A Comprehensive Analysis of FUT8 Overexpressing Prostate Cancer Cells Reveals the Role of EGFR in Castration Resistance


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
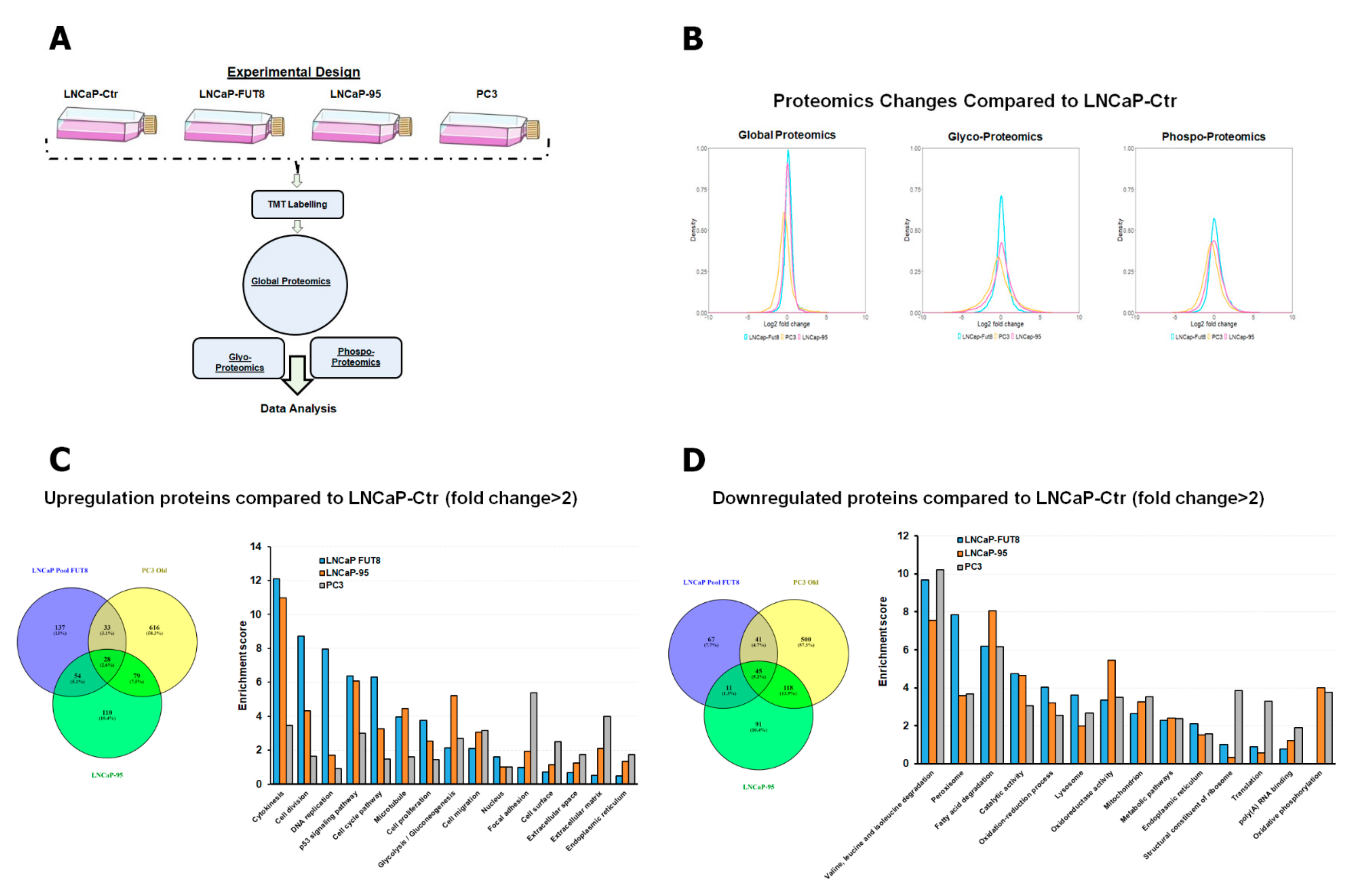
2.1. Characterization of FUT8 Expressing Prostate Cancer Cells using LC MS/MS Mass Spectrometry
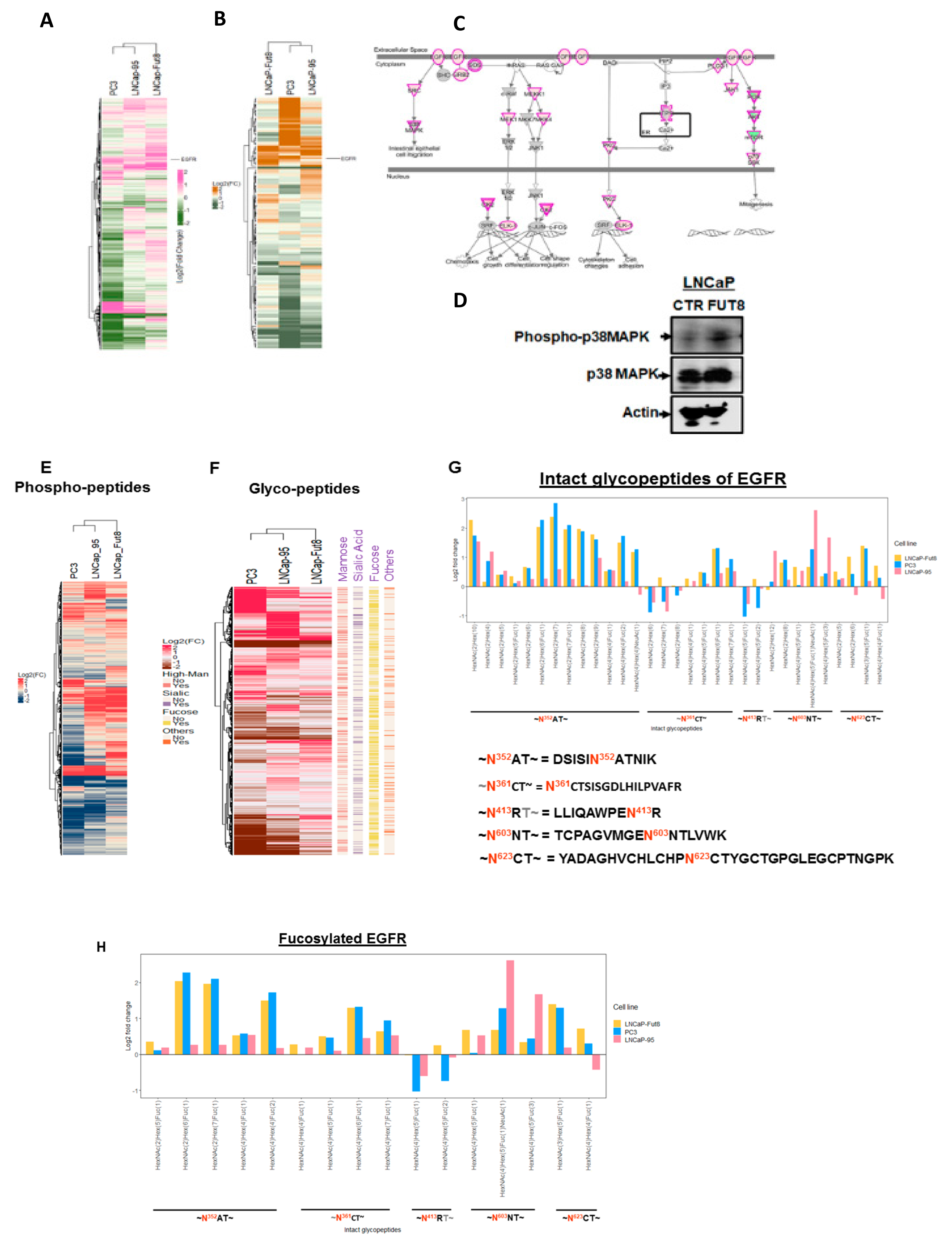
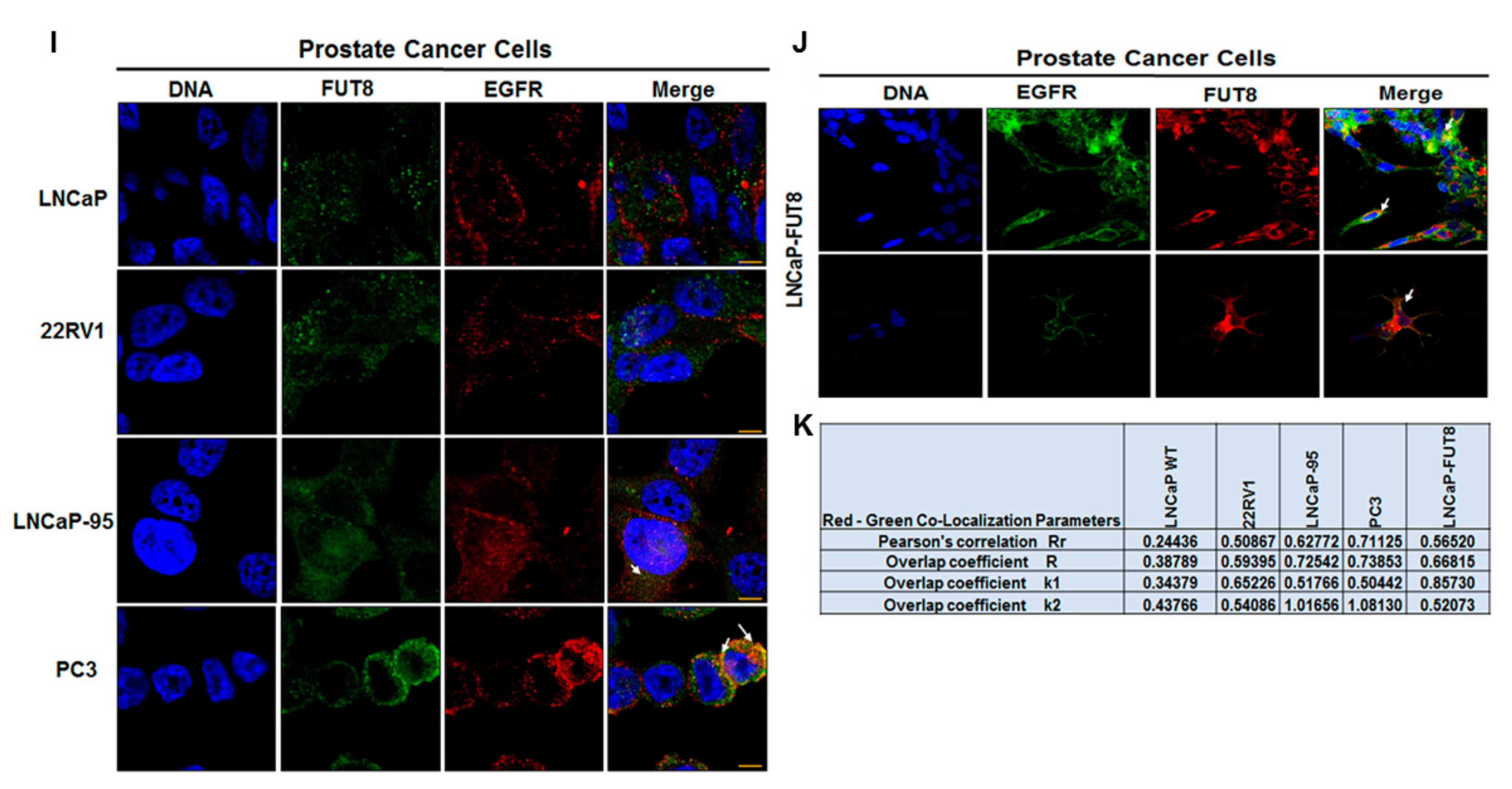
2.2. Cell Surface Receptors and FUT8 Overexpression
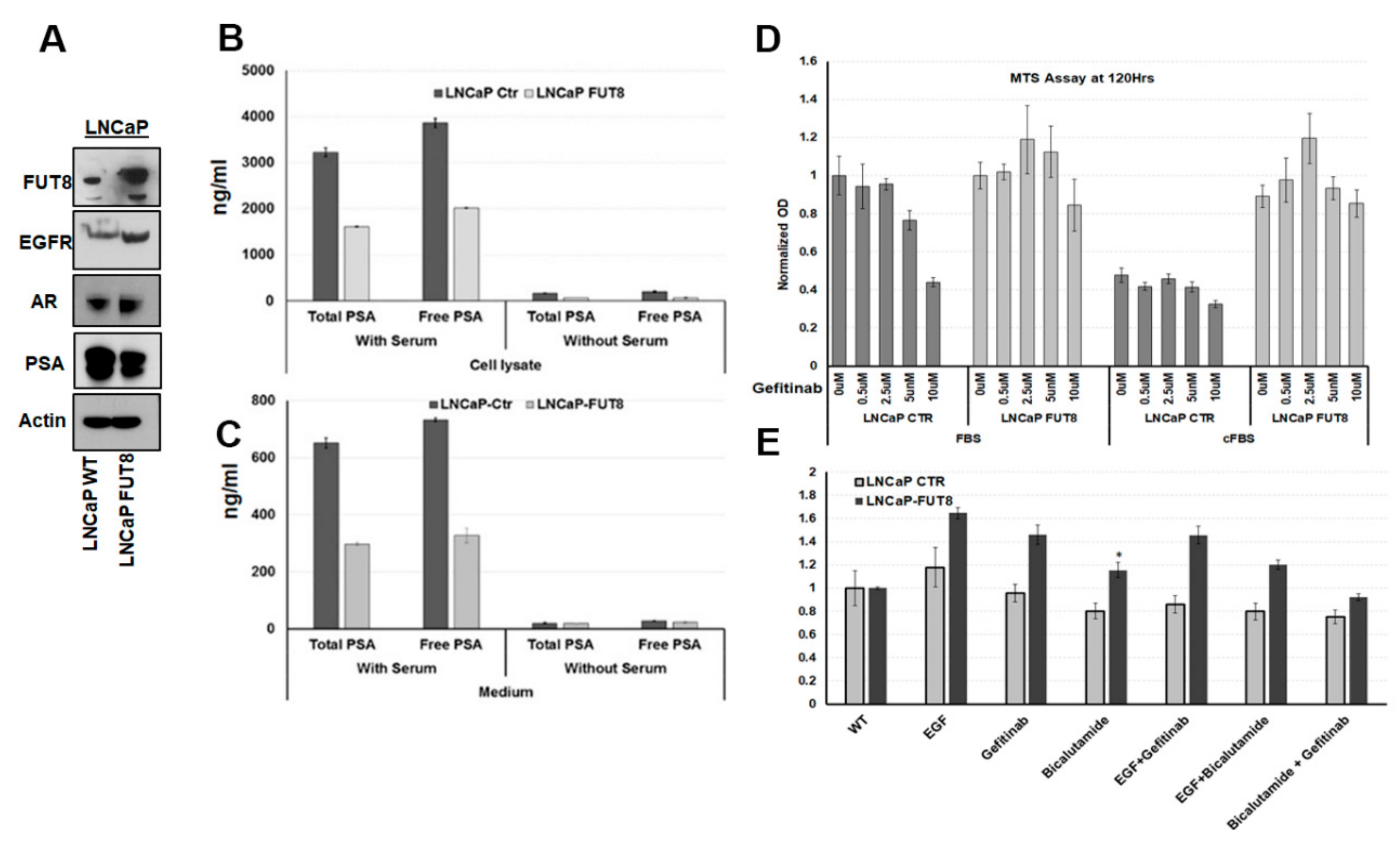
2.3. Overexpression of FUT8 Suppress the PSA Production and Increase Drug Resistance in Prostate Cancer Cells
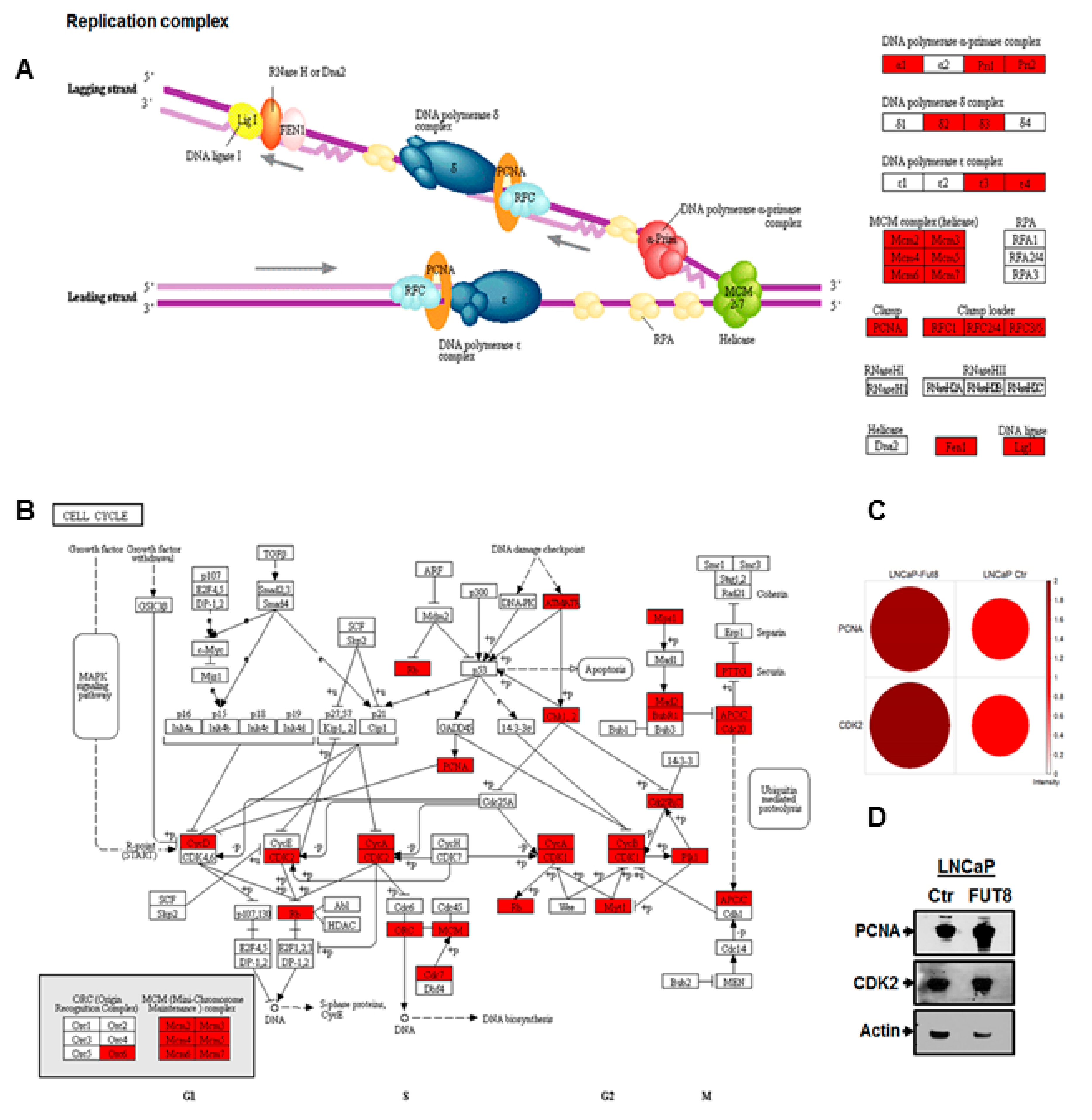
2.4. Overexpression of FUT8 Promotes DNA Replication and Cell Cycle Progression
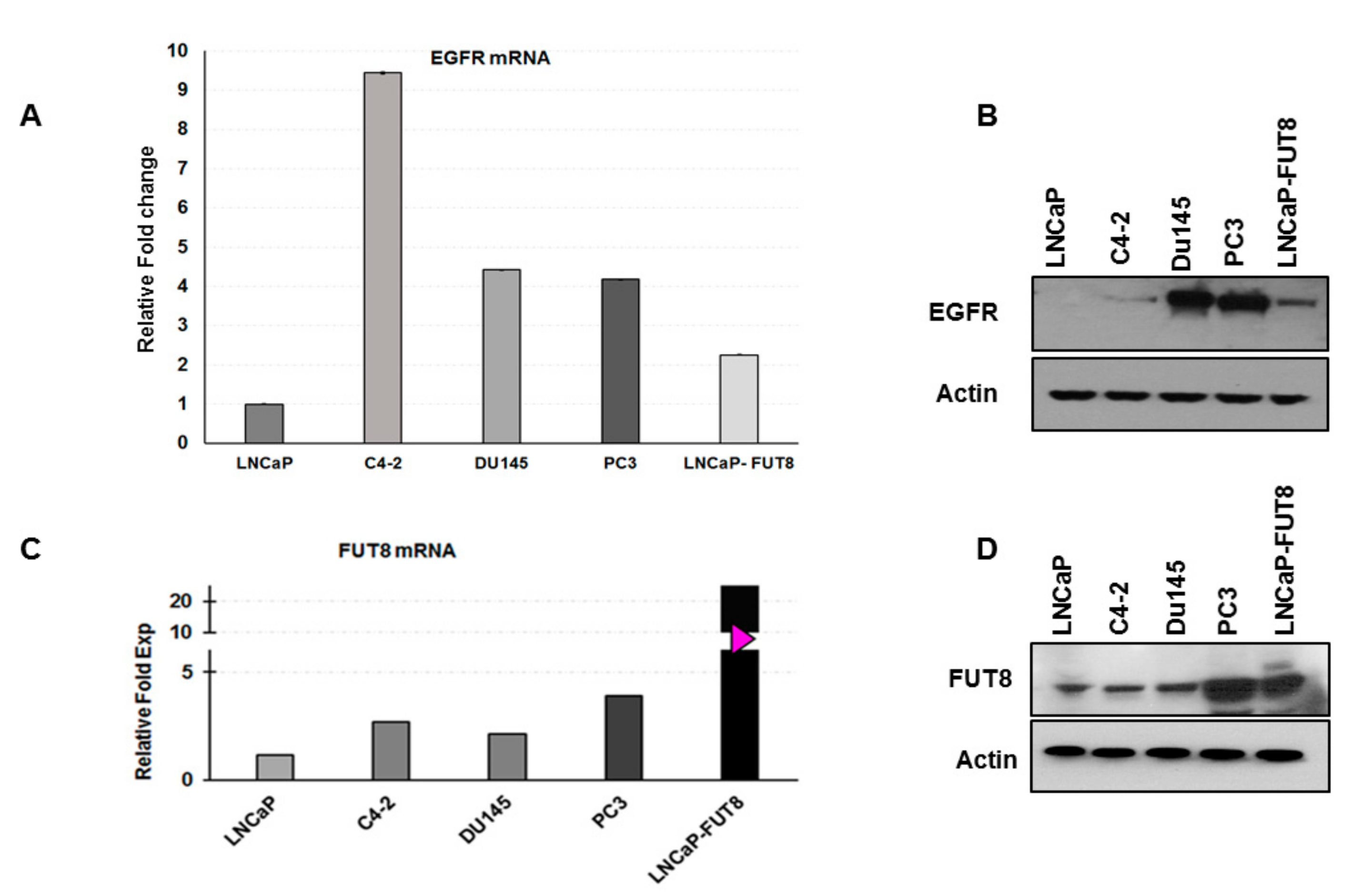
2.5. Regulation of EGFR Expression by FUT8
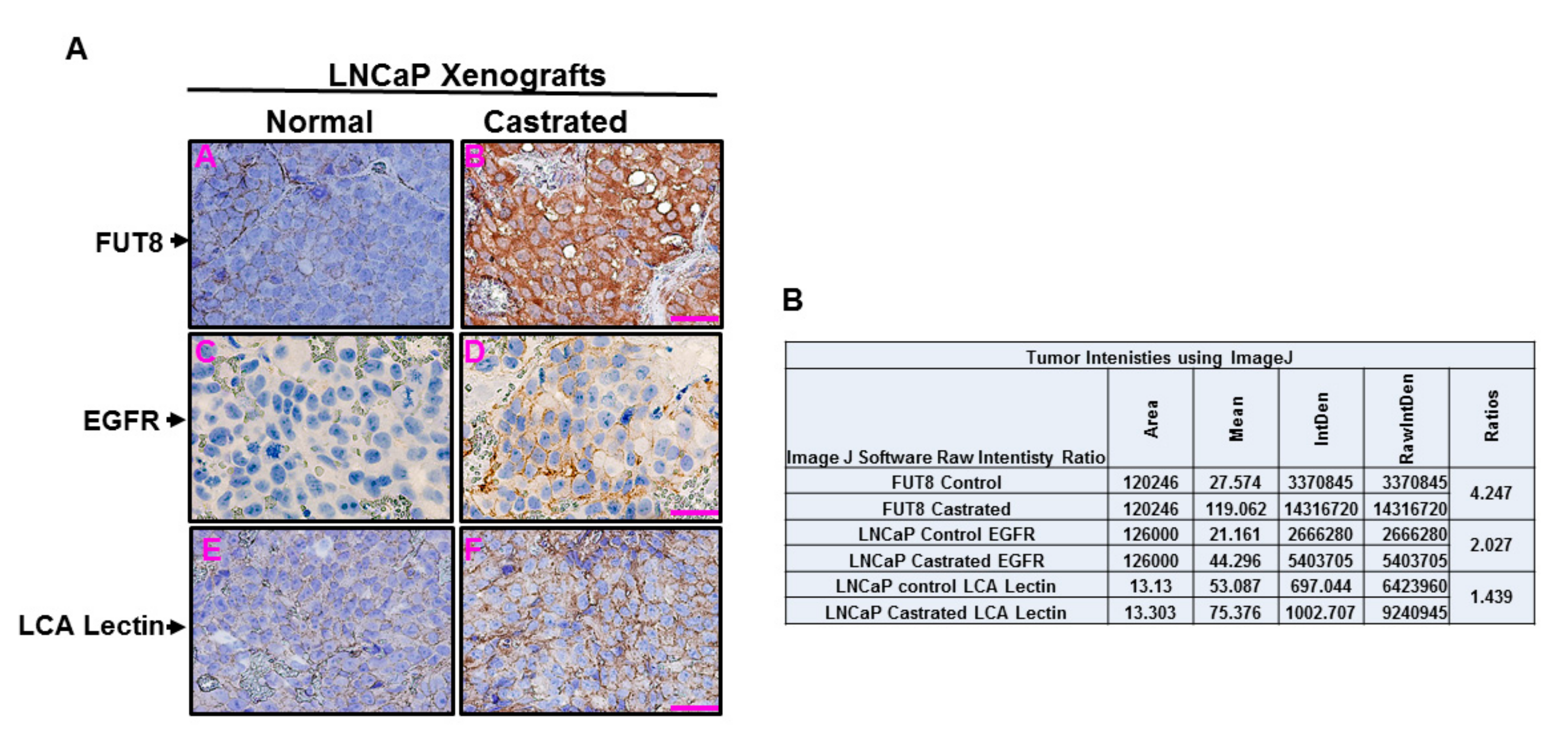
2.6. Castration-Induced FUT8 Overexpression Correlates with EGFR Expression in Xenograft Model
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Western Blot Analysis
4.3. Protein and Peptide Extraction from Cells for Proteomic Analysis
4.4. TMT Labeling of Tryptic Peptides from Prostate Cancer Cell Lines
4.5. Chromatography Fractionation
4.6. Mass Spectrometric Analysis
4.7. Protein Expression Data Analysis
4.8. qRT-PCR Analysis
- FUT8-Forward: GACAGAACTGGTTCAGCGGAGA
- FUT8-Reverse: GCAGTAGACCACATGATGGAGC
- Actin-Forward: CACCATTGGCAATGAGCGGTTC
- Actin-Reverse: AGGTCTTTGCGGATGTCCACGT
- EGFR-Forward: AACACCCTGGTCTGGAAGTACG
- EGFR-Reverse: TCGTTGGACAGCCTTCAAGACC
