Genomic Characterization of Non-Invasive Differentiated-Type Gastric Cancer in the Japanese Population

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Subsection
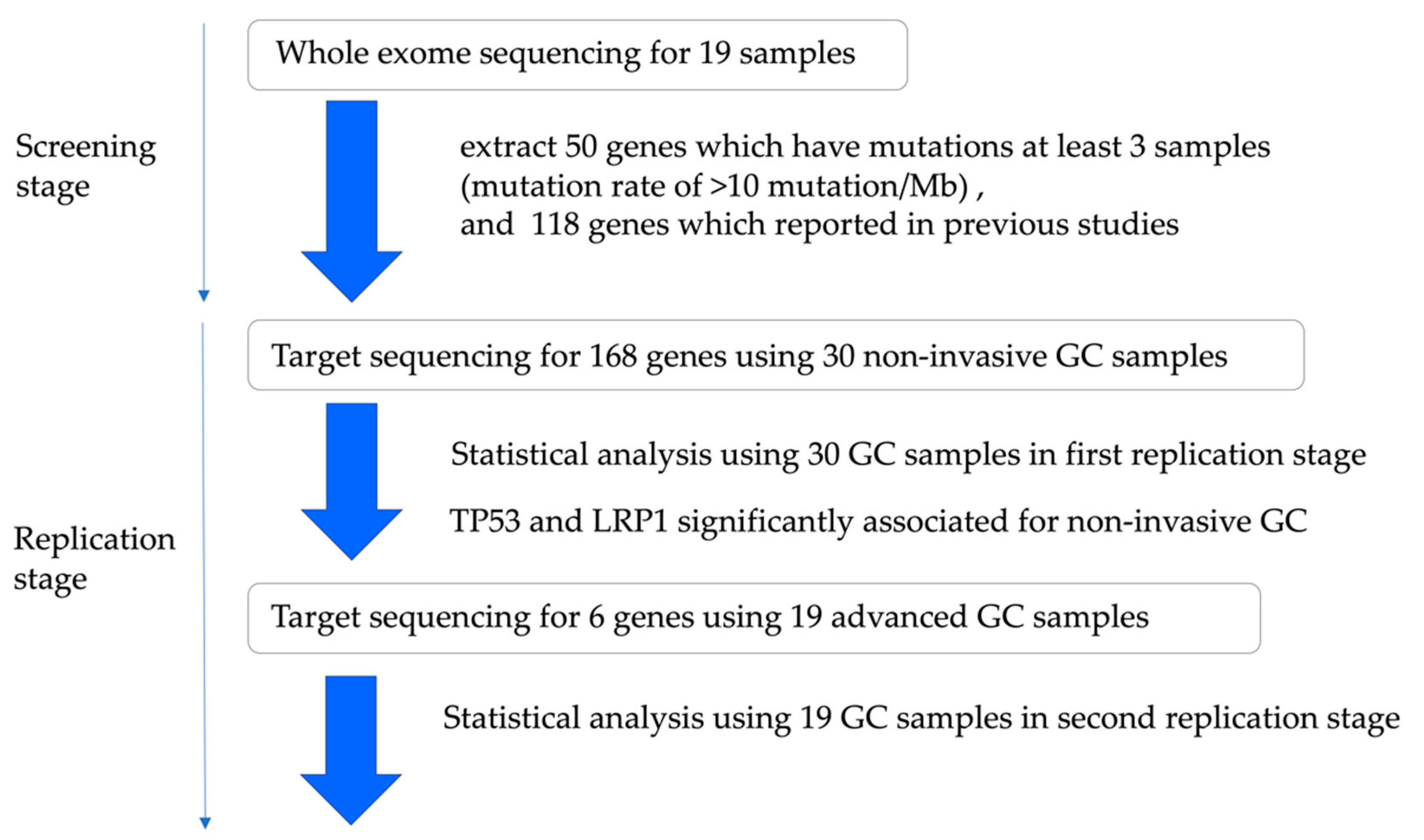
2.1.1. Study Design and Patient Characteristics
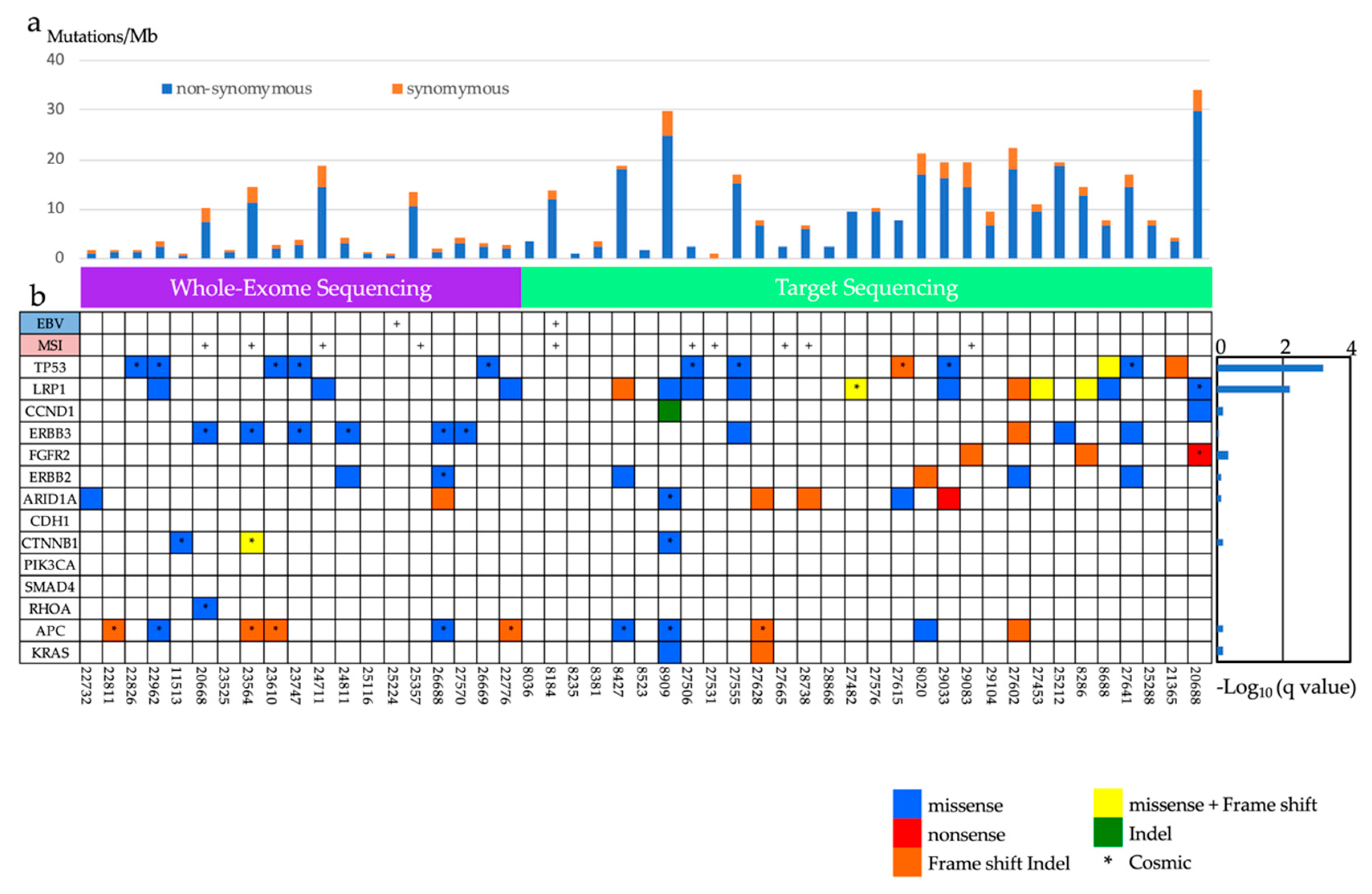
2.1.2. In the Screening Stage
2.1.3. The Analysis of Significantly Associated Genes for Different-Type Non-Invasive GC
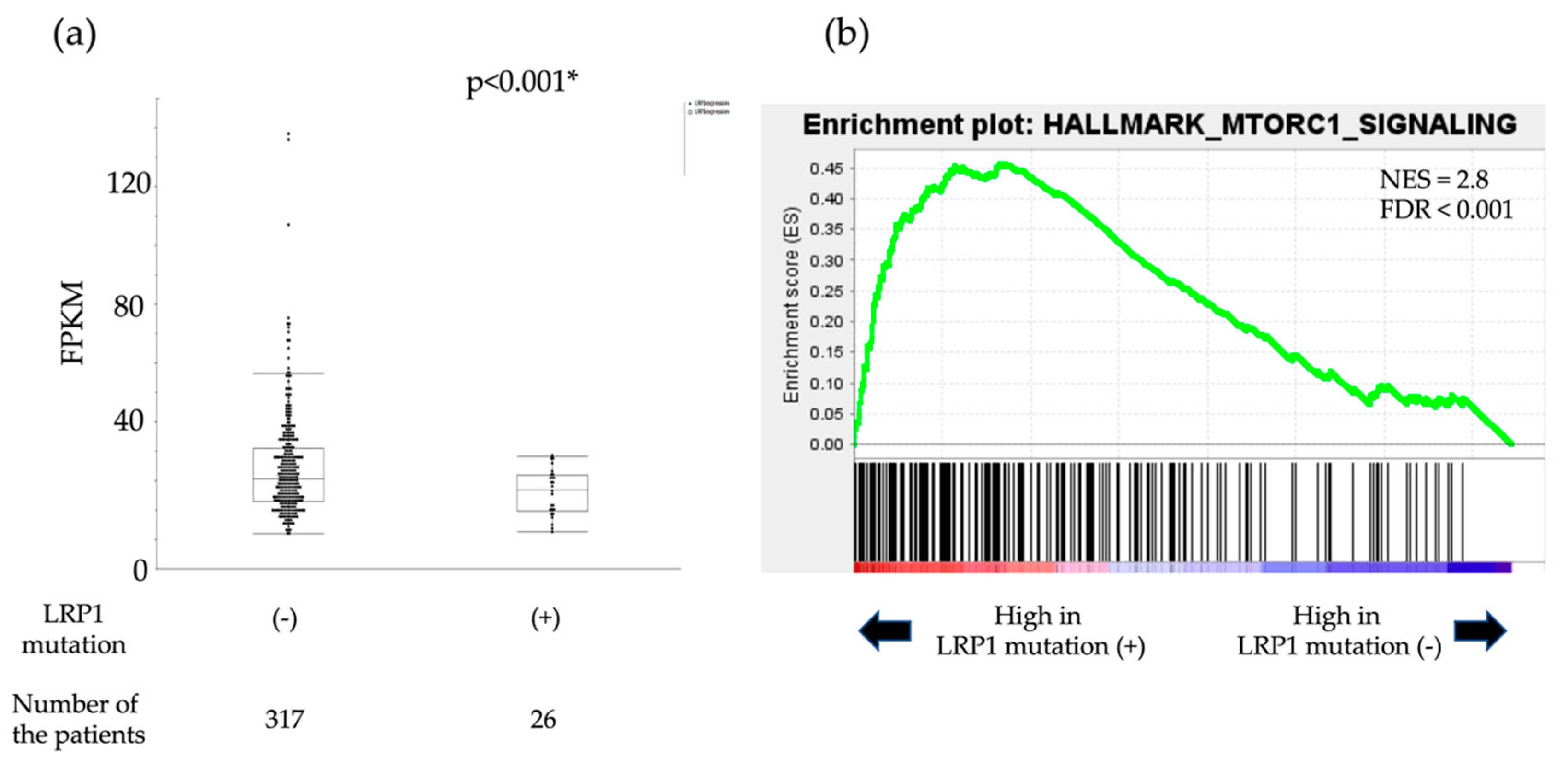
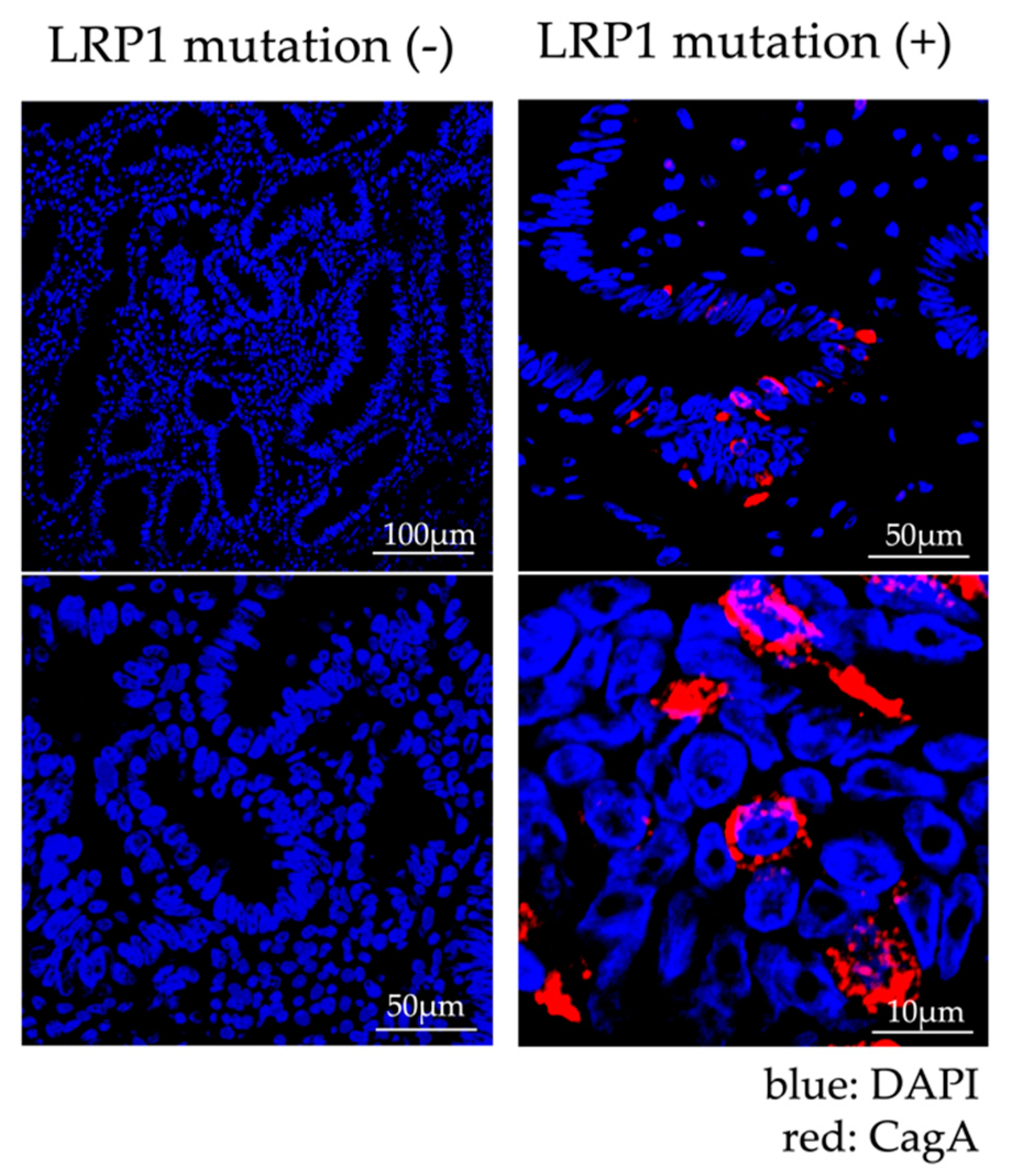
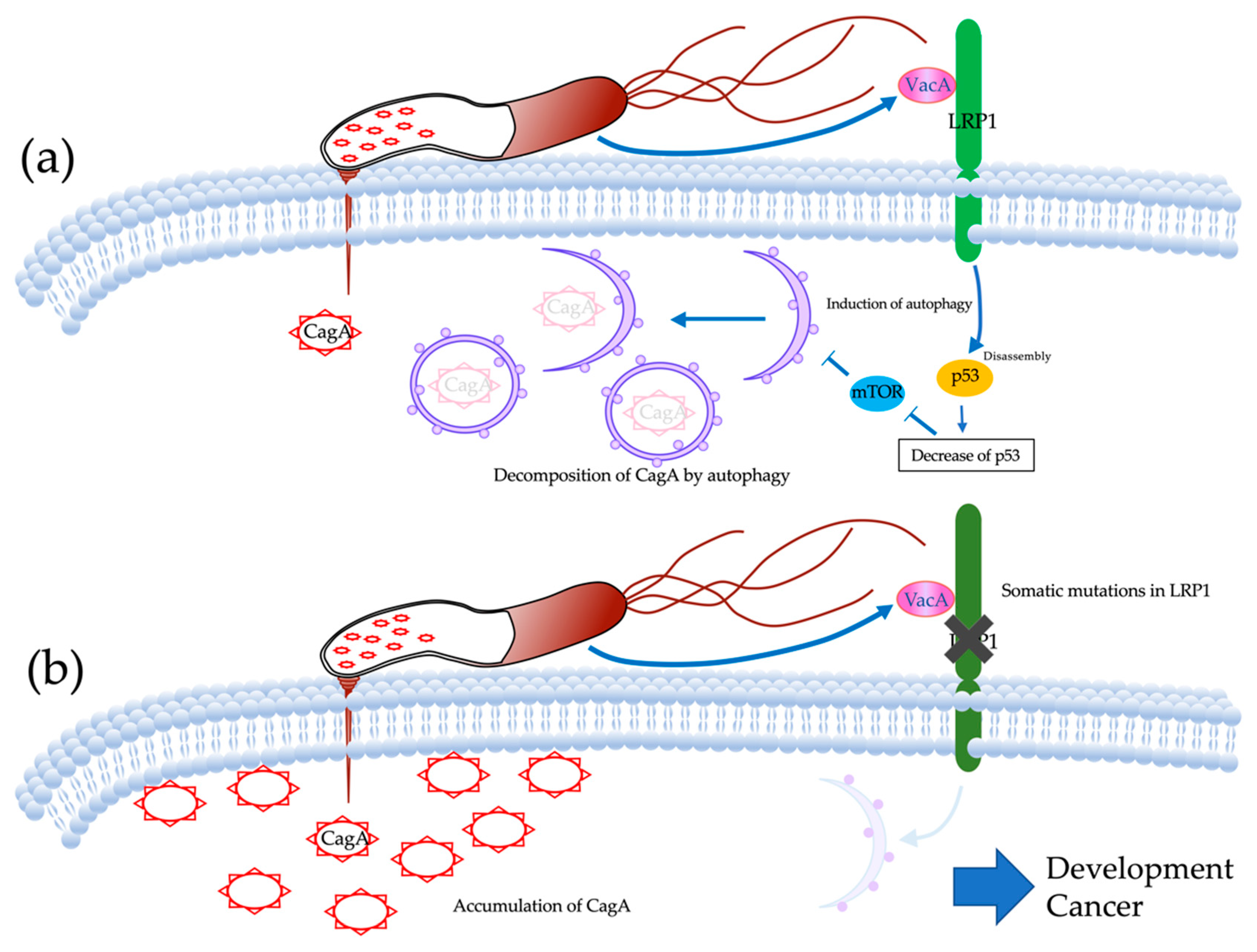
2.1.4. Analysis of Expression of LRP1 and Accumulation of CagA
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Evaluation of Hp Infection
4.3. Purification of DNA from Fresh Frozen and FFPE Tissue
4.4. Preparation and Sequencing of DNA Exome-Seq and Target-Seq Libraries
4.5. Sequence Analysis
4.6. Mutation Validation by Sanger Sequencing
4.7. Epstein-Barr Virus (EBV)
4.8. MSI Analyses
4.9. Analysis of Mutation Signature
4.10. Identification of Significantly Mutated Genes
4.11. LRP1 mRNA Analyses
4.12. Bioinformatics Analysis
4.13. Gene Set Enrichment Analysis
4.14. Immunofluorescence Staining of CagA
