Globular Adiponectin Inhibits Breast Cancer Cell Growth through Modulation of Inflammasome Activation: Critical Role of Sestrin2 and AMPK Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Globular Adiponectin Inhibits Inflammasomes Activation in Cancer Cells
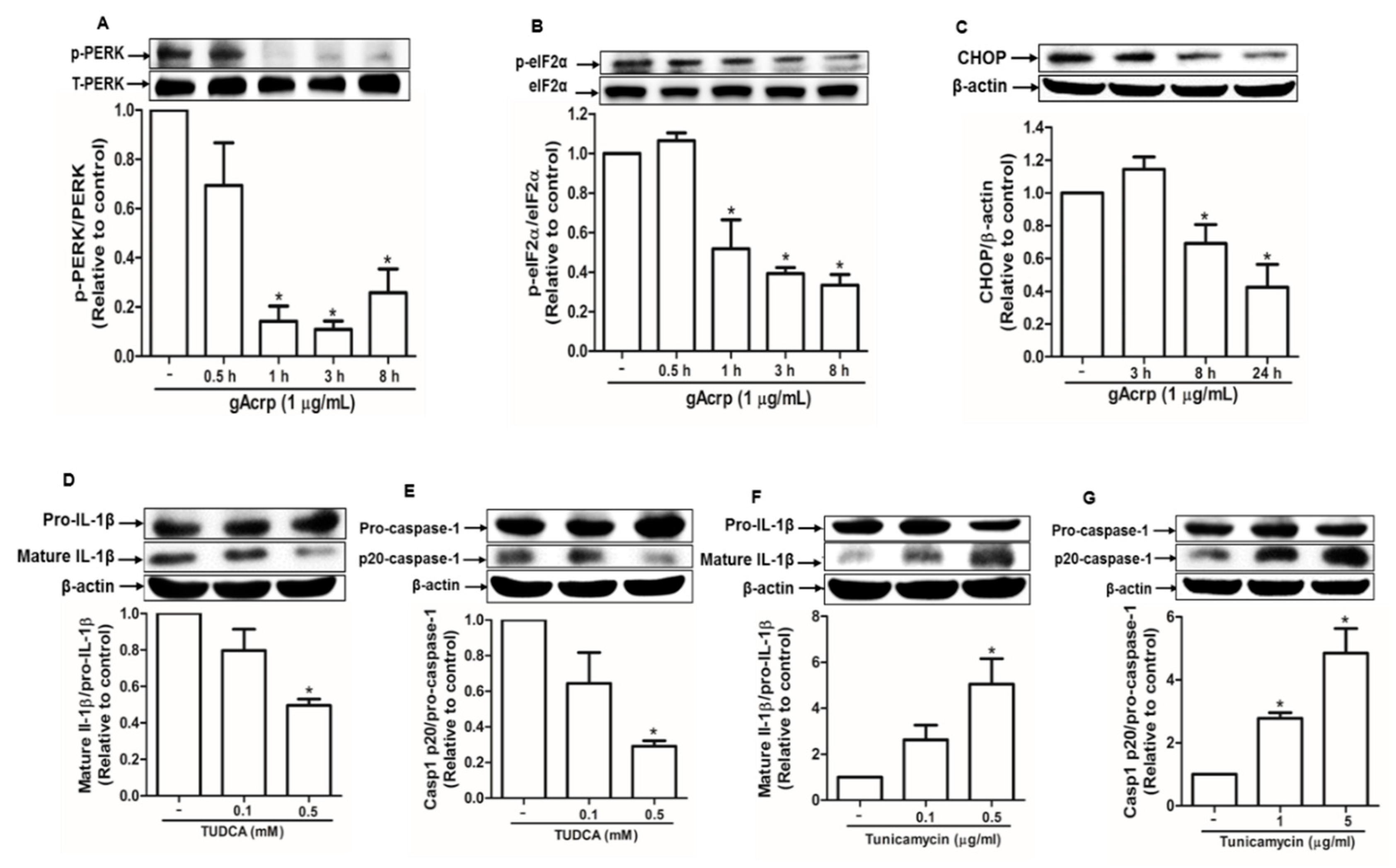
2.2. Modulation of Endoplasmic Reticulum Stress Is Implicated in the Suppression of the Inflammasome Activation by Globular Adiponectin in Breast Cancer Cells
2.3. AMPK Plays an Integral Role in the Modulation of Inflammasomes Activation and ER Stress by Globular Adiponectin in Breast Cancer Cells
2.4. Sestrin2 Acts as an Upstream Signaling Molecule for AMPK Activation and Mediates the Modulatory Effects of Globular Adiponectin on ER Stress and Inflammasomes
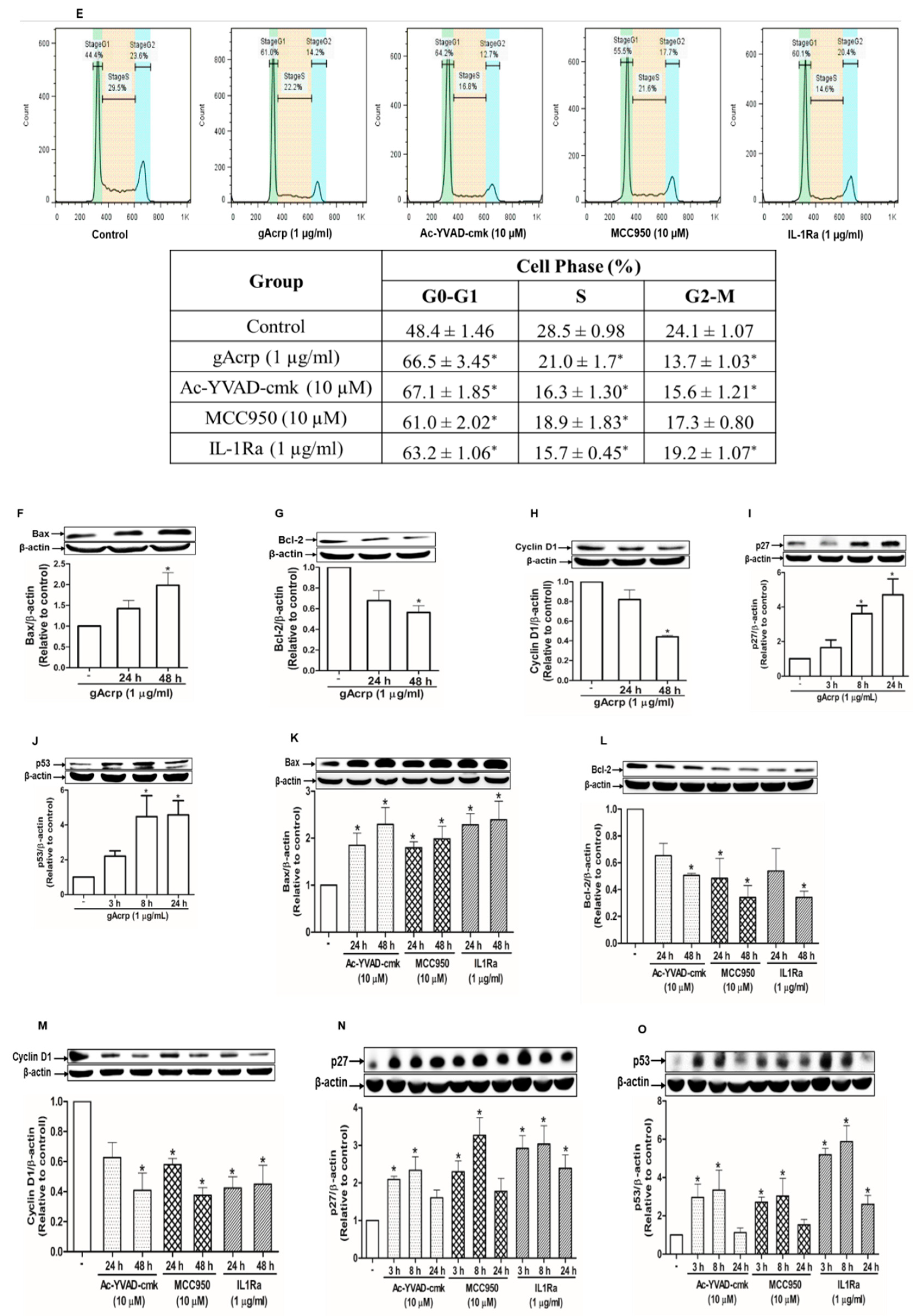
2.5. Suppression of Breast Cancer Cell Growth by Globular Adiponectin is Mediated through Modulation of Inflammasomes Activation
2.6. In Vivo Effects of Globular Adiponectin on NLRP3 Inflammasome Activation that Mediates Tumor Growth in an MCF-7 Tumor Xenograft Model
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Caspase-7 Enzyme Activity Assay
4.5. Cell Cycle Analysis
4.6. RNA Isolation, Reverse Transcription (RT) and Quantitative PCR (qPCR)
4.7. Western blot Analysis
4.8. Transient Transfection with Small Interfering RNA (siRNA)
4.9. Immunoprecipitation Assay
4.10. Immunocytochemistry and Immunohistochemistry
4.11. Development of MCF-7 Tumor Xenograft Model
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Divella, R.; De Luca, R.; Abbate, I.; Naglieri, E.; Daniele, A. Obesity and cancer: The role of adipose tissue and adipo-cytokines-induced chronic inflammation. J. Cancer 2016, 7, 2346–2359. [Google Scholar] [CrossRef] [PubMed]
- Rajala, M.W.; Scherer, P.E. Minireview: The adipocyte—At the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology 2003, 144, 3765–3773. [Google Scholar] [CrossRef] [PubMed]
- Waki, H.; Yamauchi, T.; Kamon, J.; Kita, S.; Ito, Y.; Hada, Y.; Uchida, S.; Tsuchida, A.; Takekawa, S.; Kadowaki, T. Generation of globular fragment of adiponectin by leukocyte elastase secreted by monocytic cell line thp-1. Endocrinology 2005, 146, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Adipocytokines: Mediators linking adipose tissue, inflammation and immunity. Nat. Rev. Immunol. 2006, 6, 772–783. [Google Scholar] [CrossRef]
