Oligo-Fucoidan Prevents M2 Macrophage Differentiation and HCT116 Tumor Progression


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
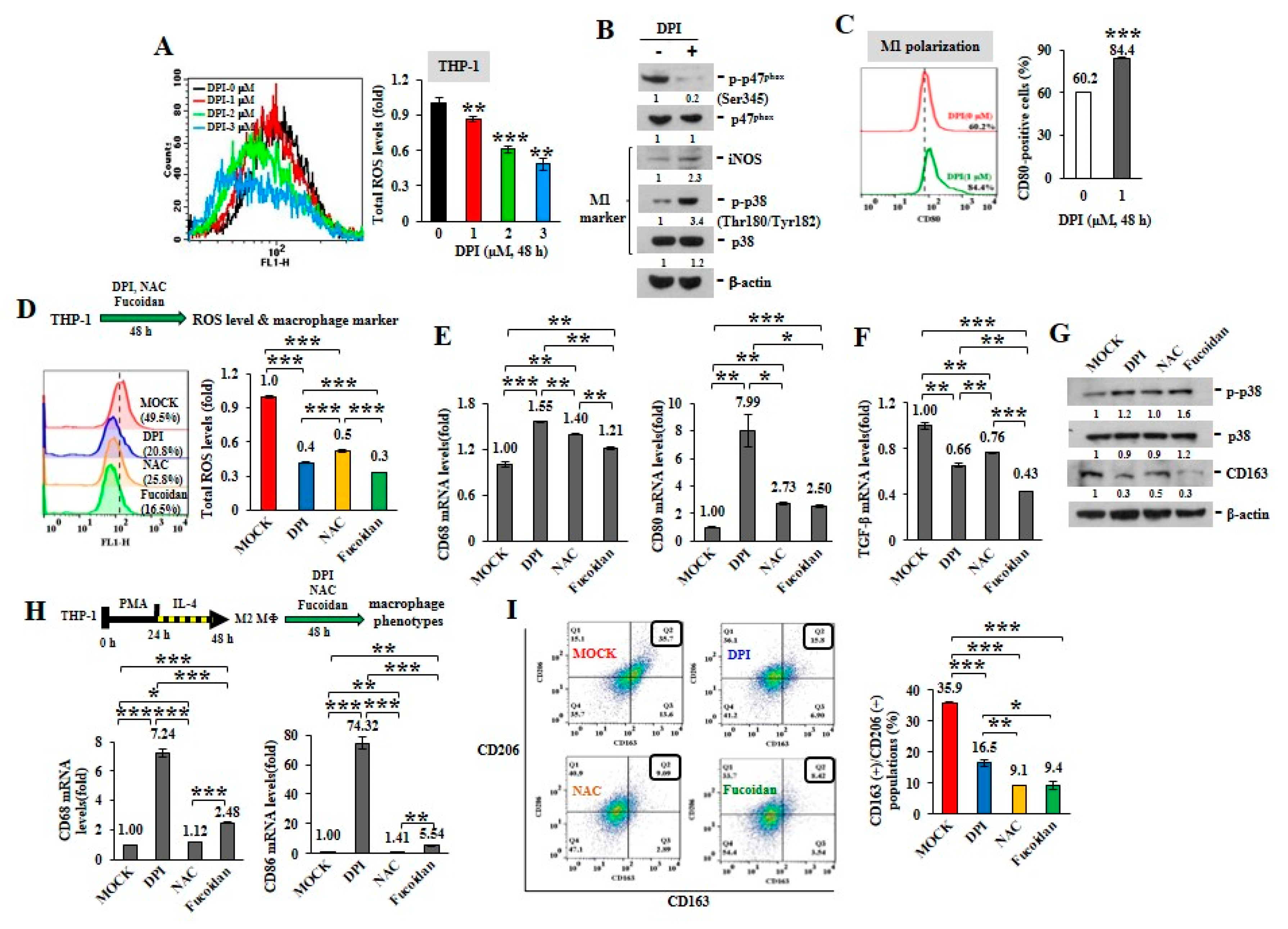
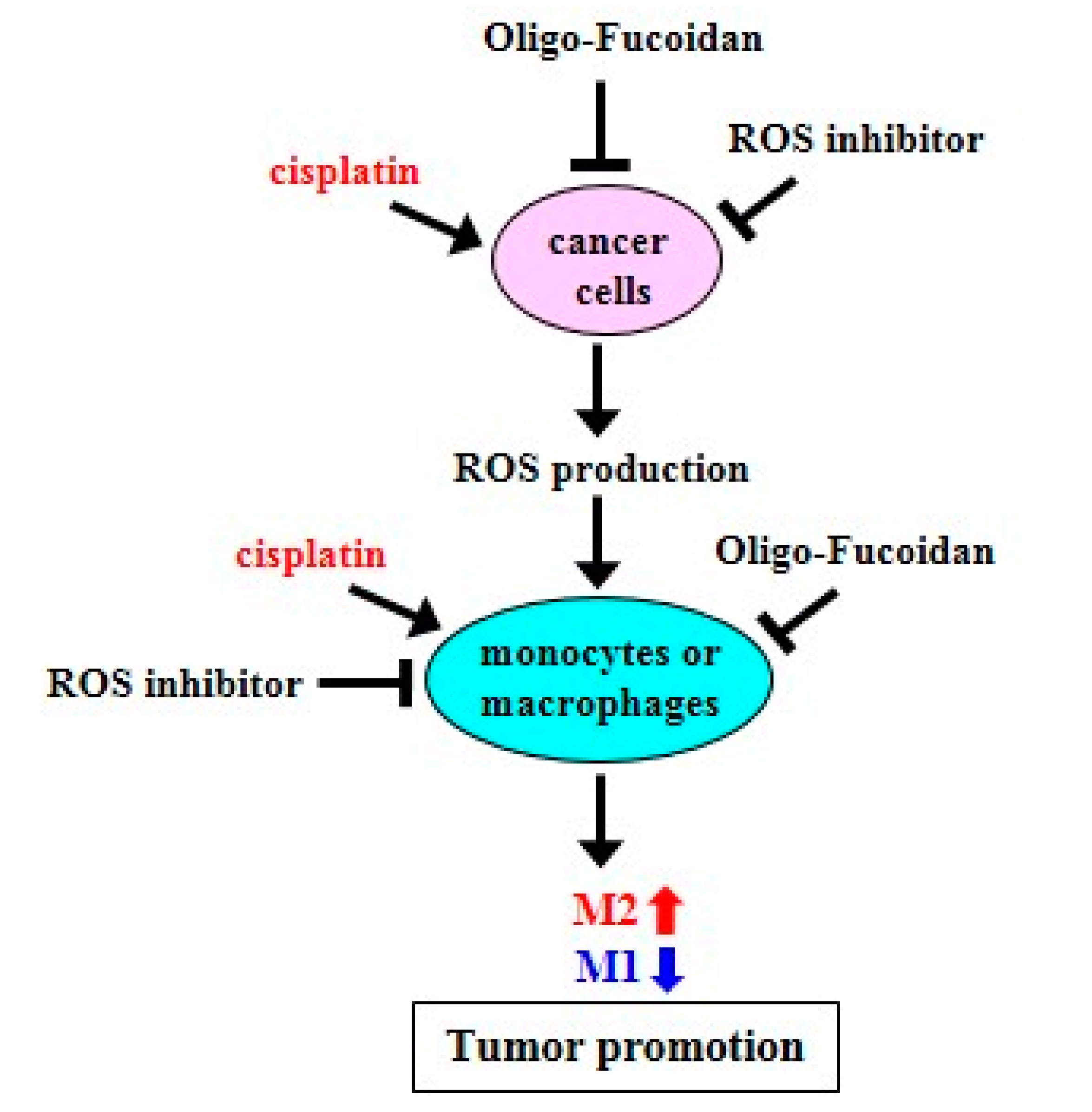