4.9. Xenograft Animal Models
4.10. Immuno-Histochemical (IHC) Staining
4.11. Fluorescence Microscopy
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gomella, L.G. Prostate cancer statistics: Anything you want them to be. Can. J. Urol. 2017, 24, 8603–8604. [Google Scholar]
- Sakr, W.A.; Grignon, D.J.; Crissman, J.D.; Heilbrun, L.K.; Cassin, B.J.; Pontes, J.J.; Haas, G.P. High grade prostatic intraepithelial neoplasia (hgpin) and prostatic adenocarcinoma between the ages of 20–69: An autopsy study of 249 cases. In Vivo 1994, 8, 439–443. [Google Scholar] [PubMed]
- Woolf, S.H. Screening for prostate cancer with prostate-specific antigen. An examination of the evidence. N. Engl. J. Med. 1995, 333, 1401–1405. [Google Scholar] [CrossRef] [PubMed]
- Stangelberger, A.; Waldert, M.; Djavan, B. Prostate cancer in elderly men. Rev. Urol. 2008, 10, 111–119. [Google Scholar]
- Zaid, U.B.; McAninch, J.W.; Glass, A.S.; Cinman, N.M.; Breyer, B.N. Presentation, management, and outcomes of complications following prostate cancer therapy. Transl. Androl. Urol. 2014, 3, 150–155. [Google Scholar] [PubMed]
- Perlmutter, M.A.; Lepor, H. Androgen deprivation therapy in the treatment of advanced prostate cancer. Rev. Urol. 2007, 9, 3–8. [Google Scholar]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef]
- Chen, C.Y.; Jan, Y.H.; Juan, Y.H.; Yang, C.J.; Huang, M.S.; Yu, C.J.; Yang, P.C.; Hsiao, M.; Hsu, T.L.; Wong, C.H. Fucosyltransferase 8 as a functional regulator of nonsmall cell lung cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 630–635. [Google Scholar] [CrossRef]
