Reduced SKP1 Expression Induces Chromosome Instability through Aberrant Cyclin E1 Protein Turnover


Abstract
1. Introduction
2. Results
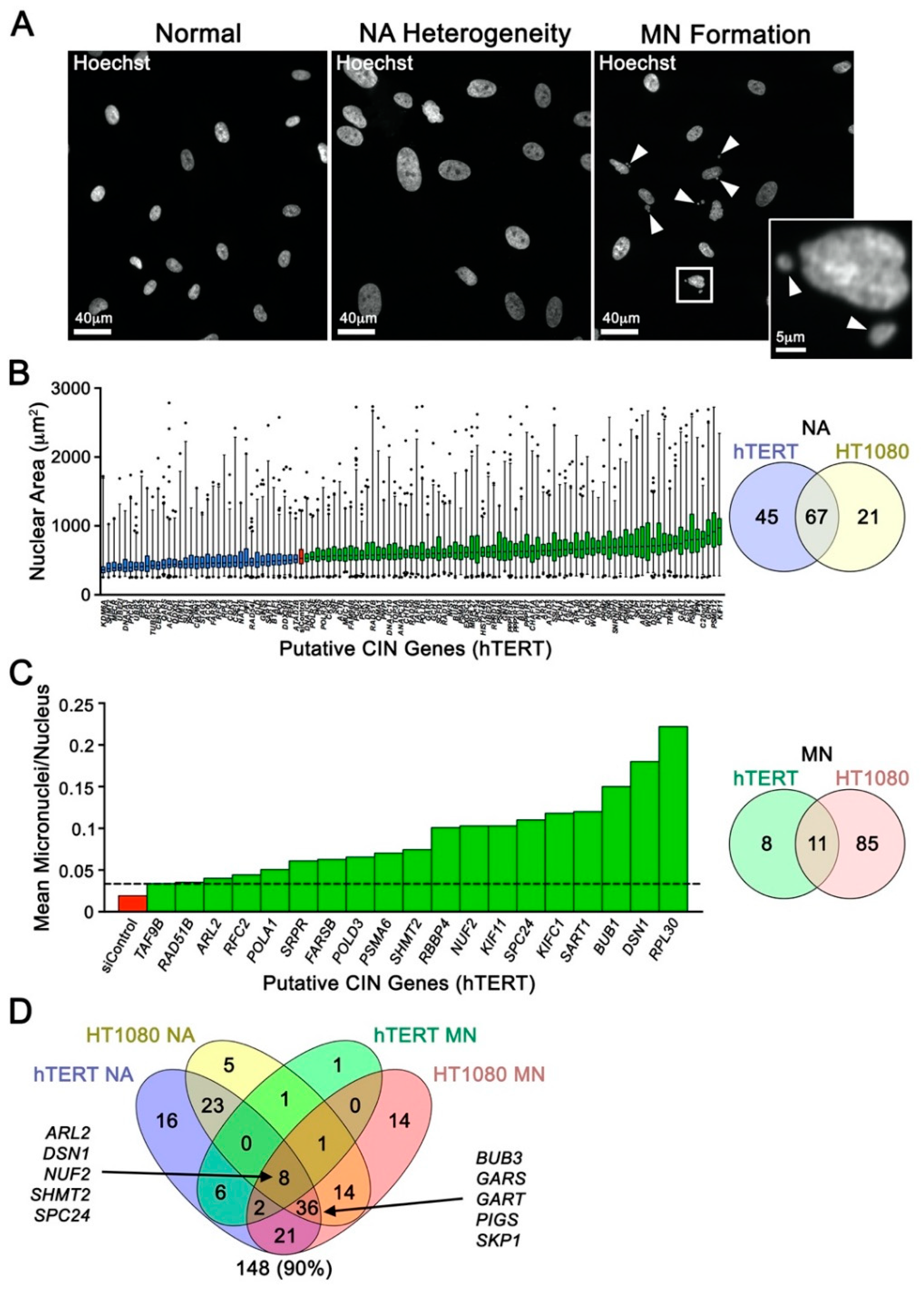
2.1. Cross-Species Analyses and Single-cell Quantitative Imaging Microscopy Identify Putative Human CIN Genes
2.2. Putative CIN Genes Exhibit Frequent Copy Number Losses that may Be Pathogenic in Cancer
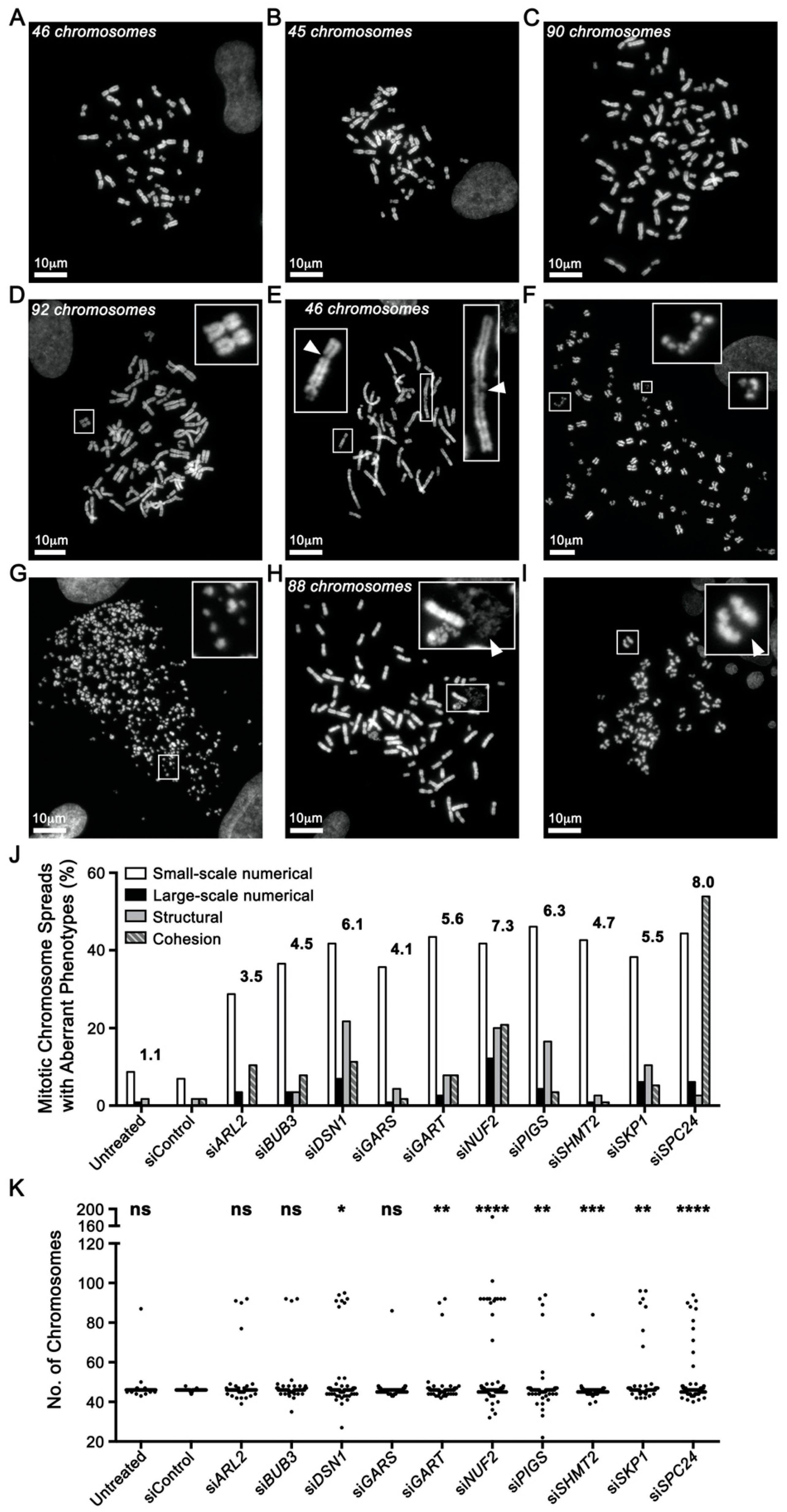
2.3. Reduced Gene Expression Drives Increases in Chromosome Aberrations
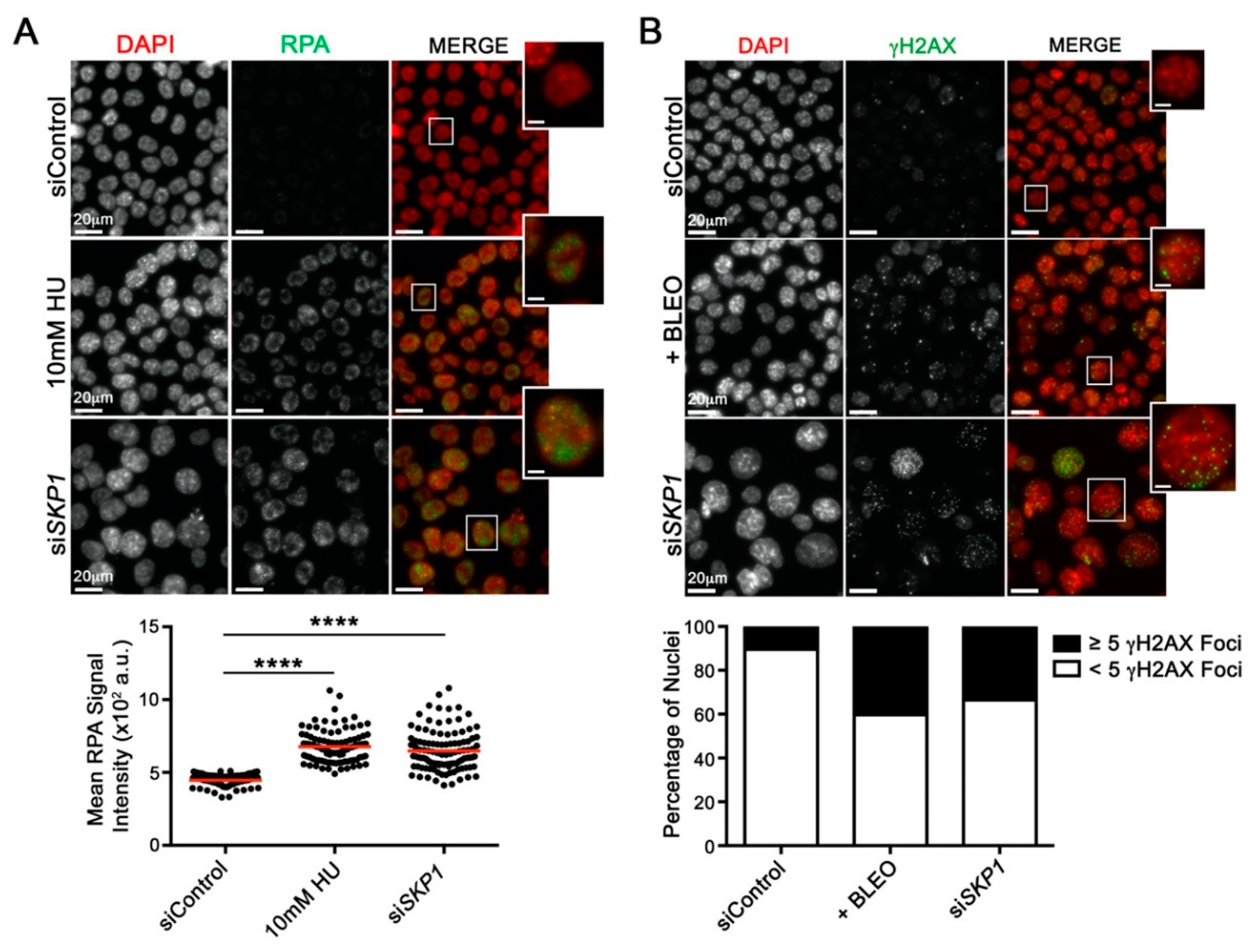
2.4. Reduced SKP1 Expression Induces Replication Stress and DNA Double-strand Breaks
2.5. SKP1 Silencing Underlies Aberrant Increases in the Oncoprotein Cyclin E1
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Identification of Candidate Human CIN Genes
4.3. Gene Silencing
4.4. ScQuantIM
4.5. Gene Alterations and Outcome Analyses in Cancer
4.6. Mitotic Chromosome Spreads and Chromosome Enumeration
4.7. Indirect Immunofluorescent Labeling and Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef]
- Geigl, J.B.; Obenauf, A.C.; Schwarzbraun, T.; Speicher, M.R. Defining ‘chromosomal instability’. Trends Genet. 2008, 24, 64–69. [Google Scholar] [CrossRef]
- McClelland, S.E.; Burrell, R.A.; Swanton, C. Chromosomal instability: A composite phenotype that influences sensitivity to chemotherapy. Cell Cycle 2009, 8, 3262–3266. [Google Scholar] [CrossRef]
- Thompson, L.L.; Jeusset, L.M.; Lepage, C.C.; McManus, K.J. Evolving Therapeutic Strategies to Exploit Chromosome Instability in Cancer. Cancers 2017, 9, 151. [Google Scholar] [CrossRef]
- Stirling, P.C.; Bloom, M.S.; Solanki-Patil, T.; Smith, S.; Sipahimalani, P.; Li, Z.; Kofoed, M.; Ben-Aroya, S.; Myung, K.; Hieter, P. The complete spectrum of yeast chromosome instability genes identifies candidate CIN cancer genes and functional roles for ASTRA complex components. PLoS Genet. 2011, 7, e1002057. [Google Scholar] [CrossRef]
- Barber, T.D.; McManus, K.; Yuen, K.W.; Reis, M.; Parmigiani, G.; Shen, D.; Barrett, I.; Nouhi, Y.; Spencer, F.; Markowitz, S.; et al. Chromatid cohesion defects may underlie chromosome instability in human colorectal cancers. Proc. Natl. Acad. Sci. USA 2008, 105, 3443–3448. [Google Scholar] [CrossRef]