2.1. ROS Inhibitors and Oligo-Fucoidan Advance M1-Like Macrophage Polarization
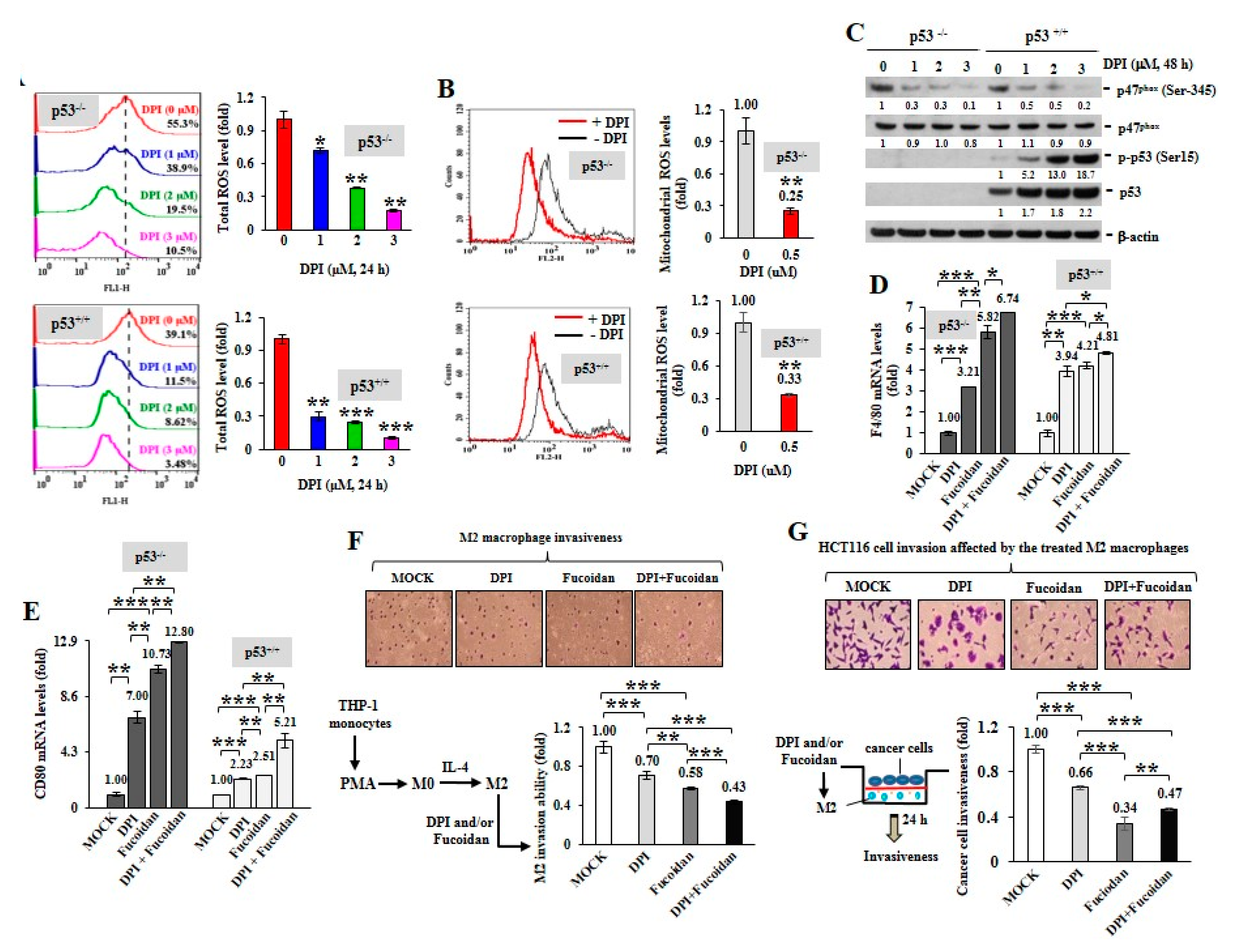