- Ma, B.; Simala-Grant, J.L.; Taylor, D.E. Fucosylation in prokaryotes and eukaryotes. Glycobiology 2006, 16, 158–184. [Google Scholar] [CrossRef]
- Sturla, L.; Fruscione, F.; Noda, K.; Miyoshi, E.; Taniguchi, N.; Contini, P.; Tonetti, M. Core fucosylation of n-linked glycans in leukocyte adhesion deficiency/congenital disorder of glycosylation iic fibroblasts. Glycobiology 2005, 15, 924–934. [Google Scholar] [CrossRef][Green Version]
- Lux, A.; Yu, X.; Scanlan, C.N.; Nimmerjahn, F. Impact of immune complex size and glycosylation on igg binding to human fcgammars. J. Immunol. 2013, 190, 4315–4323. [Google Scholar] [CrossRef] [PubMed]
- Raymond, C.; Robotham, A.; Spearman, M.; Butler, M.; Kelly, J.; Durocher, Y. Production of alpha2,6-sialylated igg1 in cho cells. mAbs 2015, 7, 571–583. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Hashimoto, H.; Okayasu, N.; Kameyama, A.; Onogi, H.; Nakagawasai, O.; Nakazawa, T.; Kurosawa, T.; Hao, Y.; Isaji, T.; et al. Alpha1,6-fucosyltransferase-deficient mice exhibit multiple behavioral abnormalities associated with a schizophrenia-like phenotype: Importance of the balance between the dopamine and serotonin systems. J. Biol. Chem. 2011, 286, 18434–18443. [Google Scholar] [CrossRef]