- Thompson, L.L.; McManus, K.J. A novel multiplexed, image-based approach to detect phenotypes that underlie chromosome instability in human cells. PLoS ONE 2015, 10, e0123200. [Google Scholar] [CrossRef]
- Lepage, C.C.; Morden, C.R.; Palmer, M.C.L.; Nachtigal, M.W.; McManus, K.J. Detecting Chromosome Instability in Cancer: Approaches to Resolve Cell-to-Cell Heterogeneity. Cancers 2019, 11, 226. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Sundin, L.J.; Guimaraes, G.J.; Deluca, J.G. The NDC80 complex proteins Nuf2 and Hec1 make distinct contributions to kinetochore-microtubule attachment in mitosis. Mol. Biol. Cell 2011, 22, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Nithianantham, S.; Le, S.; Seto, E.; Jia, W.; Leary, J.; Corbett, K.D.; Moore, J.K.; Al-Bassam, J. Tubulin cofactors and Arl2 are cage-like chaperones that regulate the soluble alphabeta-tubulin pool for microtubule dynamics. eLife 2015, 4, e08811. [Google Scholar] [CrossRef] [PubMed]
- Kudalkar, E.M.; Scarborough, E.A.; Umbreit, N.T.; Zelter, A.; Gestaut, D.R.; Riffle, M.; Johnson, R.S.; MacCoss, M.J.; Asbury, C.L.; Davis, T.N. Regulation of outer kinetochore Ndc80 complex-based microtubule attachments by the central kinetochore Mis12/MIND complex. Proc. Natl. Acad. Sci. USA 2015, 112, E5583–E5589. [Google Scholar] [CrossRef] [PubMed]
- Dahms, T.E.; Sainz, G.; Giroux, E.L.; Caperelli, C.A.; Smith, J.L. The apo and ternary complex structures of a chemotherapeutic target: Human glycinamide ribonucleotide transformylase. Biochemistry 2005, 44, 9841–9850. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.D.; Quintero, C.M.; Stover, P.J. Identification of a de novo thymidylate biosynthesis pathway in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 15163–15168. [Google Scholar] [CrossRef]
- Motley, W.W.; Talbot, K.; Fischbeck, K.H. GARS axonopathy: Not every neuron’s cup of tRNA. Trends Neurosci. 2010, 33, 59–66. [Google Scholar] [CrossRef]
