.; Alder, H.; He, G.; Vecchione, A.; Croce, C.M.; et al. Integrated microRNA and mRNA signatures associated with survival in triple negative breast cancer. PLoS ONE 2013, 8, e55910. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, A.; Scott, J.G.; Harris, A.L.; Buffa, F.M. Pan-cancer characterisation of microRNA across cancer hallmarks reveals microRNA-mediated downregulation of tumour suppressors. Nat. Commun. 2018, 9, 5228. [Google Scholar] [CrossRef] [PubMed]
- Zaka, M.U.H.; Peng, Y.; Sutton, C.W. Integrated microarray analytics for the discovery of gene signatures for triple-negative breast cancer. In Proceedings of the 2014 14th UK Workshop on Computational Intelligence (UKCI), Bradford, UK, 8–10 September 2014. [Google Scholar] [CrossRef]
- Turner, N.; Lambros, M.B.; Horlings, H.M.; Pearson, A.; Sharpe, R.; Natrajan, R.; Geyer, F.C.; van Kouwenhove, M.; Kreike, B.; Mackay, A.; et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene 2010, 29, 2013. [Google Scholar] [CrossRef]
- Kuo, W.H.; Chang, Y.Y.; Lai, L.C.; Tsai, M.H.; Hsiao, C.K.; Chang, K.J.; Chuang, E.Y. Molecular characteristics and metastasis predictor genes of triple-negative breast cancer: A clinical study of triple-negative breast carcinomas. PLoS ONE 2012, 7, e45831. [Google Scholar] [CrossRef]
- Wang, D.Y.; Done, S.J.; McCready, D.R.; Boerner, S.; Kulkarni, S.; Leong, W.L. A new gene expression signature, the ClinicoMolecular Triad Classification, may improve prediction and prognostication of breast cancer at the time of diagnosis. Breast Cancer Res. 2011, 13, R92. [Google Scholar] [CrossRef]
- Nielsen, J.A.; Lau, P.; Maric, D.; Barker, J.L.; Hudson, L.D. Integrating microRNA and mRNA expression profiles of neuronal progenitors to identify regulatory networks underlying the onset of cortical neurogenesis. BMC Neurosci. 2009, 10, 98. [Google Scholar] [CrossRef]
- Wang, Y.; Blelloch, R. Cell cycle regulation by microRNAs in stem cells. Results Probl. Cell Differ. 2011, 53, 459. [Google Scholar] [PubMed]
- Subramanian, S.; Steer, C.J. MicroRNAs as gatekeepers of apoptosis. J. Cell. Physiol. 2010, 223, 289. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Garcia, I.; Miska, E.A. MicroRNA functions in animal development and human disease. Development 2005, 132, 4653. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W. Data submission and quality in microarray-based microRNA profiling. Clin. Chem. 2013, 59, 392. [Google Scholar] [CrossRef] [PubMed]
- Sablok, G.; Milev, I.; Minkov, G.; Minkov, I.; Varotto, C.; Yahubyan, G.; Baev, V. isomiRex: Web-based identification of microRNAs, isomiR variations and differential expression using next-generation sequencing datasets. FEBS Lett. 2013, 587, 2629. [Google Scholar] [CrossRef] [PubMed]
- Taccioli, C.; Fabbri, E.; Visone, R.; Volinia, S.; Calin, G.A.; Fong, L.Y.; Gambari, R.; Bottoni, A.; Acunzo, M.; Hagan, J.; et al. UCbase & miRfunc: A database of ultraconserved sequences and microRNA function. Nucleic Acids Res. 2009, 37, D41. [Google Scholar]
