Mass Spectrometry-Based Identification of MHC-Associated Peptides

Abstract
1. Introduction: Neoantigens in Cancer Immunotherapy
2. MHC I and II Complexes
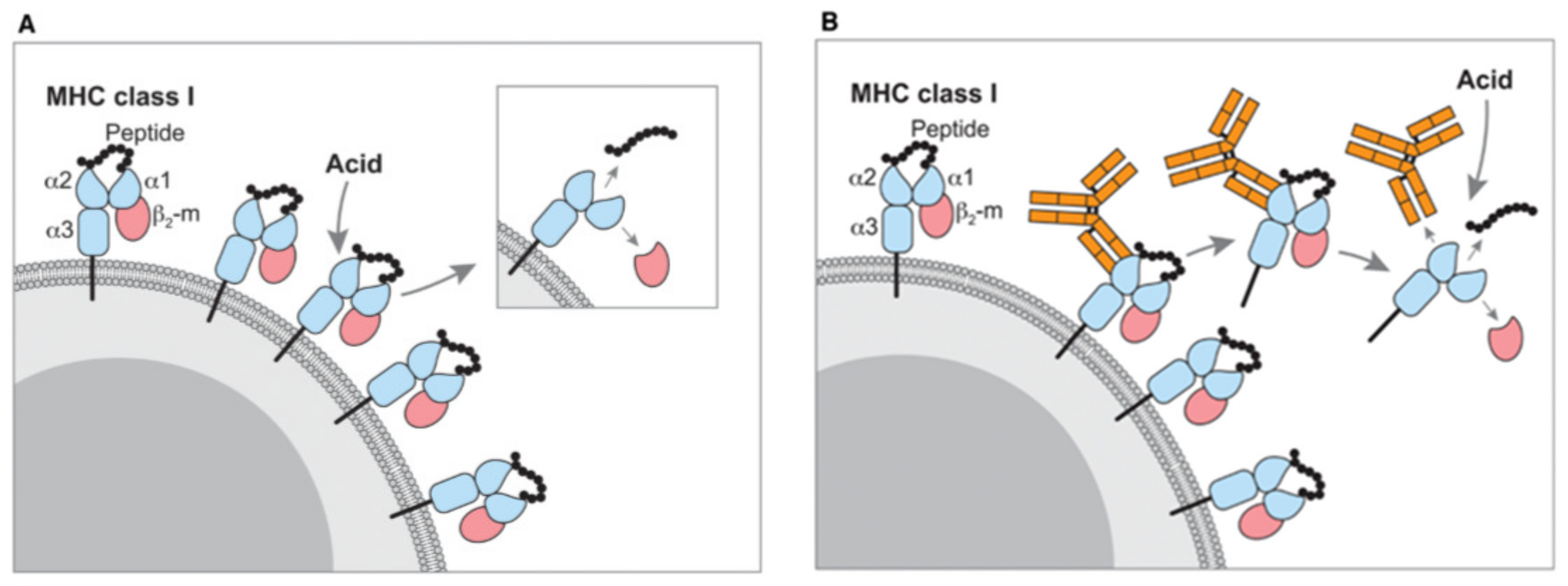
2.1. Isolation of the Immunopeptides from MHC Complexes
2.2. Detection and Identification of Immunopeptides with Mass Spectrometry
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, X.; Qi, Y.; Zhang, Q.; Liu, W. Application of mass spectrometry-based MHC immunopeptidome profiling in neoantigen identification for tumor immunotherapy. Biomed. Pharmacother. 2019, 120. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Caron, E.; Kowalewski, D.J.; Koh, C.C.; Sturm, T.; Schuster, H.; Aebersold, R. Analysis of Major Histocompatibility Complex (MHC) Immunopeptidomes Using Mass Spectrometry. Mol. Cell. Proteomics 2015, 14, 3105–3117. [Google Scholar] [CrossRef]
- Zhang, B.; Whiteaker, J.R.; Hoofnagle, A.N.; Baird, G.S.; Rodland, K.D.; Paulovich, A.G. Clinical potential of mass spectrometry-based proteogenomics. Nat. Rev. Clin. Oncol. 2019, 16, 256–268. [Google Scholar] [CrossRef]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Migliorini, D.; Dutoit, V.; Allard, M.; Grandjean Hallez, N.; Marinari, E.; Widmer, V.; Philippin, G.; Corlazzoli, F.; Gustave, R.; Kreutzfeldt, M.; et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro-Oncology 2019, 21, 923–933. [Google Scholar] [CrossRef]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Dotti, G.; Yee, C.; Goff, S.L. An Update on Adoptive T-Cell Therapy and Neoantigen Vaccines. Am. Soc. Clin. Oncol. Educ. B. 2019, e70–e78. [Google Scholar] [CrossRef] [PubMed]
- Hunt, D.F.; Henderson, R.A.; Shabanowitz, J.; Sakaguchi, K.; Michel, H.; Sevilir, N.; Cox, A.L.; Appella, E.; Engelhard, V.H. Characterization of peptides bound to the class I MHC molecule HLA-A2.1 by mass spectrometry. Science 1992, 255, 1261–1263. [Google Scholar] [CrossRef] [PubMed]
- Creech, A.L.; Ting, Y.S.; Goulding, S.P.; Sauld, J.F.K.; Barthelme, D.; Rooney, M.S.; Addona, T.A.; Abelin, J.G. The Role of Mass Spectrometry and Proteogenomics in the Advancement of HLA Epitope Prediction. Proteomics 2018, 18, e1700259. [Google Scholar] [CrossRef] [PubMed]
- Freudenmann, L.K.; Marcu, A.; Stevanović, S. Mapping the tumour human leukocyte antigen (HLA) ligandome by mass spectrometry. Immunology 2018, 154, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Bassani-Sternberg, M.; Pletscher-Frankild, S.; Jensen, L.J.; Mann, M. Mass spectrometry of human leukocyte antigen class i peptidomes reveals strong effects of protein abundance and turnover on antigen presentation. Mol. Cell. Proteomics 2015, 14, 658–673. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M. Mass spectrometry based immunopeptidomics for the discovery of cancer neoantigens. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2018; Volume 1719, pp. 209–221. [Google Scholar]