- Wang, X.; Inoue, S.; Gu, J.; Miyoshi, E.; Noda, K.; Li, W.; Mizuno-Horikawa, Y.; Nakano, M.; Asahi, M.; Takahashi, M.; et al. Dysregulation of tgf-beta1 receptor activation leads to abnormal lung development and emphysema-like phenotype in core fucose-deficient mice. Proc. Natl. Acad. Sci. USA 2005, 102, 15791–15796. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, W.; Ikeda, Y.; Miyagawa, J.I.; Taniguchi, M.; Miyoshi, E.; Sheng, Y.; Ekuni, A.; Ko, J.H.; Yamamoto, Y.; et al. Ectopic expression of alpha1,6 fucosyltransferase in mice causes steatosis in the liver and kidney accompanied by a modification of lysosomal acid lipase. Glycobiology 2001, 11, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, M.A.; Weinberg, J.M. New wrinkles in old receptors: Core fucosylation is yet another target to inhibit tgf-beta signaling. Kidney Int. 2013, 84, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Itoh, S.; Wang, X.; Isaji, T.; Miyoshi, E.; Kariya, Y.; Miyazaki, K.; Kawasaki, N.; Taniguchi, N.; Gu, J. Deletion of core fucosylation on alpha3beta1 integrin down-regulates its functions. J. Biol. Chem. 2006, 281, 38343–38350. [Google Scholar] [CrossRef]
- Osumi, D.; Takahashi, M.; Miyoshi, E.; Yokoe, S.; Lee, S.H.; Noda, K.; Nakamori, S.; Gu, J.; Ikeda, Y.; Kuroki, Y.; et al. Core fucosylation of e-cadherin enhances cell-cell adhesion in human colon carcinoma widr cells. Cancer Sci. 2009, 100, 888–895. [Google Scholar] [CrossRef]
- Hoti, N.; Yang, S.; Hu, Y.; Shah, P.; Haffner, M.C.; Zhang, H. Overexpression of alpha (1,6) fucosyltransferase in the development of castration-resistant prostate cancer cells. Prostate Cancer Prostatic Dis. 2018, 21, 137–146. [Google Scholar] [CrossRef]