- Griffiths-Jones, S. miRBase: The microRNA sequence database. Methods Mol. Biol. 2006, 342, 129. [Google Scholar]
- Yerukala Sathipati, S.; Ho, S.Y. Identifying a miRNA signature for predicting the stage of breast cancer. Sci. Rep. 2018, 8, 16138. [Google Scholar] [CrossRef]
- Kertesz, M.; Iovino, N.; Unnerstall, U.; Gaul, U.; Segal, E. The role of site accessibility in microRNA target recognition. Nat. Genet. 2007, 39, 1278. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes are MicroRNA Targets. Cell 2005, 120, 15. [Google Scholar] [CrossRef]
- Bossel Ben-Moshe, N.; Avraham, R.; Kedmi, M.; Zeisel, A.; Yitzhaky, A.; Yarden, Y.; Domany, E. Context-specific microRNA analysis: Identification of functional microRNAs and their mRNA targets. Nucleic Acids Res. 2012, 40, 10614. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; De Micheli, G. Prediction of regulatory modules comprising microRNAs and target genes. Bioinformatics 2005, 21 (Suppl. S2), ii93. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.-Z.; Zheng, T.-S.; Chen, X.; Wang, J.-B.; Zhang, W.-H.; Pan, S.-H.; Jiang, H.-C.; Liu, L.-X. microRNA expression alteration after arsenic trioxide treatment in HepG-2 cells. J. Gastroenterol. Hepatol. 2011, 26, 186. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Yang, A.C.; Huang, C.C.; Liu, M.E.; Liou, Y.J.; Wu, J.C.; Tsai, S.J.; Lin, C.P.; Hong, C.J. The association of RAB18 gene polymorphism (rs3765133) with cerebellar volume in healthy adults. Cerebellum 2014, 13, 616. [Google Scholar] [CrossRef]
- Dunham, A.; Matthews, L.H.; Burton, J.; Ashurst, J.L.; Howe, K.L.; Ashcroft, K.J.; Beare, D.M.; Burford, D.C.; Hunt, S.E.; Griffiths-Jones, S.; et al. The DNA sequence and analysis of human chromosome 13. Nature 2004, 428, 522. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, E.; Kume, A.; Maeda, Y.; Sakai, H.; Sakane, F. Diacylglycerol kinase gamma is a novel anionic phospholipid binding protein with a selective binding preference. Biochem. Biophys. Res. Commun. 2014, 444, 617. [Google Scholar] [CrossRef] [PubMed]
- Wijchers, P.J.; Hoekman, M.F.; Burbach, J.P.; Smidt, M.P. Cloning and analysis of the murine Foxi2 transcription factor. Biochim. Biophys. Acta 2005, 1731, 133. [Google Scholar] [CrossRef]
- Gui, J.; Hu, Z.; Tsai, C.Y.; Ma, T.; Song, Y.; Morales, A.; Huang, L.H.; Dmitrovsky, E.; Craig, R.W.; Usherwood, E.J. MCL1 enhances the survival of CD8+ memory T Cells after viral infection. J. Virol. 2015, 89, 2405. [Google Scholar] [CrossRef]
- Soderlund Leifler, K.; Asklid, A.; Fornander, T.; Stenmark Askmalm, M. The RAD51 135G>C polymorphism is related to the effect of adjuvant therapy in early breast cancer. J. Cancer Res. Clin. Oncol. 2015, 141, 797. [Google Scholar] [CrossRef]
- Tanno, Y.; Susumu, H.; Kawamura, M.; Sugimura, H.; Honda, T.; Watanabe, Y. The inner centromere-shugoshin network prevents chromosomal instability. Science 2015, 349, 1237. [Google Scholar] [CrossRef]