- Bassani-Sternberg, M.; Bräunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Rozanov, D.V.; Rozanov, N.D.; Chiotti, K.E.; Reddy, A.; Wilmarth, P.A.; David, L.L.; Cha, S.W.; Woo, S.; Pevzner, P.; Bafna, V.; et al. MHC class I loaded ligands from breast cancer cell lines: A potential HLA-I-typed antigen collection. J. Proteomics 2018, 176, 13–23. [Google Scholar] [CrossRef]
- Rapp, C.; Warta, R.; Stamova, S.; Nowrouzi, A.; Geisenberger, C.; Gal, Z.; Roesch, S.; Dettling, S.; Juenger, S.; Bucur, M.; et al. Identification of T cell target antigens in glioblastoma stem-like cells using an integrated proteomics-based approach in patient specimens. Acta Neuropathol. 2017, 134, 297–316. [Google Scholar] [CrossRef]
- Faridi, P.; Purcell, A.W.; Croft, N.P. In Immunopeptidomics We Need a Sniper Instead of a Shotgun. Proteomics 2018, 18, 1–4. [Google Scholar] [CrossRef]
- Shunji Sugawara; Toru Abo; Katsuo Kumagai A simple method to eliminate the antigenicity of surface class I MHC molecules from the membrane of viable cells by acid treatment at pH 3. J. Immunol. Methods 1987, 100, 83–90. [CrossRef]
- Storkus, W.J.; Zeh, H.J.; Salter, R.D.; Lotze, M.T. Identification of T-Cell epitopes: Rapid isolation of class i-presented peptides from viable cells by mild acid elution. J. Immunother. 1993, 14, 94–103. [Google Scholar] [CrossRef]
- Rötzschke, O.; Falk, K.; Deres, K.; Schild, H.; Norda, M.; Metzger, J.; Jung, G.; Rammensee, H.G. Isolation and analysis of naturally processed viral peptides as recognized by cytotoxic T cells. Nature 1990, 348, 252–254. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, F.R.; Delamarre, L.; Jhunjhunwala, S.; Modrusan, Z.; Phung, Q.T.; Elias, J.E.; Lill, J.R. Building proteomic tool boxes to monitor MHC class I and class II peptides. Proteomics 2017, 17, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Lanoix, J.; Durette, C.; Courcelles, M.; Cossette, É.; Comtois-Marotte, S.; Hardy, M.P.; Côté, C.; Perreault, C.; Thibault, P. Comparison of the MHC I Immunopeptidome Repertoire of B-Cell Lymphoblasts Using Two Isolation Methods. Proteomics 2018, 18, 1–13. [Google Scholar] [CrossRef]
- Chong, C.; Marino, F.; Pak, H.; Racle, J.; Daniel, R.T.; Müller, M.; Gfeller, D.; Coukos, G.; Bassani-Sternberg, M. High-throughput and sensitive immunopeptidomics platform reveals profound IFNγ-mediated remodeling of the HLA ligandome. Mol. Cell. Proteomics 2018, 17, 533–548. [Google Scholar] [CrossRef]
- Purcell, A.W.; Ramarathinam, S.H.; Ternette, N. Mass spectrometry – based identi fi cation of MHC-bound peptides for immunopeptidomics. Nat. Protoc. 2019, 14, 1687–1707. [Google Scholar] [CrossRef]
- Gebreselassie, D.; Spiegel, H.; Vukmanovic, S. Sampling of Major Histocompatibility Complex Class I-Associated Peptidome Suggests Relatively Looser Global Association of HLA-B*5101 With Peptides. Hum. Immunol. 2006, 67, 894–906. [Google Scholar] [CrossRef]
- Hassan, C.; Kester, M.G.D.; Oudgenoeg, G.; de Ru, A.H.; Janssen, G.M.C.; Drijfhout, J.W.; Spaapen, R.M.; Jiménez, C.R.; Heemskerk, M.H.M.; Falkenburg, J.H.F.; et al. Accurate quantitation of MHC-bound peptides by application of isotopically labeled peptide MHC complexes. J. Proteomics 2014, 109, 240–244. [Google Scholar] [CrossRef]