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ferlay, J. GLOBOCAN 2018 v1.0, Cancer Incidence and Mortality Worldwide. IARC. Available online: http://globocan.iarc.fr (accessed on 20 December 2019).
- Guggenheim, D.E.; Shah, M.A. Gastric cancer epidemiology and risk factors. J. Surg. Oncol. 2013, 107, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Okamoto, S.; Yamamoto, S.; Matsumura, N.; Yamaguchi, S.; Yamakido, M.; Taniyama, K.; Sasaki, N.; Schlemper, R.J. Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 2001, 345, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Russell, R.M. Nutrition and gastric cancer risk: An update. Nutr. Rev. 2008, 66, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.M.; Kim, E.H. Epstein-Barr Virus and Gastric Cancer Risk: A Meta-analysis With Meta-regression of Case-control Studies. J. Prev. Med. Public Health 2016, 49, 97–107. [Google Scholar] [CrossRef]
- Correa, P. Human gastric carcinogenesis: A multistep and multifactorial process--First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992, 52, 6735–6740. [Google Scholar]
- Matsumoto, Y.; Marusawa, H.; Kinoshita, K.; Endo, Y.; Kou, T.; Morisawa, T.; Azuma, T.; Okazaki, I.M.; Honjo, T.; Chiba, T. Helicobacter pylori infection triggers aberrant expression of activation- induced cytidine deaminase in gastric epithelium. Nat. Med. 2007, 13, 470–476. [Google Scholar] [CrossRef]