- Fujita, M.; Kinoshita, T. GPI-anchor remodeling: Potential functions of GPI-anchors in intracellular trafficking and membrane dynamics. Biochim. Biophys. Acta 2012, 1821, 1050–1058. [Google Scholar] [CrossRef]
- Chandra Dantu, S.; Nathubhai Kachariya, N.; Kumar, A. Molecular dynamics simulations elucidate the mode of protein recognition by Skp1 and the F-box domain in the SCF complex. Proteins 2016, 84, 159–171. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjostedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Asbaghi, Y.; Thompson, L.L.; Lichtensztejn, Z.; McManus, K.J. KIF11 silencing and inhibition induces chromosome instability that may contribute to cancer. Genes Chromosomes Cancer 2017, 56, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Guppy, B.J.; McManus, K.J. Mitotic accumulation of dimethylated lysine 79 of histone H3 is important for maintaining genome integrity during mitosis in human cells. Genetics 2015, 199, 423–433. [Google Scholar] [CrossRef] [PubMed]
- McManus, K.J.; Barrett, I.J.; Nouhi, Y.; Hieter, P. Specific synthetic lethal killing of RAD54B-deficient human colorectal cancer cells by FEN1 silencing. Proc. Natl. Acad. Sci. USA 2009, 106, 3276–3281. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Kwon, M.; Mannino, M.; Yang, N.; Renda, F.; Khodjakov, A.; Pellman, D. Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature 2018, 561, 551–555. [Google Scholar] [CrossRef]
- Luijten, M.N.H.; Lee, J.X.T.; Crasta, K.C. Mutational game changer: Chromothripsis and its emerging relevance to cancer. Mutat. Res. 2018, 777, 29–51. [Google Scholar] [CrossRef]
- Maher, C.A.; Wilson, R.K. Chromothripsis and human disease: Piecing together the shattering process. Cell 2012, 148, 29–32. [Google Scholar] [CrossRef]
- Jones, M.J.; Jallepalli, P.V. Chromothripsis: Chromosomes in crisis. Dev. Cell 2012, 23, 908–917. [Google Scholar] [CrossRef]
- Forment, J.V.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Rev. Cancer 2012, 12, 663–670. [Google Scholar] [CrossRef]
- Silverman, J.S.; Skaar, J.R.; Pagano, M. SCF ubiquitin ligases in the maintenance of genome stability. Trends Biochem. Sci. 2012, 37, 66–73. [Google Scholar] [CrossRef]