- Lai, F.; Godley, L.A.; Joslin, J.; Fernald, A.A.; Liu, J.; Espinosa, R., 3rd; Zhao, N.; Pamintuan, L.; Till, B.G.; Larson, R.A.; et al. Transcript map and comparative analysis of the 1.5-Mb commonly deleted segment of human 5q31 in malignant myeloid diseases with a del(5q). Genomics 2001, 71, 235. [Google Scholar] [CrossRef] [PubMed]
- Adamo, A.; Atashpaz, S.; Germain, P.L.; Zanella, M.; D’Agostino, G.; Albertin, V.; Chenoweth, J.; Micale, L.; Fusco, C.; Unger, C.; et al. 7q11.23 dosage-dependent dysregulation in human pluripotent stem cells affects transcriptional programs in disease-relevant lineages. Nat. Genet. 2015, 47, 132. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, H.; Wang, J.; Wang, D.; Yao, A.; Li, Q. Prognostic and biological significance of microRNA-127 expression in human breast cancer. Dis. Markers 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Formosa, A.; Markert, E.K.; Lena, A.M.; Italiano, D.; Finazzi-Agro, E.; Levine, A.J.; Bernardini, S.; Garabadgiu, A.V.; Melino, G.; Candi, E. MicroRNAs, miR-154, miR-299-5p, miR-376a, miR-376c, miR-377, miR-381, miR-487b, miR-485-3p, miR-495 and miR-654-3p, mapped to the 14q32.31 locus, regulate proliferation, apoptosis, migration and invasion in metastatic prostate cancer cells. Oncogene 2014, 33, 5173. [Google Scholar] [CrossRef] [PubMed]
- Crippa, E.; Lusa, L.; De Cecco, L.; Marchesi, E.; Calin, G.A.; Radice, P.; Manoukian, S.; Peissel, B.; Daidone, M.G.; Gariboldi, M.; et al. miR-342 regulates BRCA1 expression through modulation of ID4 in breast cancer. PLoS ONE 2014, 9, e87039. [Google Scholar] [CrossRef]
- Higuchi, C.; Nakatsuka, A.; Eguchi, J.; Teshigawara, S.; Kanzaki, M.; Katayama, A.; Yamaguchi, S.; Takahashi, N.; Murakami, K.; Ogawa, D.; et al. Identification of circulating miR-101, miR-375 and miR-802 as biomarkers for type 2 diabetes. Metab. Clin. Exp. 2015, 64, 489. [Google Scholar] [CrossRef]
- Wach, S.; Al-Janabi, O.; Weigelt, K.; Fischer, K.; Greither, T.; Marcou, M.; Theil, G.; Nolte, E.; Holzhausen, H.J.; Stohr, R.; et al. The combined serum levels of miR-375 and urokinase plasminogen activator receptor are suggested as diagnostic and prognostic biomarkers in prostate cancer. Int. J. Cancer. J. Int. Cancer 2015, 137, 1406. [Google Scholar] [CrossRef]
- Zhang, J.X.; Mai, S.J.; Huang, X.X.; Wang, F.W.; Liao, Y.J.; Lin, M.C.; Kung, H.F.; Zeng, Y.X.; Xie, D. MiR-29c mediates epithelial-to-mesenchymal transition in human colorectal carcinoma metastasis via PTP4A and GNA13 regulation of beta-catenin signaling. Ann. Oncol. 2014, 25, 2196. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593. [Google Scholar] [CrossRef]
- Schreiber, M.; Muller, W.J.; Singh, G.; Graham, F.L. Comparison of the effectiveness of adenovirus vectors expressing cyclin kinase inhibitors p16INK4A, p18INK4C, p19INK4D, p21(WAF1/CIP1) and p27KIP1 in inducing cell cycle arrest, apoptosis and inhibition of tumorigenicity. Oncogene 1999, 18, 1663. [Google Scholar] [CrossRef]
- Wang, S.C.; Nakajima, Y.; Yu, Y.L.; Xia, W.; Chen, C.T.; Yang, C.C.; McIntush, E.W.; Li, L.Y.; Hawke, D.H.; Kobayashi, R.; et al. Tyrosine phosphorylation controls PCNA function through protein stability. Nat. Cell Biol. 2006, 8, 1359. [Google Scholar] [CrossRef] [PubMed]