- Ohnishi, S.; Ma, N.; Thanan, R.; Pinlaor, R.; Hammam, O.; Murata, M.; Kawanishi, M. DNA damage in inflammation-related carcinogenesis and cancer stem cells. Oxid. Med. Cell. Longev. 2007, 13, 470–476. [Google Scholar] [CrossRef]
- Chin, L.; Andersen, J.N.; Futreal, P.A. Cancer genomics: From discovery science to personalized medicine. Nat. Med. 2011, 17, 297–303. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Zang, Z.J.; Cutcutache, I.; Poon, S.L.; Zhang, S.L.; McPherson, J.R.; Tao, J.; Rajasegaran, V.; Heng, H.L.; Deng, N.; Gan, A.; et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat. Genet. 2012, 44, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Rokutan, H.; Abe, H.; Nakamura, H.; Ushiku, T.; Arakawa, E.; Hosoda, F.; Yachida, S.; Tsuji, Y.; Fujishiro, M.; Koike, K.; et al. Initial and crucial genetic events in intestinal-type gastric intramucosal neoplasia. J. Pathol. 2019, 247, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Yahiro, K.; Satoh, M.; Nakano, M.; Hisatsune, J.; Isomoto, H.; Sap, J.; Suzuki, H.; Nomura, F.; Noda, M.; Moss, J.; et al. Low-density Lipoprotein Receptor-related Protein-1 (LRP1) Mediates Autophagy and Apoptosis Caused by Helicobacter pylori VacA. J. Biol. Chem. 2012, 287, 31104–31115. [Google Scholar] [CrossRef]