- D’Angiolella, V.; Donato, V.; Forrester, F.M.; Jeong, Y.T.; Pellacani, C.; Kudo, Y.; Saraf, A.; Florens, L.; Washburn, M.P.; Pagano, M. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 2012, 149, 1023–1034. [Google Scholar] [CrossRef]
- Walter, D.; Hoffmann, S.; Komseli, E.S.; Rappsilber, J.; Gorgoulis, V.; Sorensen, C.S. SCF(Cyclin F)-dependent degradation of CDC6 suppresses DNA re-replication. Nat. Commun. 2016, 7, 10530. [Google Scholar] [CrossRef] [PubMed]
- Neelsen, K.J.; Zanini, I.M.; Mijic, S.; Herrador, R.; Zellweger, R.; Ray Chaudhuri, A.; Creavin, K.D.; Blow, J.J.; Lopes, M. Deregulated origin licensing leads to chromosomal breaks by rereplication of a gapped DNA template. Genes Dev. 2013, 27, 2537–2542. [Google Scholar] [CrossRef] [PubMed]
- Vesela, E.; Chroma, K.; Turi, Z.; Mistrik, M. Common Chemical Inductors of Replication Stress: Focus on Cell-Based Studies. Biomolecules 2017, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Machida, Y.J.; Dutta, A. The APC/C inhibitor, Emi1, is essential for prevention of rereplication. Genes Dev. 2007, 21, 184–194. [Google Scholar] [CrossRef]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef]
- Siu, K.T.; Rosner, M.R.; Minella, A.C. An integrated view of cyclin E function and regulation. Cell Cycle 2012, 11, 57–64. [Google Scholar] [CrossRef]
- Nakayama, K.; Nagahama, H.; Minamishima, Y.A.; Matsumoto, M.; Nakamichi, I.; Kitagawa, K.; Shirane, M.; Tsunematsu, R.; Tsukiyama, T.; Ishida, N.; et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J. 2000, 19, 2069–2081. [Google Scholar] [CrossRef]
- Ekholm-Reed, S.; Mendez, J.; Tedesco, D.; Zetterberg, A.; Stillman, B.; Reed, S.I. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J. Cell Biol. 2004, 165, 789–800. [Google Scholar] [CrossRef]
- Jones, R.M.; Mortusewicz, O.; Afzal, I.; Lorvellec, M.; Garcia, P.; Helleday, T.; Petermann, E. Increased replication initiation and conflicts with transcription underlie Cyclin E-induced replication stress. Oncogene 2013, 32, 3744–3753. [Google Scholar] [CrossRef]