- Andl, C.D.; Mizushima, T.; Nakagawa, H.; Oyama, K.; Harada, H.; Chruma, K.; Herlyn, M.; Rustgi, A.K. Epidermal growth factor receptor mediates increased cell proliferation, migration, and aggregation in esophageal keratinocytes in vitro and in vivo. J. Biol. Chem. 2003, 278, 1824. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.; Christiansen, J.; Lykke-Andersen, J.; Johnsen, A.H.; Wewer, U.M.; Nielsen, F.C. A family of insulin-like growth factor II mRNA-binding proteins represses translation in late development. Mol. Cell. Biol. 1999, 19, 1262. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature 2014, 508, 103. [Google Scholar] [CrossRef]
- Lewis, G.H.; Subhawong, A.P.; Nassar, H.; Vang, R.; Illei, P.B.; Park, B.H.; Argani, P. Relationship between molecular subtype of invasive breast carcinoma and expression of gross cystic disease fluid protein 15 and mammaglobin. Am. J. Clin. Pathol. 2011, 135, 587. [Google Scholar] [CrossRef]
- Roll, J.D.; Rivenbark, A.G.; Sandhu, R.; Parker, J.S.; Jones, W.D.; Carey, L.A.; Livasy, C.A.; Coleman, W.B. Dysregulation of the epigenome in triple-negative breast cancers: Basal-like and claudin-low breast cancers express aberrant DNA hypermethylation. Exp. Mol. Pathol. 2013, 95, 276. [Google Scholar] [CrossRef]
- Rouleau, N.; Domans’kyi, A.; Reeben, M.; Moilanen, A.M.; Havas, K.; Kang, Z.; Owen-Hughes, T.; Palvimo, J.J.; Janne, O.A. Novel ATPase of SNF2-like protein family interacts with androgen receptor and modulates androgen-dependent transcription. Mol. Biol. Cell 2002, 13, 2106. [Google Scholar] [CrossRef]
- Dang, D.N.; Raj, G.; Sarode, V.; Molberg, K.H.; Vadlamudi, R.K.; Peng, Y. Significantly increased PELP1 protein expression in primary and metastatic triple-negative breast carcinoma: Comparison with GATA3 expression and PELP1’s potential role in triple-negative breast carcinoma. Hum. Pathol. 2015, 46, 1829. [Google Scholar] [CrossRef]
- Hashimoto, K.; Tsuda, H.; Koizumi, F.; Shimizu, C.; Yonemori, K.; Ando, M.; Kodaira, M.; Yunokawa, M.; Fujiwara, Y.; Tamura, K. Activated PI3K/AKT and MAPK pathways are potential good prognostic markers in node-positive, triple-negative breast cancer. Ann. Oncol. 2014, 25, 1973. [Google Scholar] [CrossRef]
- Guiu, S.; Charon-Barra, C.; Vernerey, D.; Fumoleau, P.; Campone, M.; Spielmann, M.; Roche, H.; Mesleard, C.; Arnould, L.; Lemonnier, J.; et al. Coexpression of androgen receptor and FOXA1 in nonmetastatic triple-negative breast cancer: Ancillary study from PACS08 trial. Future Oncol. 2015, 11, 2283. [Google Scholar] [CrossRef]
- Purrington, K.S.; Slager, S.; Eccles, D.; Yannoukakos, D.; Fasching, P.A.; Miron, P.; Carpenter, J.; Chang-Claude, J.; Martin, N.G.; Montgomery, G.W.; et al. Genome-wide association study identifies 25 known breast cancer susceptibility loci as risk factors for triple-negative breast cancer. Carcinogenesis 2014, 35, 1012. [Google Scholar] [CrossRef] [PubMed]