- Zhou, H.; Tan, S.; Li, H.; Lin, X. Expression and significance of EBV, ARID1A and PIK3CA in gastric carcinoma. Mol. Med. Rep. 2019, 10, 2125–2136. [Google Scholar] [CrossRef]
- Esteller, M. CpG island hypermethylation and tumor suppressor genes: A booming present, a brighter future. Oncogene 2002, 21, 5427–5440. [Google Scholar] [CrossRef]
- Ushijima, T.; Sasako, M. Focus on gastric cancer. Cancer Cell 2004, 5, 121–125. [Google Scholar] [CrossRef]
- Lillis, A.P.; Van Duyn, L.B.; Murphy-Ullrich, J.E.; Strickland, D.K. LDL receptor-related protein 1: Unique tissue-specific functions revealed by selective gene knockout studies. Physiol. Rev. 2008, 88, 887–918. [Google Scholar] [CrossRef]
- Van Gool, B.; Dedieu, S.; Emonard, H.; Roebroek, A.J. The Matricellular Receptor LRP1 Forms an Interface for Signaling and Endocytosis in Modulation of the Extracellular Tumor Environment. Front. Pharmacol. 2015, 6, 271. [Google Scholar] [CrossRef]
- Amos, S.; Mut, M.; diPierro, C.G.; Carpenter, J.E.; Xiao, A.; Kohutek, Z.A.; Redpath, G.T.; Zhao, Y.; Wang, J.; Shaffrey, M.E.; et al. Protein kinase C-α-mediated regulation of low-density lipoprotein receptor related protein and urokinase increases astrocytoma invasion. Cancer Res. 2007, 67, 10241–10251. [Google Scholar] [CrossRef]