- Komov, L.; Kadosh, D.M.; Barnea, E.; Milner, E.; Hendler, A.; Admon, A. Cell Surface MHC Class I Expression Is Limited by the Availability of Peptide-Receptive “Empty” Molecules Rather than by the Supply of Peptide Ligands. Proteomics 2018, 18. [Google Scholar] [CrossRef]
- Caron, E.; Espona, L.; Kowalewski, D.J.; Schuster, H.; Ternette, N.; Alpízar, A.; Schittenhelm, R.B.; Ramarathinam, S.H.; Arlehamn, C.S.L.; Koh, C.C.; et al. An open-source computational and data resource to analyze digital maps of immunopeptidomes. Elife 2015, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Khodadoust, M.S.; Olsson, N.; Wagar, L.E.; Haabeth, O.A.W.; Chen, B.; Swaminathan, K.; Rawson, K.; Liu, C.L.; Steiner, D.; Lund, P.; et al. Antigen presentation profiling reveals recognition of lymphoma immunoglobulin neoantigens. Nature 2017, 543, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.-J.; et al. Checkpoint Blockade Cancer Immunotherapy Targets Tumour- Specific Mutant Antigens. Nature 2015, 515, 577–581. [Google Scholar]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef]
- Ebrahimi-nik, H.; Bassani-sternberg, M.; Srivastava, P.K.; Ebrahimi-nik, H.; Michaux, J.; Corwin, W.L.; Keller, G.L.J.; Shcheglova, T.; Pak, H.; Coukos, G.; et al. Mass spectrometry – driven exploration reveals nuances of neoepitope-driven tumor rejection Find the latest version: Mass spectrometry – driven exploration reveals nuances of neoepitope-driven tumor rejection. JCI Insight 2019, 4, e129152. [Google Scholar] [CrossRef]
- Löffler, M.W.; Mohr, C.; Bichmann, L.; Freudenmann, L.K.; Walzer, M.; Schroeder, C.M.; Trautwein, N.; Hilke, F.J.; Zinser, R.S.; Mühlenbruch, L.; et al. Multi-omics discovery of exome-derived neoantigens in hepatocellular carcinoma. Genome Med. 2019, 11, 1–16. [Google Scholar] [CrossRef]
- Newey, A.; Griffiths, B.; Michaux, J.; Pak, H.S.; Stevenson, B.J.; Woolston, A.; Semiannikova, M.; Spain, G.; Barber, L.J.; Matthews, N.; et al. Immunopeptidomics of colorectal cancer organoids reveals a sparse HLA class I neoantigen landscape and no increase in neoantigens with interferon or MEK-inhibitor treatment. J. Immunother. cancer 2019, 7, 309. [Google Scholar] [CrossRef]
- Chen, R.; Fauteux, F.; Foote, S.; Stupak, J.; Tremblay, T.L.; Gurnani, K.; Fulton, K.M.; Weeratna, R.D.; Twine, S.M.; Li, J. Chemical Derivatization Strategy for Extending the Identification of MHC Class i Immunopeptides. Anal. Chem. 2018, 90, 11409–11416. [Google Scholar] [CrossRef]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, É.; Bonneil, É.; Laverdure, J.P.; Gendron, P.; Courcelles, M.; Hardy, M.P.; Côté, C.; et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Apcher, S.; Daskalogianni, C.; Fåhraeus, R. Pioneer translation products as an alternative source for MHC-I antigenic peptides. Mol. Immunol. 2015, 68, 68–71. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Antón, L.C.; Bennink, J.R. Defective ribosomal products (DRiPs): a major source of antigenic peptides for MHC class I molecules? J. Immunol. 1996, 157, 1823–1826. [Google Scholar] [PubMed]