- Kotoula, V.; Kalogeras, K.T.; Kouvatseas, G.; Televantou, D.; Kronenwett, R.; Wirtz, R.M.; Fountzilas, G. Sample parameters affecting the clinical relevance of RNA biomarkers in translational breast cancer research. Virchows Arch. Int. J. Pathol. 2013, 462, 141. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fan, Y.; Ding, X.; Xu, B.; Ma, F.; Yuan, P.; Wang, J.; Zhang, P.; Li, Q.; Luo, Y. Prognostic Significance of Single Progesterone Receptor Positivity: A Comparison Study of Estrogen Receptor Negative/Progesterone Receptor Positive/Her2 Negative Primary Breast Cancer With Triple Negative Breast Cancer. Medicine 2015, 94, e2066. [Google Scholar] [CrossRef] [PubMed]
- Chiu, W.W.; Chamley, L.W. Human seminal plasma prolactin-inducible protein is an immunoglobulin G-binding protein. J. Reprod. Immunol. 2003, 60, 97. [Google Scholar] [CrossRef]
- Enerly, E.; Steinfeld, I.; Kleivi, K.; Leivonen, S.K.; Aure, M.R.; Russnes, H.G.; Ronneberg, J.A.; Johnsen, H.; Navon, R.; Rodland, E.; et al. miRNA-mRNA integrated analysis reveals roles for miRNAs in primary breast tumors. PLoS ONE 2011, 6, e16915. [Google Scholar] [CrossRef]
- Blenkiron, C.; Goldstein, L.D.; Thorne, N.P.; Spiteri, I.; Chin, S.F.; Dunning, M.J.; Barbosa-Morais, N.L.; Teschendorff, A.E.; Green, A.R.; Ellis, I.O.; et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007, 8, R214. [Google Scholar] [CrossRef]
- De Rinaldis, E.; Gazinska, P.; Mera, A.; Modrusan, Z.; Fedorowicz, G.M.; Burford, B.; Gillett, C.; Marra, P.; Grigoriadis, A.; Dornan, D.; et al. Integrated genomic analysis of triple-negative breast cancers reveals novel microRNAs associated with clinical and molecular phenotypes and sheds light on the pathways they control. BMC Genom. 2013, 14, 643. [Google Scholar] [CrossRef]
- Moi, L.; Braaten, T.; Al-Shibli, K.; Lund, E.; Busund, L.-T.R. Differential expression of the miR-17-92 cluster and miR-17 family in breast cancer according to tumor type; results from the Norwegian Women and Cancer (NOWAC) study. J. Transl. Med. 2019, 17, 334. [Google Scholar] [CrossRef]
- Kabekkodu, S.P.; Shukla, V.; Varghese, V.K.; Adiga, D.; Vethil Jishnu, P.; Chakrabarty, S.; Satyamoorthy, K. Cluster miRNAs and cancer: Diagnostic, prognostic and therapeutic opportunities. WIREs RNA 2020, 11, e1563. [Google Scholar] [CrossRef]
- Endo, Y.; Toyama, T.; Takahashi, S.; Yoshimoto, N.; Iwasa, M.; Asano, T.; Fujii, Y.; Yamashita, H. miR-1290 and its potential targets are associated with characteristics of estrogen receptor alpha-positive breast cancer. Endocr. Relat. Cancer 2013, 20, 91. [Google Scholar] [CrossRef]
- Shou, J.; Lai, Y.; Xu, J.; Huang, J. Prognostic value of FOXA1 in breast cancer: A systematic review and meta-analysis. Breast 2016, 27, 35. [Google Scholar] [CrossRef] [PubMed]
- Hollern, D.P.; Honeysett, J.; Cardiff, R.D.; Andrechek, E.R. The E2F transcription factors regulate tumor development and metastasis in a mouse model of metastatic breast cancer. Mol. Cell. Biol. 2014, 34, 3229. [Google Scholar] [CrossRef]