- Hatakeyama, M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat. Rev. Cancer 2004, 4, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Higashi, H.; Tsutsumi, R.; Muto, S.; Sugiyama, T.; Azuma, T.; Asaka, M.; Hatakeyama, M. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 2002, 295, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Ohta, T.; Hatakeyama, M. Stability of Helicobacter pylori CagA oncoprotein in human gastric epithelial cells. FEBS Lett. 2009, 583, 2414–2418. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Suzuki, H.; Saya, H.; Hatakeyama, M.; Hirayama, T.; Hirata, K.; Nagano, O.; Matsuzaki, J.; Hibi, T. Reactive oxygen species-induced autophagic degradation of Helicobacter pylori CagA is specifically suppressed in cancer stem-like cells. Cell Host Microbe 2012, 12, 764–777. [Google Scholar] [CrossRef] [PubMed]
- Terebiznik, M.R.; Raju, D.; Vázquez, C.L.; Torbricki, K.; Kulkarni, R.; Blanke, S.R.; Yoshimori, T.; Colombo, M.I.; Jones, N.L. Effect of Helicobacter pylori’s vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy 2009, 5, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Raju, D.; Hussey, S.; Ang, M.; Terebiznik, M.R.; Sibony, M.; Galindo-Mata, E.; Gupta, V.; Blanke, S.R.; Delgado, A.; Romero-Gallo, J.; et al. Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology 2012, 142, 1160–1171. [Google Scholar] [CrossRef]
- Van Doorn, L.J.; Figueiredo, C.; Mégraud, F.; Pena, S.; Midolo, P.; Queiroz, D.M.; Carneiro, F.; Vanderborght, B.; Pegado, M.D.; Sanna, R.; et al. Geographic Distribution of vacA Allelic Types of Helicobacter pylori. Gastroenterology 1999, 116, 823–830. [Google Scholar] [CrossRef]
- Ito, Y.; Azuma, T.; Ito, S.; Miyaji, H.; Hirai, M.; Yamazaki, Y.; Sato, F.; Kato, T.; Kohli, Y.; Kuriyama, M. Analysis and Typing of the vacA Gene from cagA-Positive Strains of Helicobacter pylori Isolated in Japan. J. Clin. Microbiol. 1997, 35, 1710–1714. [Google Scholar] [CrossRef]
- Takaoka, S.; Hirotsu, Y.; Ohyama, H.; Mochizuki, H.; Amemiya, K.; Oyama, T.; Ashizawa, H.; Yoshimura, D.; Nakagomi, K.; Hosoda, K.; et al. Molecular subtype switching in early-stage gastric cancers with multiple occurrences. J. Gastroenterol. 2019, 54, 674–686. [Google Scholar] [CrossRef]