- Faridi, P.; Li, C.; Ramarathinam, S.H.; Vivian, J.P.; Illing, P.T.; Mifsud, N.A.; Ayala, R.; Song, J.; Gearing, L.J.; Hertzog, P.J.; et al. A subset of HLA-I peptides are not genomically templated: Evidence for cis- and trans-spliced peptide ligands. Sci. Immunol. 2018, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Laumont, C.M.; Daouda, T.; Laverdure, J.P.; Bonneil, É.; Caron-Lizotte, O.; Hardy, M.P.; Granados, D.P.; Durette, C.; Lemieux, S.; Thibault, P.; et al. Global proteogenomic analysis of human MHC class I-associated peptides derived from non-canonical reading frames. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Fortier, M.H.; Caron, É.; Hardy, M.P.; Voisin, G.; Lemieux, S.; Perreault, C.; Thibault, P. The MHC class I peptide repertoire is molded by the transcriptome. J. Exp. Med. 2008, 205, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garijo, A.; Fajardo, C.A.; Gros, A. Determinants for Neoantigen Identification. Front. Immunol. 2019, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, V.; Fatho, M.; Gentilini, C.; Frye, R.A.; Lifke, A.; Ferel, D.; Wölfel, C.; Huber, C.; Wölfel, T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. USA 2005, 102, 16013–16018. [Google Scholar] [CrossRef]
- Zhao, W.; Sher, X. Systematically benchmarking peptide-MHC binding predictors: From synthetic to naturally processed epitopes. PLoS Comput. Biol. 2018, 14, 1–28. [Google Scholar] [CrossRef]
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019, 11, 1–21. [Google Scholar] [CrossRef]
- The problem with neoantigen prediction. Nat. Biotechnol. 2017, 35, 97. [CrossRef]
- Ghosh, M.; Gauger, M.; Marcu, A.; Nelde, A.; Denk, M.; Schuster, H.; Rammensee, H.; Stevanovic, S. Validation of a high-performance liquid chromatography-tandem mass spectrometry immunopeptidomics assay for the identification of HLA class I ligands suitable for pharmaceutical therapies. bioRxiv 2019, 821249. [Google Scholar] [CrossRef]
- Fritsche, J.; Rakitsch, B.; Hoffgaard, F.; Römer, M.; Schuster, H.; Kowalewski, D.J.; Priemer, M.; Stos-Zweifel, V.; Hörzer, H.; Satelli, A.; et al. Translating Immunopeptidomics to Immunotherapy-Decision-Making for Patient and Personalized Target Selection. Proteomics 2018, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Demmers, L.C.; Heck, A.J.R.; Wu, W. Pre-fractionation Extends but also Creates a Bias in the Detectable HLA Class I Ligandome. J. Proteome Res. 2019, 18, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Hassan, C.; Kester, M.G.D.; De Ru, A.H.; Hombrink, P.; Drijfhout, J.W.; Nijveen, H.; Leunissen, J.A.M.; Heemskerk, M.H.M.; Falkenburg, J.H.F.; Van Veelen, P.A. The human leukocyte antigen-presented ligandome of B lymphocytes. Mol. Cell. Proteomics 2013, 12, 1829–1843. [Google Scholar] [CrossRef]