- Fruebis, J.; Tsao, T.S.; Javorschi, S.; Ebbets-Reed, D.; Erickson, M.R.; Yen, F.T.; Bihain, B.E.; Lodish, H.F. Proteolytic cleavage product of 30-kda adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc. Natl. Acad. Sci. USA 2001, 98, 2005–2010. [Google Scholar] [CrossRef]
- Berg, A.H.; Combs, T.P.; Scherer, P.E. Acrp30/adiponectin: An adipokine regulating glucose and lipid metabolism. Trends Endocrinol. Metab. 2002, 13, 84–89. [Google Scholar] [CrossRef]
- Grossmann, M.E.; Nkhata, K.J.; Mizuno, N.K.; Ray, A.; Cleary, M.P. Effects of adiponectin on breast cancer cell growth and signaling. Br. J. Cancer 2008, 98, 370–379. [Google Scholar] [CrossRef]
- Katira, A.; Tan, P.H. Evolving role of adiponectin in cancer-controversies and update. Cancer Biol. Med. 2016, 13, 101–119. [Google Scholar] [CrossRef]
- Dalamaga, M.; Diakopoulos, K.N.; Mantzoros, C.S. The role of adiponectin in cancer: A review of current evidence. Endocr. Rev. 2012, 33, 547–594. [Google Scholar] [CrossRef]
- Danthala, M.; Rajesh, G.; Gundeti, S.; Raju, G.; Chandran, P.; Srinivas, M. Obesity and breast cancer: Association of serum adiponectin, leptin, and adiponectin-leptin ratio as risk biomarkers. Indian J. Med. Paediatr. Oncol. 2018, 39, 292–296. [Google Scholar] [CrossRef]
- Shrestha, A.; Nepal, S.; Kim, M.J.; Chang, J.H.; Kim, S.H.; Jeong, G.S.; Jeong, C.H.; Park, G.H.; Jung, S.; Lim, J.; et al. Critical role of ampk/foxo3a axis in globular adiponectin-induced cell cycle arrest and apoptosis in cancer cells. J. Cell. Physiol. 2016, 231, 357–369. [Google Scholar] [CrossRef]
- Cui, E.; Guo, H.; Shen, M.; Yu, H.; Gu, D.; Mao, W.; Wang, X. Adiponectin inhibits migration and invasion by reversing epithelial-mesenchymal transition in non-small cell lung carcinoma. Oncol. Rep. 2018, 40, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Nigro, E.; Schettino, P.; Polito, R.; Scudiero, O.; Monaco, M.L.; De Palma, G.D.; Daniele, A. Adiponectin and colon cancer: Evidence for inhibitory effects on viability and migration of human colorectal cell lines. Mol. Cell. Biochem. 