- Kuhn, E.; Wang, T.L.; Doberstein, K.; Bahadirli-Talbott, A.; Ayhan, A.; Sehdev, A.S.; Drapkin, R.; Kurman, R.J.; Shih Ie, M. CCNE1 amplification and centrosome number abnormality in serous tubal intraepithelial carcinoma: Further evidence supporting its role as a precursor of ovarian high-grade serous carcinoma. Mod. Pathol. 2016, 29, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Ayhan, A.; Kuhn, E.; Wu, R.C.; Ogawa, H.; Bahadirli-Talbott, A.; Mao, T.L.; Sugimura, H.; Shih, I.M.; Wang, T.L. CCNE1 copy-number gain and overexpression identify ovarian clear cell carcinoma with a poor prognosis. Mod. Pathol. 2017, 30, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Bayard, Q.; Meunier, L.; Peneau, C.; Renault, V.; Shinde, J.; Nault, J.C.; Mami, I.; Couchy, G.; Amaddeo, G.; Tubacher, E.; et al. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat. Commun. 2018, 9, 5235. [Google Scholar] [CrossRef] [PubMed]
- Stronach, E.A.; Paul, J.; Timms, K.M.; Hughes, E.; Brown, K.; Neff, C.; Perry, M.; Gutin, A.; El-Bahrawy, M.; Steel, J.H.; et al. Biomarker Assessment of HR Deficiency, Tumor BRCA1/2 Mutations, and CCNE1 Copy Number in Ovarian Cancer: Associations with Clinical Outcome Following Platinum Monotherapy. Mol. Cancer Res. 2018, 16, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Karst, A.M.; Jones, P.M.; Vena, N.; Ligon, A.H.; Liu, J.F.; Hirsch, M.S.; Etemadmoghadam, D.; Bowtell, D.D.; Drapkin, R. Cyclin E1 deregulation occurs early in secretory cell transformation to promote formation of fallopian tube-derived high-grade serous ovarian cancers. Cancer Res. 2014, 74, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Ohishi, K.; Inoue, N.; Kinoshita, T. PIG-S and PIG-T, essential for GPI anchor attachment to proteins, form a complex with GAA1 and GPI8. EMBO J. 2001, 20, 4088–4098. [Google Scholar] [CrossRef]
- Meyerson, M.; Pellman, D. Cancer genomes evolve by pulverizing single chromosomes. Cell 2011, 144, 9–10. [Google Scholar] [CrossRef]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Rode, A.; Maass, K.K.; Willmund, K.V.; Lichter, P.; Ernst, A. Chromothripsis in cancer cells: An update. Int. J. Cancer 2016, 138, 2322–2333. [Google Scholar] [CrossRef]