- Escobar, H.; Reyes-Vargas, E.; Jensen, P.E.; Delgado, J.C.; Crockett, D.K. Utility of characteristic QTOF MS/MS fragmentation for MHC class i peptides. J. Proteome Res. 2011, 10, 2494–2507. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, M.S.; van der Heeft, E.; de Jong, A.P.J.M. On-line derivatization of peptides for improved sequence analysis by micro-column liquid chromatography coupled with electrospray ionization-tandem mass spectrometry. Rapid Commun. Mass Spectrom. 1997, 11, 1271–1278. [Google Scholar] [CrossRef]
- Mommen, G.P.M.; Frese, C.K.; Meiring, H.D.; Gaans-van Den Brink, J.; De Jong, A.P.J.M.; Van Els, C.A.C.M.; Heck, A.J.R. Expanding the detectable HLA peptide repertoire using electron-transfer/ higher-energy collision dissociation (EThcD). Proc. Natl. Acad. Sci. USA 2014, 111, 4507–4512. [Google Scholar] [CrossRef] [PubMed]
- Frese, C.K.; Altelaar, A.F.M.; Van Den Toorn, H.; Nolting, D.; Griep-Raming, J.; Heck, A.J.R.; Mohammed, S. Toward full peptide sequence coverage by dual fragmentation combining electron-transfer and higher-energy collision dissociation tandem mass spectrometry. Anal. Chem. 2012, 84, 9668–9673. [Google Scholar] [CrossRef]
- Tan, C.T.; Croft, N.P.; Dudek, N.L.; Williamson, N.A.; Purcell, A.W. Direct quantitation of MHC-bound peptide epitopes by selected reaction monitoring. Proteomics 2011, 11, 2336–2340. [Google Scholar] [CrossRef]
- Malaker, S.A.; Ferracane, M.J.; Depontieu, F.R.; Zarling, A.L.; Shabanowitz, J.; Bai, D.L.; Topalian, S.L.; Engelhard, V.H.; Hunt, D.F. Identification and Characterization of Complex Glycosylated Peptides Presented by the MHC Class II Processing Pathway in Melanoma. J. Proteome Res. 2017, 16, 228–237. [Google Scholar] [CrossRef]
- Mohammed, F.; Stones, D.H.; Zarling, A.L.; Willcox, C.R.; Shabanowitz, J.; Cummings, K.L.; Hunt, D.F.; Cobbold, M.; Engelhard, V.H.; Willcox, B.E. The antigenic identity of human class I MHC phosphopeptides is critically dependent upon phosphorylation status. Oncotarget 2017, 8, 54160–54172. [Google Scholar] [CrossRef]
- Zarling, A.L.; Polefrone, J.M.; Evans, A.M.; Mikesh, L.M.; Shabanowitz, J.; Lewis, S.T.; Engelhard, V.H.; Hunt, D.F. Identification of class I MHC-associated phosphopeptides as targets for cancer immunotherapy. Proc. Natl. Acad. Sci. USA 2006, 103, 14889–14894. [Google Scholar] [CrossRef] [PubMed]
- Solleder, M.; Guillaume, P.; Racle, J.; Michaux, J.; Pak, H.-S.; Muller, M.; Coukos, G.; Bassani-Sternberg, M.; Gfeller, D. Mass spectrometry based immunopeptidomics leads to robust predictions of phosphorylated HLA class I ligands. bioRxiv 2019, 836189. [Google Scholar] [CrossRef] [PubMed]
- Engelhard, V.H.; Altrich-Vanlith, M.; Ostankovitch, M.; Zarling, A.L. Post-translational modifications of naturally processed MHC-binding epitopes. Curr. Opin. Immunol. 2006, 18, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Seungjin Na; Eunok Paek SOFTWARE EYES FOR PROTEIN POST-TRANSLATIONAL MODIFICATIONS. Mass Spectrom. Rev. 2015, 133–147.
- Depristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinforma. 2013, 43. [Google Scholar] [CrossRef]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2017, 201178. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Di Marco, M.; Schuster, H.; Backert, L.; Ghosh, M.; Rammensee, H.-G.; Stevanović, S. Unveiling the Peptide Motifs of HLA-C and HLA-G from Naturally Presented Peptides and Generation of Binding Prediction Matrices. J. Immunol. 2017, 199, 2639–2651. [Google Scholar] [CrossRef]
- Khodadoust, M.S.; Olsson, N.; Chen, B.; Sworder, B.; Shree, T.; Liu, C.L.; Zhang, L.; Czerwinski, D.K.; Davis, M.M.; Levy, R.; et al. B-cell lymphomas present immunoglobulin neoantigens. Blood 2019, 133, 878–881. [Google Scholar] [CrossRef]
- Andreatta, M.; Nicastri, A.; Peng, X.; Hancock, G.; Dorrell, L.; Ternette, N.; Nielsen, M. MS-Rescue: A Computational Pipeline to Increase the Quality and Yield of Immunopeptidomics Experiments. Proteomics 2019, 19, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.P.; Konda, P.; Kowalewski, D.J.; Schuster, H.; Clements, D.; Kim, Y.; Cohen, A.M.; Sharif, T.; Nielsen, M.; Stevanovic, S.; et al. MHC-I Ligand Discovery Using Targeted Database Searches of Mass Spectrometry Data: Implications for T-Cell Immunotherapies. J. Proteome Res. 2017, 16, 1806–1816. [Google Scholar] [CrossRef] [PubMed]
- Bichmann, L.; Nelde, A.; Ghosh, M.; Heumos, L.; Mohr, C.; Peltzer, A.; Kuchenbecker, L.; Sachsenberg, T.; Walz, J.S.; Stevanović, S.; et al. MHCquant: Automated and reproducible data analysis for immunopeptidomics. J. Proteome Res. 2019. [Google Scholar] [CrossRef] [PubMed]
- Erhard, F.; Halenius, A.; Zimmermann, C.; L’Hernault, A.; Kowalewski, D.J.; Weekes, M.P.; Stevanovic, S.; Zimmer, R.; Dölken, L. Improved Ribo-seq enables identification of cryptic translation events. Nat. Methods 2018, 15, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Liepe, J.; Ovaa, H.; Mishto, M. Why do proteases mess up with antigen presentation by re-shuffling antigen sequences? Curr. Opin. Immunol. 2018, 52, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Muth, T.; Hartkopf, F.; Vaudel, M.; Renard, B.Y. A Potential Golden Age to Come—Current Tools, Recent Use Cases, and Future Avenues for De Novo Sequencing in Proteomics. Proteomics 2018, 18, 1–14. [Google Scholar] [CrossRef]
- Tran, N.H.; Qiao, R.; Xin, L.; Chen, X.; Shan, B.; Li, M. Identifying neoantigens for cancer vaccines by personalized deep learning of individual immunopeptidomes. bioRxiv 2019, 620468. [Google Scholar]
- Karunratanakul, K.; Tang, H.-Y.; Speicher, D.W.; Chuangsuwanich, E.; Sriswasdi, S. Uncovering Thousands of New Peptides with Sequence-Mask-Search Hybrid De Novo Peptide Sequencing Framework. Mol. Cell. Proteomics Cell. Proteomics 2019, 18, 2478–2491. [Google Scholar] [CrossRef]
- Hundal, J.; Kiwala, S.; McMichael, J.; Miller, C.A.; Xia, H.; Wollam, A.T.; Liu, C.J.; Zhao, S.; Feng, Y.-Y.; Graubert, A.P.; et al. pVACtools: A computational toolkit to identify and visualize cancer neoantigens. Cancer Immunol. Res. 2020. canimm.0401.2019. [Google Scholar] [CrossRef]
- Hundal, J.; Kiwala, S.; Feng, Y.Y.; Liu, C.J.; Govindan, R.; Chapman, W.C.; Uppaluri, R.; Swamidass, S.J.; Griffith, O.L.; Mardis, E.R.; et al. Accounting for proximal variants improves neoantigen prediction. Nat. Genet. 2019, 51, 175–179. [Google Scholar] [CrossRef]