- Wang, X.; Chen, J.; Li, Q.K.; Peskoe, S.B.; Zhang, B.; Choi, C.; Platz, E.A.; Zhang, H. Overexpression of alpha (1,6) fucosyltransferase associated with aggressive prostate cancer. Glycobiology 2014, 24, 935–944. [Google Scholar] [CrossRef]
- Wang, Y.; Fukuda, T.; Isaji, T.; Lu, J.; Gu, W.; Lee, H.H.; Ohkubo, Y.; Kamada, Y.; Taniguchi, N.; Miyoshi, E.; et al. Loss of α1,6-fucosyltransferase suppressed liver regeneration: Implication of core fucose in the regulation of growth factor receptor-mediated cellular signaling. Sci. Rep. 2015, 5, 8264. [Google Scholar] [CrossRef] [PubMed]
- Hakariya, T.; Shida, Y.; Sakai, H.; Kanetake, H.; Igawa, T. Egfr signaling pathway negatively regulates psa expression and secretion via the pi3k-akt pathway in lncap prostate cancer cells. Biochem. Biophys. Res. Commun. 2006, 342, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Hoti, N.; Chowdhury, W.; Hsieh, J.T.; Sachs, M.D.; Lupold, S.E.; Rodriguez, R. Valproic acid, a histone deacetylase inhibitor, is an antagonist for oncolytic adenoviral gene therapy. Mol. Ther. 2006, 14, 768–778. [Google Scholar] [CrossRef]
- Hoti, N.; Li, Y.; Chen, C.L.; Chowdhury, W.H.; Johns, D.C.; Xia, Q.; Kabul, A.; Hsieh, J.T.; Berg, M.; Ketner, G.; et al. Androgen receptor attenuation of ad5 replication: Implications for the development of conditionally replication competent adenoviruses. Mol. Ther. 2007, 15, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Koundrioukoff, S.; Jonsson, Z.O.; Hasan, S.; de