- Cai, H.; Kumar, N.; Bagheri, H.C.; von Mering, C.; Robinson, M.D.; Baudis, M. Chromothripsis-like patterns are recurring but heterogeneously distributed features in a survey of 22,347 cancer genome screens. BMC Genomics 2014, 15, 82. [Google Scholar] [CrossRef]
- Tan, Y.; Sun, D.; Jiang, W.; Klotz-Noack, K.; Vashisht, A.A.; Wohlschlegel, J.; Widschwendter, M.; Spruck, C. PP2A-B55beta antagonizes cyclin E1 proteolysis and promotes its dysregulation in cancer. Cancer Res. 2014, 74, 2006–2014. [Google Scholar] [CrossRef] [PubMed]
- Kanska, J.; Zakhour, M.; Taylor-Harding, B.; Karlan, B.Y.; Wiedemeyer, W.R. Cyclin E as a potential therapeutic target in high grade serous ovarian cancer. Gynecol. Oncol. 2016, 143, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Aziz, K.; Limzerwala, J.F.; Sturmlechner, I.; Hurley, E.; Zhang, C.; Jeganathan, K.B.; Nelson, G.; Bronk, S.; Fierro Velasco, R.O.; van Deursen, E.J.; et al. Ccne1 Overexpression Causes Chromosome Instability in Liver Cells and Liver Tumor Development in Mice. Gastroenterology 2019, 157, 210–226. [Google Scholar] [CrossRef] [PubMed]
- Sajesh, B.V.; Bailey, M.; Lichtensztejn, Z.; Hieter, P.; McManus, K.J. Synthetic lethal targeting of superoxide dismutase 1 selectively kills RAD54B-deficient colorectal cancer cells. Genetics 2013, 195, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Lepage, C.C.; Thompson, L.L.; Larson, B.; McManus, K.J. An automated, single cell quantitative imaging microscopy approach to assess micronucleus formation, genotoxicity and chromosome instability. Cells 2020, 9, 344. [Google Scholar] [CrossRef] [PubMed]
- Baergen, A.K.; Jeusset, L.M.; Lichtensztejn, Z.; McManus, K.J. Diminished Condensin Gene Expression Drives Chromosome Instability That May Contribute to Colorectal Cancer Pathogenesis. Cancers 2019, 11, 1066. [Google Scholar] [CrossRef] [PubMed]
- Leylek, T.; Jeusset, L.M.; Lichtensztejn, Z.; McManus, K.J. Reduced expression of Genes Regulating cohesion induces chromosome instability that May promote cancer and impact patient outcomes. Sci. Rep. 2020, 10, 592. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Protein 1 | Species | Catalog No. | [WB] 2 | [IIF] 3 |