2018, 448, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Park, P.H. Autophagy induction: A critical event for the modulation of cell death/survival and inflammatory responses by adipokines. Arch. Pharm. Res. 2018, 41, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.J.; Nagaraju, G.P.; Nagalingam, A.; Muniraj, N.; Kuppusamy, P.; Walker, A.; Woo, J.; Gyorffy, B.; Gabrielson, E.; Saxena, N.K.; et al. Adipoq/adiponectin induces cytotoxic autophagy in breast cancer cells through stk11/lkb1-mediated activation of the ampk-ulk1 axis. Autophagy 2017, 13, 1386–1403. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Li, X.; Liu, Y.; Xia, Y.; Chang, R.; Zhang, C. Inflammasome inhibitors: Promising therapeutic approaches against cancer. J. Hematol. Oncol. 2019, 12, 64. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Kong, H.; Wang, Y.; Zeng, X.; Wang, Z.; Wang, H.; Xie, W. Differential expression of inflammasomes in lung cancer cell lines and tissues. Tumour Biol. 2015, 36, 7501–7513. [Google Scholar] [CrossRef]
- Zhai, Z.; Liu, W.; Kaur, M.; Luo, Y.; Domenico, J.; Samson, J.M.; Shellman, Y.G.; Norris, D.A.; Dinarello, C.A.; Spritz, R.A.; et al. Nlrp1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene 2017, 36, 3820–3830. [Google Scholar] [CrossRef]
- Wang, H.; Luo, Q.; Feng, X.; Zhang, R.; Li, J.; Chen, F. Nlrp3 promotes tumor growth and metastasis in human oral squamous cell carcinoma. BMC Cancer 2018, 18, 500. [Google Scholar] [CrossRef]
- Ershaid, N.; Sharon, Y.; Doron, H.; Raz, Y.; Shani, O.; Cohen, N.; Monteran, L.; Leider-Trejo, L.; Ben-Shmuel, A.; Yassin, M.; et al. Nlrp3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat. Commun. 2019, 10, 4375. [Google Scholar] [CrossRef]
- Kolb, R.; Phan, L.; Borcherding, N.; Liu, Y.; Yuan, F.; Janowski, A.M.; Xie, Q.; Markan, K.R.; Li, W.; Potthoff, M.J.; et al. Obesity-associated nlrc4 inflammasome activation drives breast cancer progression. Nat. Commun. 2016, 7, 13007. [Google Scholar] [CrossRef]
- Kantono, M.; Guo, B. Inflammasomes and cancer: The dynamic role of the inflammasome in tumor development. Front. Immunol. 2017, 8, 1132. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Man, S.M.; Kanneganti, T.-D. Inflammasomes and cancer. Cancer Immunol. Res. 2017, 5, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, S.; Qi, J.; Chen, Z.; Wu, Y.; Guo, J.; Wang, K.; Sun, X.; Zheng, J. Cleavage of gsdme by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019, 10, 193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.-C.; Li, C.-G.; Wang, Y.-F.; Xu, L.-H.; He, X.-H.; Zeng, Q.-Z.; Zeng, C.-Y.; Mai, F.-Y.; Hu, B.; Ouyang, D.-Y. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in a549 lung cancer cells via caspase-3/gsdme activation. Apoptosis 2019, 24, 312–325. [Google Scholar] [CrossRef]