- Peng, F.; Zhang, Y.; Wang, R.; Zhou, W.; Zhao, Z.; Liang, H.; Qi, L.; Zhao, W.; Wang, H.; Wang, C.; et al. Identification of differentially expressed miRNAs in individual breast cancer patient and application in personalized medicine. Oncogenesis 2016, 5, e194. [Google Scholar] [CrossRef] [PubMed]
- Tai, M.C.; Kajino, T.; Nakatochi, M.; Arima, C.; Shimada, Y.; Suzuki, M.; Miyoshi, H.; Yatabe, Y.; Yanagisawa, K.; Takahashi, T. miR-342-3p regulates MYC transcriptional activity via direct repression of E2F1 in human lung cancer. Carcinogenesis 2015, 36, 1464. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, T.; Cao, Z.; Huang, H.; Li, J.; Liu, W.; Liu, S.; You, L.; Zhou, L.; Zhang, T.; et al. MiR-497 downregulation contributes to the malignancy of pancreatic cancer and associates with a poor prognosis. Oncotarget 2014, 5, 6983. [Google Scholar] [CrossRef]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36 (Suppl. S1), D154. [Google Scholar] [CrossRef]
- Sethupathy, P.; Corda, B.; Hatzigeorgiou, A.G. TarBase: A comprehensive database of experimentally supported animal microRNA targets. RNA 2006, 12, 192. [Google Scholar] [CrossRef]
- Xiao, F.; Zuo, Z.; Cai, G.; Kang, S.; Gao, X.; Li, T. miRecords: An integrated resource for microRNA-target interactions. Nucleic Acids Res 2009, 37, D105. [Google Scholar] [CrossRef]
- Lopes, C.T.; Franz, M.; Kazi, F.; Donaldson, S.L.; Morris, Q.; Bader, G.D. Cytoscape Web: An interactive web-based network browser. Bioinformatics 2010, 26, 2347. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Sidiropoulos, K.; Garapati, P.; Gillespie, M.; Hausmann, K.; Haw, R.; Jassal, B.; Jupe, S.; Korninger, F.; McKay, S.; et al. The Reactome pathway Knowledgebase. Nucleic Acids Res. 2016, 44, D481. [Google Scholar] [CrossRef] [PubMed]
- Krizman, D.B.; Wagner, L.; Lash, A.; Strausberg, R.L.; Emmert-Buck, M.R. The Cancer Genome Anatomy Project: EST sequencing and the genetics of cancer progression. Neoplasia 1999, 1, 101. [Google Scholar] [CrossRef] [PubMed]
- Bockhorn, J.; Dalton, R.; Nwachukwu, C.; Huang, S.; Prat, A.; Yee, K.; Chang, Y.F.; Huo, D.; Wen, Y.; Swanson, K.E.; et al. MicroRNA-30c inhibits human breast tumour chemotherapy resistance by regulating TWF1 and IL-11. Nat. Commun. 2013, 4, 1393. [Google Scholar] [CrossRef] [PubMed]
- Buffa, F.M.; Camps, C.; Winchester, L.; Snell, C.E.; Gee, H.E.; Sheldon, H.; Taylor, M.; Harris, A.L.; Ragoussis, J. microRNA-associated progression pathways and potential therapeutic targets identified by integrated mRNA and microRNA expression profiling in breast cancer. Cancer Res. 2011, 71, 5635. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| miRNAs | HR | Lower | Higher | p-Value | TNBC vs Non-TNBC | ||
|---|---|---|---|---|---|---|---|
| Regulation | p-Value | Type | |||||
| hsa-miR-29c | 0.72 | 0.52 | 1 | 0.0469 | Up | 1.73E-16 | Protective |
| hsa-miR-342-3p | 0.52 | 0.31 | 0.89 | 0.0162 | Up | 2.99E-12 | Protective |
| hsa-miR-342-5p | 0.3 | 0.1 | 0.93 | 0.0356 | Up | 1.49E-09 | Protective |
| hsa-let-7c | 0.63 | 0.41 | 0.98 | 0.0411 | Up | 4.25E-06 | Protective |
| hsa-miR-19b-1 * | 0 | 0 | 0.69 | 0.0374 | Down | 9.29E-06 | Protective |