- Mizuguchi, A.; Takai, A.; Shimizu, T.; Matsumoto, T.; Kumagai, K.; Miyamoto, S.; Seno, H.; Marusawa, H. Genetic features of multicentric/multifocal intramucosal gastric carcinoma. Int. J. Cancer 2018, 143, 1923–1934. [Google Scholar]
- Yoshida, T.; Yamaguchi, T.; Maekawa, S.; Takano, S.; Kuno, T.; Tanaka, K.; Iwamoto, F.; Tsukui, Y.; Kobayashi, S.; Asakawa, Y.; et al. Identification of early genetic changes in well-differentiated intramucosal gastric carcinoma by target deep sequencing. Gastric Cancer 2019, 22, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: Development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar] [PubMed]
- Hirano, D.; Urabe, Y.; Tanaka, S.; Nakamura, K.; Ninomiya, Y.; Yuge, R.; Hayashi, R.; Oka, S.; Kitadai, Y.; Shimamoto, F.; et al. Early-stage serrated adenocarcinomas are divided into several molecularly distinct subtypes. PLoS ONE 2019, 14, e0211477. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Gay, M.; Vila-Casadesús, M.; Franch-Expósito, S.; Hernández-Illán, E.; Lozano, J.; Castellví-Bel, S. Mutational Signatures in Cancer (MuSiCa): A web application to implement mutational signatures analysis in cancer samples. BMC Bioinform. 