- Pizato, N.; Luzete, B.C.; Kiffer, L.F.M.V.; Corrêa, L.H.; de Oliveira Santos, I.; Assumpção, J.A.F.; Ito, M.K.; Magalhães, K.G. Omega-3 docosahexaenoic acid induces pyroptosis cell death in triple-negative breast cancer cells. Sci. Rep. 2018, 8, 1952. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, E.H.; Pun, N.T.; Chang, J.H.; Kim, J.A.; Jeong, J.H.; Choi, D.Y.; Kim, S.H.; Park, P.H. Globular adiponectin inhibits lipopolysaccharide-primed inflammasomes activation in macrophages via autophagy induction: The critical role of ampk signaling. Int. J. Mol. Sci. 2017, 18, 1275. [Google Scholar] [CrossRef]
- Wang, F.; Liu, Y.; Yang, W.; Yuan, J.; Mo, J. Adiponectin inhibitor nlrp3 inflammasome by modulating the ampk-ros pathway. Int. J. Clin. Exp. Pathol. 2018, 11, 3338–3347. [Google Scholar]
- Ho, A.; Cho, C.S.; Namkoong, S.; Cho, U.S.; Lee, J.H. Biochemical basis of sestrin physiological activities. Trends Biochem. Sci. 2016, 41, 621–632. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Wu, X.Q.; Deng, R.; Sun, T.; Feng, G.K.; Zhu, X.F. Upregulation of sestrin 2 expression via jnk pathway activation contributes to autophagy induction in cancer cells. Cell. Signal. 2013, 25, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.-L.; Fang, M.; Fu, Z.-X.; Zhang, S.-R.; Guo, J.-B.; Wang, R.; Lv, Z.-B.; Xiong, Y.-F. Sestrin 2 suppresses cells proliferation through ampk/mtorc1 pathway activation in colorectal cancer. Oncotarget 2017, 8, 49318–49328. [Google Scholar] [CrossRef] [PubMed]
- Sanli, T.; Linher-Melville, K.; Tsakiridis, T.; Singh, G. Sestrin2 modulates ampk subunit expression and its response to ionizing radiation in breast cancer cells. PLoS ONE 2012, 7, e32035. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Bae, S.H.; Ryu, J.C.; Kwon, Y.; Oh, J.H.; Kwon, J.; Moon, J.S.; Kim, K.; Miyawaki, A.; Lee, M.G.; et al. Sesn2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting nlrp3 activation in macrophages. Autophagy 2016, 12, 1272–1291. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Qin, Y.; Wang, Y.; Meng, S.; Xian, H.; Che, H.; Lv, J.; Li, Y.; Yu, Y.; Bai, Y.; et al. Metformin inhibits the nlrp3 inflammasome via ampk/mtor-dependent effects in diabetic cardiomyopathy. Int. J. Biol. Sci. 2019, 15, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- Cordero, M.D.; Williams, M.R.; Ryffel, B. Amp-activated protein kinase regulation of the nlrp3 inflammasome during aging. Trends Endocrinol. Metab. 2018, 29, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, J.; Li, S.; Li, Y.; Wang, X.; Liu, B.; Fu, Q.; Ma, S. Curcumin attenuates glutamate neurotoxicity in the hippocampus by suppression of er stress-associated txnip/nlrp3 inflammasome activation in a manner dependent on ampk. Toxicol. Appl. Pharmacol. 2015, 286, 53–63. [Google Scholar] [CrossRef]
- Bae, H.R.; Kim, D.H.; Park, M.H.; Lee, B.; Kim, M.J.; Lee, E.K.; Chung, K.W.; Kim, S.M.; Im, D.S.; Chung, H.Y. Beta-hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via ampk activation. Oncotarget 2016, 7, 66444–66454. [Google Scholar] [CrossRef]
- Park, H.W.; Park, H.; Ro, S.H.; Jang, I.; Semple, I.A.; Kim, D.N.; Kim, M.; Nam, M.; Zhang, D.; Yin, L.; et al. Hepatoprotective role of sestrin2 against chronic er stress. Nat. Commun. 2014, 5, 4233. [Google Scholar] [CrossRef]
- Wei, Q.; Guo, P.; Mu, K.; Zhang, Y.; Zhao, W.; Huai, W.; Qiu, Y.; Li, T.; Ma, X.; Liu, Y.; et al. Estrogen suppresses hepatocellular carcinoma cells through erbeta-mediated upregulation of the nlrp3 inflammasome. Lab. Investig. 2015, 95, 804–816. [Google Scholar] [CrossRef]
- Sharma, D.; Kanneganti, T.-D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Huang, C.-F.; Li, Y.; Deng, W.-W.; Mao, L.; Wu, L.; Zhang, W.-F.; Zhang, L.; Zhi, S. Blockage of the nlrp3 inflammasome by mcc950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell. Mol. Life Sci. 2018, 75, 2045–2058. [Google Scholar]
- Siterman, M.; Lengier, S.; Zadik, L.; Ofir, N.; Nachmias, N. Mcc950 a novel inhibitor of nlrp3 inflammasome reduces migration and invasion of lung adenocarcinoma in-vitro. Am. J. Respir. Crit. Care Med. 2017, 195, A7558. [Google Scholar]
- Sun, Y.; Guo, Y. Expression of caspase-1 in breast cancer tissues and its effects on cell proliferation, apoptosis and invasion. Oncol. Lett. 2018, 15, 6431–6435. [Google Scholar] [CrossRef]
- Schlosser, S.; Gansauge, F.; Ramadani, M.; Beger, H.G.; Gansauge, S. Inhibition of caspase-1 induces cell death in pancreatic carcinoma cells and potentially modulates expression levels of bcl-2 family proteins. FEBS Lett. 2001, 491, 104–108. [Google Scholar] [CrossRef]