Jong, R.N.; van der Vliet, P.C.; Hottiger, M.O.; Hubscher, U. A direct interaction between proliferating cell nuclear antigen (pcna) and cdk2 targets pcna-interacting proteins for phosphorylation. J. Biol. Chem. 2000, 275, 22882–22887. [Google Scholar] [CrossRef]
- Nelson, P.S.; Clegg, N.; Arnold, H.; Ferguson, C.; Bonham, M.; White, J.; Hood, L.; Lin, B. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc. Natl. Acad. Sci. USA 2002, 99, 11890–11895. [Google Scholar] [CrossRef]
- Thompson, C.J.; Tam, N.N.; Joyce, J.M.; Leav, I.; Ho, S.M. Gene expression profiling of testosterone and estradiol-17 beta-induced prostatic dysplasia in noble rats and response to the antiestrogen ici 182,780. Endocrinology 2002, 143, 2093–2105. [Google Scholar] [CrossRef]
- Pignon, J.C.; Koopmansch, B.; Nolens, G.; Delacroix, L.; Waltregny, D.; Winkler, R. Androgen receptor controls egfr and erbb2 gene expression at different levels in prostate cancer cell lines. Cancer Res. 2009, 69, 2941–2949. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, W.; Hu, Y.; Hoti, N.; Liu, Y.; Shah, P.; Sun, S.; Clark, D.; Thomas, S.; Zhang, H. Site-specific fucosylation analysis identifying glycoproteins associated with aggressive prostate cancer cell lines using tandem affinity enrichments of intact glycopeptides followed by mass spectrometry. Anal. Chem. 2017, 89, 7623–7630. [Google Scholar] [CrossRef]