2018, 19, 224. [Google Scholar] [CrossRef]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, K.; Urabe, Y.; Kagemoto, K.; Yuge, R.; Hayashi, R.; Ono, A.; Hayes, C.N.; Oka, S.; Ito, M.; Nishisaka, T.; et al. Genomic Characterization of Non-Invasive Differentiated-Type Gastric Cancer in the Japanese Population. Cancers 2020, 12, 510. https://doi.org/10.3390/cancers12020510
Nakamura K, Urabe Y, Kagemoto K, Yuge R, Hayashi R, Ono A, Hayes CN, Oka S, Ito M, Nishisaka T, et al. Genomic Characterization of Non-Invasive Differentiated-Type Gastric Cancer in the Japanese Population. Cancers. 2020; 12(2):510. https://doi.org/10.3390/cancers12020510
Chicago/Turabian StyleNakamura, Koki, Yuji Urabe, Kenichi Kagemoto, Ryo Yuge, Ryohei Hayashi, Atsushi Ono, C. Nelson Hayes, Shiro Oka, Masanori Ito, Takashi Nishisaka, and et al. 2020. "Genomic Characterization of Non-Invasive Differentiated-Type Gastric Cancer in the Japanese Population" Cancers 12, no. 2: 510. https://doi.org/10.3390/cancers12020510
APA StyleNakamura, K., Urabe, Y., Kagemoto, K., Yuge, R., Hayashi, R., Ono, A., Hayes, C. N., Oka, S., Ito, M., Nishisaka, T., Tanabe, K., Arihiro, K., Ohdan, H., Tanaka, S., & Chayama, K. (2020). Genomic Characterization of Non-Invasive Differentiated-Type Gastric Cancer in the Japanese Population. Cancers, 12(2), 510. https://doi.org/10.3390/cancers12020510