|---|---|---|---|---|
| α-Tubulin * | Mouse | Abcam, ab7291 | 1:4000 | - |
| ARL2 | Rabbit | Abcam; ab183510 | 1:5000 | - |
| BUB3 | Rabbit | Abcam; ab4180 | 1:5000 | - |
| CCNE1 | Rabbit | Abcam; ab133266 | 1:5000 | 1:200 |
| Cyclophilin B * | Rabbit | Abcam; ab16045 | 1:50,000 | - |
| DSN1 | Rabbit | Thermo Fisher; PA534879 | 1:1000 | - |
| GARS | Rabbit | Abcam; ab42905 | 1:5000 | - |
| GART | Rabbit | Abcam; ab169550 | 1:5000 | - |
| γH2AX | Rabbit | Abcam; ab2893 | - | 1:1000 |
| NUF2 | Rabbit | Abcam; ab176556 | 1:20,000 | - |
| PIGS | Rabbit | Abcam; ab113817 | 1:5000 | - |
| RPA32/RPA2 | Mouse | Abcam; ab2175 | - | 1:200 |
| SHMT2 | Rabbit | Abcam; ab180786 | 1:1000 | - |
| SKP1 | Mouse | Abcam; ab124473 | 1:5000 | - |
| SPC24 | Rabbit | Abcam; ab157184 | 1:5000 | - |
| Anti-Rabbit HRP | Goat | Jackson Immunoresearch; 111-035-114 | 1:10,000 | - |
| Anti-Mouse HRP | Goat | Jackson Immunoresearch; 115-035-146 | 1:10,000 | - |
| Anti-Rabbit AlexFluor488 | Goat | Abcam; ab150081 | - | 1:200 |
| Anti-Mouse Cy3 | Goat | Abcam; ab150117 | - | 1:200 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thompson, L.L.; Baergen, A.K.; Lichtensztejn, Z.; McManus, K.J. Reduced SKP1 Expression Induces Chromosome Instability through Aberrant Cyclin E1 Protein Turnover. Cancers 2020, 12, 531. https://doi.org/10.3390/cancers12030531
Thompson LL, Baergen AK, Lichtensztejn Z, McManus KJ. Reduced SKP1 Expression Induces Chromosome Instability through Aberrant Cyclin E1 Protein Turnover. Cancers. 2020; 12(3):531. https://doi.org/10.3390/cancers12030531
Chicago/Turabian StyleThompson, Laura L., Allison K. Baergen, Zelda Lichtensztejn, and Kirk J. McManus. 2020. "Reduced SKP1 Expression Induces Chromosome Instability through Aberrant Cyclin E1 Protein Turnover" Cancers 12, no. 3: 531. https://doi.org/10.3390/cancers12030531
APA StyleThompson, L. L., Baergen, A. K., Lichtensztejn, Z., & McManus, K. J. (2020). Reduced SKP1 Expression Induces Chromosome Instability through Aberrant Cyclin E1 Protein Turnover. Cancers, 12(3), 531. https://doi.org/10.3390/cancers12030531