- Jones, H.E.; Goddard, L.; Gee, J.M.; Hiscox, S.; Rubini, M.; Barrow, D.; Knowlden, J.M.; Williams, S.; Wakeling, A.E.; Nicholson, R.I. Insulin-like growth factor-i receptor signalling and acquired resistance to gefitinib (zd1839; iressa) in human breast and prostate cancer cells. Endocr. Relat. Cancer 2004, 11, 793–814. [Google Scholar] [CrossRef]
- Jameson, M.J.; Beckler, A.D.; Taniguchi, L.E.; Allak, A.; Vanwagner, L.B.; Lee, N.G.; Thomsen, W.C.; Hubbard, M.A.; Thomas, C.Y. Activation of the insulin-like growth factor-1 receptor induces resistance to epidermal growth factor receptor antagonism in head and neck squamous carcinoma cells. Mol. Cancer Ther. 2011, 10, 2124–2134. [Google Scholar] [CrossRef] [PubMed]
- Pflug, B.R.; Reiter, R.E.; Nelson, J.B. Caveolin expression is decreased following androgen deprivation in human prostate cancer cell lines. Prostate 1999, 40, 269–273. [Google Scholar] [CrossRef]
- Hoti, N.; Yang, S.; Aiyetan, P.; Kumar, B.; Hu, Y.; Clark, D.; Eroglu, A.U.; Shah, P.; Johnson, T.; Chowdery, W.H.; et al. Overexpression of exportin-5 overrides the inhibitory effect of mirnas regulation control and stabilize proteins via posttranslation modifications in prostate cancer. Neoplasia 2017, 19, 817–829. [Google Scholar] [CrossRef]
- Johnson, T.J.; Hoti, N.; Liu, C.; Chowdhury, W.H.; Li, Y.; Zhang, Y.; Lupold, S.E.; Deweese, T.; Rodriguez, R. Bicalutamide-activated oncolytic adenovirus for the adjuvant therapy of high-risk prostate cancer. Cancer Gene Ther. 2013, 20, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Tang, L.C.; Krug, K.; Clark, D.J.; Gritsenko, M.A.; Chen, L.; Clauser, K.R.; Clauss, T.R.; Shah, P.; Gillette, M.A.; et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc. 2018, 13, 1632–1661. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Höti, N.; Lih, T.-S.; Pan, J.; Zhou, Y.; Yang, G.; Deng, A.; Chen, L.; Dong, M.; Yang, R.-B.; Tu, C.-F.; et al. A Comprehensive Analysis of FUT8 Overexpressing Prostate Cancer Cells Reveals the Role of EGFR in Castration Resistance. Cancers 2020, 12, 468. https://doi.org/10.3390/cancers12020468
Höti N, Lih T-S, Pan J, Zhou Y, Yang G, Deng A, Chen L, Dong M, Yang R-B, Tu C-F, et al. A Comprehensive Analysis of FUT8 Overexpressing Prostate Cancer Cells Reveals the Role of EGFR in Castration Resistance. Cancers. 2020; 12(2):468. https://doi.org/10.3390/cancers12020468
Chicago/Turabian StyleHöti, Naseruddin, Tung-Shing Lih, Jianbo Pan, Yangying Zhou, Ganglong Yang, Ashely Deng, Lijun Chen, Mingmimg Dong, Ruey-Bing Yang, Cheng-Fen Tu, and et al. 2020. "A Comprehensive Analysis of FUT8 Overexpressing Prostate Cancer Cells Reveals the Role of EGFR in Castration Resistance" Cancers 12, no. 2: 468. https://doi.org/10.3390/cancers12020468
APA StyleHöti, N., Lih, T.-S., Pan, J., Zhou, Y., Yang, G., Deng, A., Chen, L., Dong, M., Yang, R.-B., Tu, C.-F., Haffner, M. C., Kay Li, Q., & Zhang, H. (2020). A Comprehensive Analysis of FUT8 Overexpressing Prostate Cancer Cells Reveals the Role of EGFR in Castration Resistance. Cancers, 12(2), 468. https://doi.org/10.3390/cancers12020468