| hsa-let-7b | 0.5 | 0.31 | 0.83 | 0.0057 | Up | 9.60E-06 | Protective |
| hsa-miR-1290 | 1.71 | 1.2 | 2.43 | 0.0022 | Down | 2.61E-04 | Risky |
| hsa-miR-369-5p | 0 | 0 | 0.42 | 0.0262 | Up | 5.27E-04 | Protective |
| hsa-miR-301b | 5.31 | 1.13 | 24.96 | 0.0324 | Down | 6.60E-04 | Risky |
| hsa-miR-630 | 1.64 | 1.17 | 2.3 | 0.0029 | Down | 2.26E-03 | Risky |
| hsa-miR-101 | 0.58 | 0.33 | 1 | 0.0486 | Up | 7.98E-03 | Protective |
| hsa-miR-1246 | 1.53 | 1.12 | 2.09 | 0.0071 | Down | 1.03E-02 | Risky |
| hsa-miR-181d | 0.31 | 0.1 | 0.95 | 0.0382 | Down | 1.13E-02 | Protective |
| hsa-miR-181c * | 0.1 | 0.01 | 0.76 | 0.0244 | Down | 1.39E-02 | Protective |
| hsa-miR-30e | 0.49 | 0.25 | 0.98 | 0.0436 | Down | 1.63E-02 | Protective |
| hsa-miR-497 | 0.51 | 0.29 | 0.9 | 0.0193 | Up | 2.30E-02 | Protective |
| hsa-miR-154 | 0.05 | 0 | 0.58 | 0.0168 | Up | 3.28E-02 | Protective |
| hsa-miR-130a | 0.5 | 0.33 | 0.78 | 0.0017 | Down | 4.22E-02 | Protective |
| miRNA | HR | Lower | Higher | p-Value | TNBC vs Non-TNBC | ||
|---|---|---|---|---|---|---|---|
| Regulation | P-Value | Type | |||||
| hsa-miR-342-3p | 0.68 | 0.5 | 0.92 | 0.0127 | Up | 2.99E-12 | Protective |
| hsa-miR-342-5p | 0.39 | 0.2 | 0.75 | 0.00415 | Up | 1.49E-09 | Protective |
| hsa-miR-193b | 1.5 | 1 | 2.25 | 0.0487 | Up | 3.23E-09 | Risky |
| hsa-miR-195 | 0.76 | 0.59 | 0.98 | 0.0325 | Up | 1.56E-03 | Protective |
| hsa-miR-155 | 0.61 | 0.41 | 0.91 | 0.0157 | Down | 7.44E-03 | Protective |
| hsa-miR-936 | 5.79 | 1.04 | 32.08 | 0.0442 | Up | 1.17E-02 | Protective |
| hsa-miR-338-3p | 0.43 | 0.19 | 0.96 | 0.0377 | Up | 1.40E-02 | Protective |
| hsa-miR-1208 | 376.22 | 10.32 | 13709.16 | 0.00111 | Down | 1.78E-02 | Risky |
| hsa-miR-497 | 0.64 | 0.44 | 0.94 | 0.021 | Up | 2.30E-02 | Protective |
| hsa-miR-146b-5p | 0.65 | 0.45 | 0.94 | 0.0212 | Down | 2.39E-02 | Protective |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaka, M.; Sutton, C.W.; Peng, Y.; Konur, S. Model-Based Integration Analysis Revealed Presence of Novel Prognostic miRNA Targets and Important Cancer Driver Genes in Triple-Negative Breast Cancers. Cancers 2020, 12, 632. https://doi.org/10.3390/cancers12030632
Zaka M, Sutton CW, Peng Y, Konur S. Model-Based Integration Analysis Revealed Presence of Novel Prognostic miRNA Targets and Important Cancer Driver Genes in Triple-Negative Breast Cancers. Cancers. 2020; 12(3):632. https://doi.org/10.3390/cancers12030632
Chicago/Turabian StyleZaka, Masood, Chris W. Sutton, Yonghong Peng, and Savas Konur. 2020. "Model-Based Integration Analysis Revealed Presence of Novel Prognostic miRNA Targets and Important Cancer Driver Genes in Triple-Negative Breast Cancers" Cancers 12, no. 3: 632. https://doi.org/10.3390/cancers12030632
APA StyleZaka, M., Sutton, C. W., Peng, Y., & Konur, S. (2020). Model-Based Integration Analysis Revealed Presence of Novel Prognostic miRNA Targets and Important Cancer Driver Genes in Triple-Negative Breast Cancers. Cancers, 12(3), 632. https://doi.org/10.3390/cancers12030632