- Hundal, J.; Carreno, B.M.; Petti, A.A.; Linette, G.P.; Griffith, O.L.; Mardis, E.R.; Griffith, M. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Schenck, R.O.; Lakatos, E.; Gatenbee, C.; Graham, T.A.; Anderson, A.R.A. NeoPredPipe: high-throughput neoantigen prediction and recognition potential pipeline. BMC Bioinform. 2019, 20, 264. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study Type | Cancer Type | Isolation Method | LC | Mass Spectrometer | Data Analysis | Sample Type and Amount | Number of Identified Peptide Ligands | Identified Neoantigens | Validation Method | Study |
|---|---|---|---|---|---|---|---|---|---|---|
| Neoantigen discovery | B-cell lymphoma | IP of MHC-I and MHC-II | 180 mm × 100 µm C18 column, 160 min gradient, DMSO as mobile phase additive | LTQ Orbitrap Elite, Combined CID+HCD | SEQUEST and PEAKS DB for database searching, PEAKS Studio de novo sequencing | 108–109 cells | >24,000 MHC-I >12,000 MHC-II | 14 | MHC affinity, existence of specific T-cells in patients | [33] |
| Confirmation of in-silico discovered neoantigens | MCA sarcoma cell lines | IP of MHC-I | Discovery: 500 mm × 50 µm C18 column, 140 min gradient, targeted: 150 mm × 75 µm C18 column, 60 min gradient | Discovery: LTQ Orbitrap XL Targeted: AB Sciex QTRAP 5500 | Discovery: MASCOT Targeted: SEQUEST, Skyline | 5 × 108 cells | 250 MHC-I | 1 confirmed | MHC tetramer staining, immunogenicity | [34] |
| Neoantigen discovery | MC-38 and TRAMP-C1 mouse tumor cell lines | IP of MHC-I | 250 mm × 75 µm column packed with 1.7 µm C18, 180 min gradient | LTQ-Orbitrap Velos | Mascot | 1 × 108 cells | 6239 for MC-38 3631 for TRAMP-C1 | 7 MC-38 0 TRAMP-C1 | In vivo immunogenicity | [35] |
| Neoantigen discovery | Meth A mouse cell line | IP of MHC-I | 450 mm × 75 µm column packed with 1.9 µm C18, 85 min gradient | Q-Exactive HF, Q-Exactive HFX | Discovery: MaxQuant. PRM verification: Skyline | 1–10 × 108 cells | 6209 | 8 confirmed | Spike-in heavy peptides, in vivo tumor rejection | [36] |
| Neoantigen discovery | Melanoma tissue | IP of MHC-I | 450 mm × 75 µm column packed with 1.9 µm C18, 90 min gradient | Q-Exactive, Q-Exactive HF | MaxQuant | 25 tissue samples ranging from 0.1 to 4 g | 78,605 MHC-I 15,009 MHC-II | 11 | In vitro immunogenicity | [18] |
| Neoantigen discovery | Hepatocellular carcinoma tissue | IP of MHC-I and MHC-II | 250 mm × 50 µm PepMap RSLC (2um C18) column, 90 min gradient | LTQ Orbitrap XL | MaxQuant | Not exactly specified | average 1403 ± 621 from single sample | 0 | [37] | |
| Comparison of purification methods | B lymphoblasts EBV transformed and leukemia xenografts | IP of MHC-I and MAE | 100 mm × 150 µm column packed with 3 µm C18, 56 min gradient | Q-Exactive HF | PEAKS | 2 × 106, 2 × 107, 108 cells | 2 × 106: 314 MAE 2016 IP 2 × 107: 2081 MAE 3931 IP 108: 2996 MAE 5093 IP | not a subject of study | [26] | |
| Neoantigen discovery | Colorectal cancer organoids | IP of MHC-I and MHC-II | 500 mm × 75 µm column with 1.9 µm C18 material. 125 min gradient for MHC-I and 90 min for MHC-II peptides | Q-Exactive HFX | MaxQuant | 3.85 × 107–108 per biological replicate. 42 biological replicates in total | average 9936 from single sample | 3 | None | [38] |