- Cleary, M.P.; Grossmann, M.E. Obesity and Breast Cancer: The Estrogen Connection. Endocrinology 2009, 150, 2537–2542. [Google Scholar] [CrossRef]
- Andò, S.; Gelsomino, L.; Panza, S.; Giordano, C.; Bonofiglio, D.; Barone, I.; Catalano, S. Obesity, Leptin and Breast Cancer: Epidemiological Evidence and Proposed Mechanisms. Cancers (Basel) 2019, 11, 62. [Google Scholar] [CrossRef] [PubMed]
- Raut, P.K.; Choi, D.Y.; Kim, S.H.; Hong, J.T.; Kwon, T.K.; Jeong, J.H.; Park, P.-H. Estrogen receptor signaling mediates leptin-induced growth of breast cancer cells via autophagy induction. Oncotarget 2017, 8, 109417–109435. [Google Scholar] [CrossRef]
- Raut, P.K.; Kim, S.-H.; Choi, D.Y.; Jeong, G.-S.; Park, P.-H. Growth of breast cancer cells by leptin is mediated via activation of the inflammasome: Critical roles of estrogen receptor signaling and reactive oxygen species production. Biochem. Pharmacol. 2019, 161, 73–88. [Google Scholar] [CrossRef]
- Renehan, A.G.; Zwahlen, M.; Egger, M. Adiposity and cancer risk: New mechanistic insights from epidemiology. Nat. Rev. Cancer 2015, 15, 484–498. [Google Scholar] [CrossRef]
- Iyengar, N.M.; Zhou, X.K.; Gucalp, A.; Morris, P.G.; Howe, L.R.; Giri, D.D.; Morrow, M.; Wang, H.; Pollak, M.; Jones, L.W.; et al. Systemic correlates of white adipose tissue inflammation in early-stage breast cancer. Clin. Cancer Res. 2016, 22, 2283. [Google Scholar] [CrossRef]
- Sánchez-Jiménez, F.; Pérez-Pérez, A.; de la Cruz-Merino, L.; Sánchez-Margalet, V. Obesity and breast cancer: Role of leptin. Front. Oncol. 2019, 9, 596. [Google Scholar] [CrossRef]
- Ahechu, P.; Zozaya, G.; Marti, P.; Hernandez-Lizoain, J.L.; Baixauli, J.; Unamuno, X.; Fruhbeck, G.; Catalan, V. Nlrp3 inflammasome: A possible link between obesity-associated low-grade chronic inflammation and colorectal cancer development. Front. Immunol. 2018, 9, 2918. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.E.; Lashinger, L.M.; Hursting, S.D. The growing challenge of obesity and cancer: An inflammatory issue. Ann. N. Y. Acad. Sci. 2011, 1229, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, R.; Joosten, L.A.; Koenen, T.; van Tits, B.; van Diepen, J.A.; van den Berg, S.A.; Rensen, P.C.; Voshol, P.J.; Fantuzzi, G.; Hijmans, A.; et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010, 12, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The nlrp3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Adijiang, A.; Vandanmagsar, B.; Burk, D.; Ravussin, A.; Dixit, V.D. Elimination of the nlrp3-asc inflammasome protects against chronic obesity-induced pancreatic damage. Endocrinology 2011, 152, 4039–4045. [Google Scholar] [CrossRef]
- Coppola, A.; Marfella, R.; Coppola, L.; Tagliamonte, E.; Fontana, D.; Liguori, E.; Cirillo, T.; Cafiero, M.; Natale, S.; Astarita, C. Effect of weight loss on coronary circulation and adiponectin levels in obese women. Int. J. Cardiol. 2009, 134, 414–416. [Google Scholar] [CrossRef]
- Li, J.; Han, X. Adipocytokines and breast cancer. Curr. Probl. Cancer 2018, 42, 208–214. [Google Scholar] [CrossRef]
- Wolin, K.Y.; Carson, K.; Colditz, G.A. Obesity and cancer. Oncologist 2010, 15, 556–565. [Google Scholar] [CrossRef]
- Ro, S.H.; Semple, I.A.; Park, H.; Park, H.W.; Kim, M.; Kim, J.S.; Lee, J.H. Sestrin2 promotes unc-51-like kinase 1 mediated phosphorylation of p62/sequestosome-1. FEBS J. 2014, 281, 3816–3827. [Google Scholar] [CrossRef] [PubMed]
- Chai, D.; Wang, G.; Zhou, Z.; Yang, H.; Yu, Z. Insulin increases sestrin 2 content by reducing its degradation through the pi 3 k/mtor signaling pathway. Int. J. Endocrinol. 2015, 2015, 505849. [Google Scholar] [CrossRef]
- Taliaferro-Smith, L.; Nagalingam, A.; Zhong, D.; Zhou, W.; Saxena, N.K.; Sharma, D. Lkb1 is required for adiponectin-mediated modulation of ampk-s6k axis and inhibition of migration and invasion of breast cancer cells. Oncogene 2009, 28, 2621–2633. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, L.; Liu, Y.; Yuan, J.; Mo, Z. Adiponectin attenuates nlrp3 inflammasome by modulating ampk-ros pathway. Diabetes 2018, 67, 1232-P. [Google Scholar] [CrossRef]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Oakes, S.A. Endoplasmic reticulum proteostasis: A key checkpoint in cancer. Am. J. Physiol. Cell Physiol. 2017, 312, C93–C102. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Wang, Y.; Wen, X.; Ma, X.-N.; Chen, W.; Huang, F.; Kou, J.; Qi, L.-W.; Liu, B.; et al. Pharmacological activation of ampk prevents drp1-mediated mitochondrial fission and alleviates endoplasmic reticulum stress-associated endothelial dysfunction. J. Mol. Cell. Cardiol. 2015, 86, 62–74. [Google Scholar] [CrossRef]
- Li, A.; Zhang, S.; Li, J.; Liu, K.; Huang, F.; Liu, B. Metformin and resveratrol inhibit drp1-mediated mitochondrial fission and prevent er stress-associated nlrp3 inflammasome activation in the adipose tissue of diabetic mice. Mol. Cell. Endocrinol. 2016, 434, 36–47. [Google Scholar] [CrossRef]
- Nepal, S.; Park, P.-H. Activation of autophagy by globular adiponectin attenuates ethanol-induced apoptosis in hepg2 cells: Involvement of ampk/foxo3a axis. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 2111–2125. [Google Scholar] [CrossRef]
- Nepal, S.; Shrestha, A.; Park, P.-H. Ubiquitin specific protease 2 acts as a key modulator for the regulation of cell cycle by adiponectin and leptin in cancer cells. Mol. Cell. Endocrinol. 2015, 412, 44–55. [Google Scholar] [CrossRef]
- Varghese, F.; Bukhari, A.B.; Malhotra, R.; De, A. Ihc profiler: An open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS ONE 2014, 9, e96801. [Google Scholar] [CrossRef] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, D.-V.; Raut, P.K.; Pandit, M.; Chang, J.-H.; Katila, N.; Choi, D.-Y.; Jeong, J.-H.; Park, P.-H. Globular Adiponectin Inhibits Breast Cancer Cell Growth through Modulation of Inflammasome Activation: Critical Role of Sestrin2 and AMPK Signaling. Cancers 2020, 12, 613. https://doi.org/10.3390/cancers12030613
Pham D-V, Raut PK, Pandit M, Chang J-H, Katila N, Choi D-Y, Jeong J-H, Park P-H. Globular Adiponectin Inhibits Breast Cancer Cell Growth through Modulation of Inflammasome Activation: Critical Role of Sestrin2 and AMPK Signaling. Cancers. 2020; 12(3):613. https://doi.org/10.3390/cancers12030613
Chicago/Turabian StylePham, Duc-Vinh, Pawan Kumar Raut, Mahesh Pandit, Jae-Hoon Chang, Nikita Katila, Dong-Young Choi, Jee-Heon Jeong, and Pil-Hoon Park. 2020. "Globular Adiponectin Inhibits Breast Cancer Cell Growth through Modulation of Inflammasome Activation: Critical Role of Sestrin2 and AMPK Signaling" Cancers 12, no. 3: 613. https://doi.org/10.3390/cancers12030613
APA StylePham, D.-V., Raut, P. K., Pandit, M., Chang, J.-H., Katila, N., Choi, D.-Y., Jeong, J.-H., & Park, P.-H. (2020). Globular Adiponectin Inhibits Breast Cancer Cell Growth through Modulation of Inflammasome Activation: Critical Role of Sestrin2 and AMPK Signaling. Cancers, 12(3), 613. https://doi.org/10.3390/cancers12030613