| Neoantigen discovery | HCT 116 cell line | MAE | 500 mm × 75 µm column packed with 1.9 µm C18 material, 120 min gradient, chemical peptide derivatization | Q-Exactive | Mascot | 2–6 × 108 cells per replicate, total 9 technical replicates | 3148 for 3 technical replicates | 10 | None | [39] |
| Neoantigen discovery | EL4, CT26 cell lines, primary leukemia and lung cancer cell lines | MAE | 150 mm × 150 µm column packed with C18 material, 56min gradient | Q-Exactive Plus, Q-Exactive HF | PEAKS | 2.5–7 × 108 cells per sample. 17 samples in total | 1875 CT26 cells 873 r EL4 cells | 4 (CT26) 2 (EL4) 2 (human samples) | Synthetic peptides, selected hits for in vivo immunogenicity | [40] |
| Neoantigen Isolation Method | Reference | |
|---|---|---|
| Advantages | Drawbacks | |
| Immunoprecipitation (IP) | ||
| Due to specificity of the antibody, IP is less likely to select contaminating peptides | Due to the binding specificity of the antibody, HLA subtype information is also lost in IP-based methods utilizing the pan MHC antibody | [25] |
| Highly specific because the MHC antibodies capture the MHC complexes, where about 90% of MHC peptides come from immunopeptidomes | IP requires extensive washes to remove contaminants/unspecific peptides and detergent, which may lead to losses of low-affinity MHC I-associated peptides | [16,29] |
| Applicable for a variety of biological samples; fresh or frozen dissociated cells and solid tissues | [26] | |
| Mild acid elution (MAE) | ||
| MAE is applicable when starting material may be limited | After MAE, further analysis is challenging due to the additional cell surface (non-MHC) contaminating peptides bound through hydrostatic interactions that may also be eluted by this acid-wash process | [23] |
| MAE allows for cell regeneration following β2M-dissociation, so it can be re-analyzed after a second perturbation | [23] | |
| This method is simple, quick, and reproducible | It extracts not only MHC I-associated peptides but also other contaminants or contaminant peptides | [22,38] |
| The MAE method can be used in situations where antibodies against MHC I molecules are not available | MAE needs viable dissociated cells to perform acid elution, which is the limitation when using fragile cells/solid tumor tissue | [45] |
| It involves fewer purification steps and no detergents | It is been used for high-throughput sequencing of the MHC I-associated peptide repertoire because it is assumed that eluted peptides contain not only MHC I-associated peptides, but also “contaminant” peptides | [45] |
| MAE introduces no bias linked to preferential loss of low-affinity peptides | [45] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kote, S.; Pirog, A.; Bedran, G.; Alfaro, J.; Dapic, I. Mass Spectrometry-Based Identification of MHC-Associated Peptides. Cancers 2020, 12, 535. https://doi.org/10.3390/cancers12030535
Kote S, Pirog A, Bedran G, Alfaro J, Dapic I. Mass Spectrometry-Based Identification of MHC-Associated Peptides. Cancers. 2020; 12(3):535. https://doi.org/10.3390/cancers12030535
Chicago/Turabian StyleKote, Sachin, Artur Pirog, Georges Bedran, Javier Alfaro, and Irena Dapic. 2020. "Mass Spectrometry-Based Identification of MHC-Associated Peptides" Cancers 12, no. 3: 535. https://doi.org/10.3390/cancers12030535
APA StyleKote, S., Pirog, A., Bedran, G., Alfaro, J., & Dapic, I. (2020). Mass Spectrometry-Based Identification of MHC-Associated Peptides. Cancers, 12(3), 535. https://doi.org/10.3390/cancers12030535