2.2. ROS Produced in Cancer Cells Influence Macrophage Differentiation
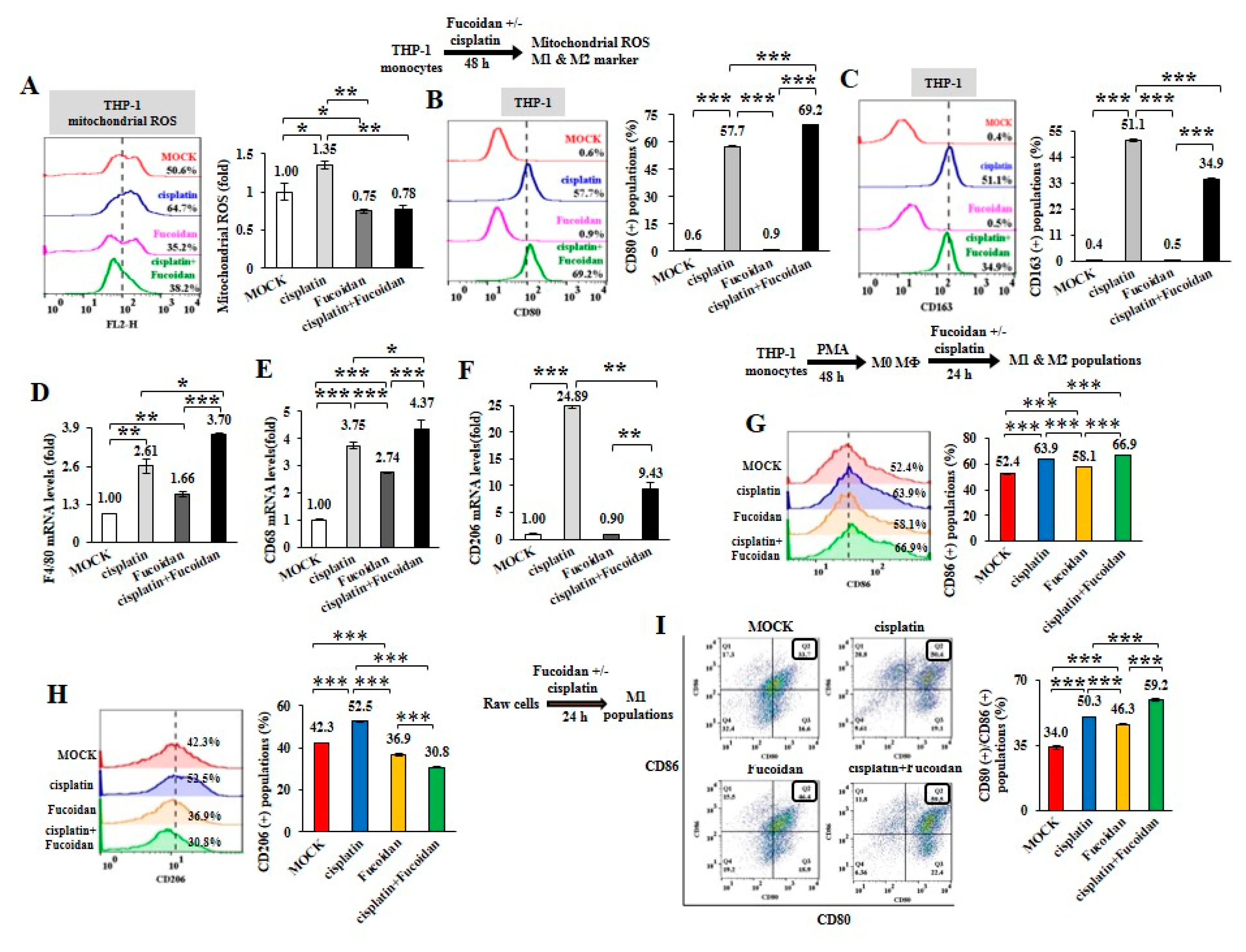
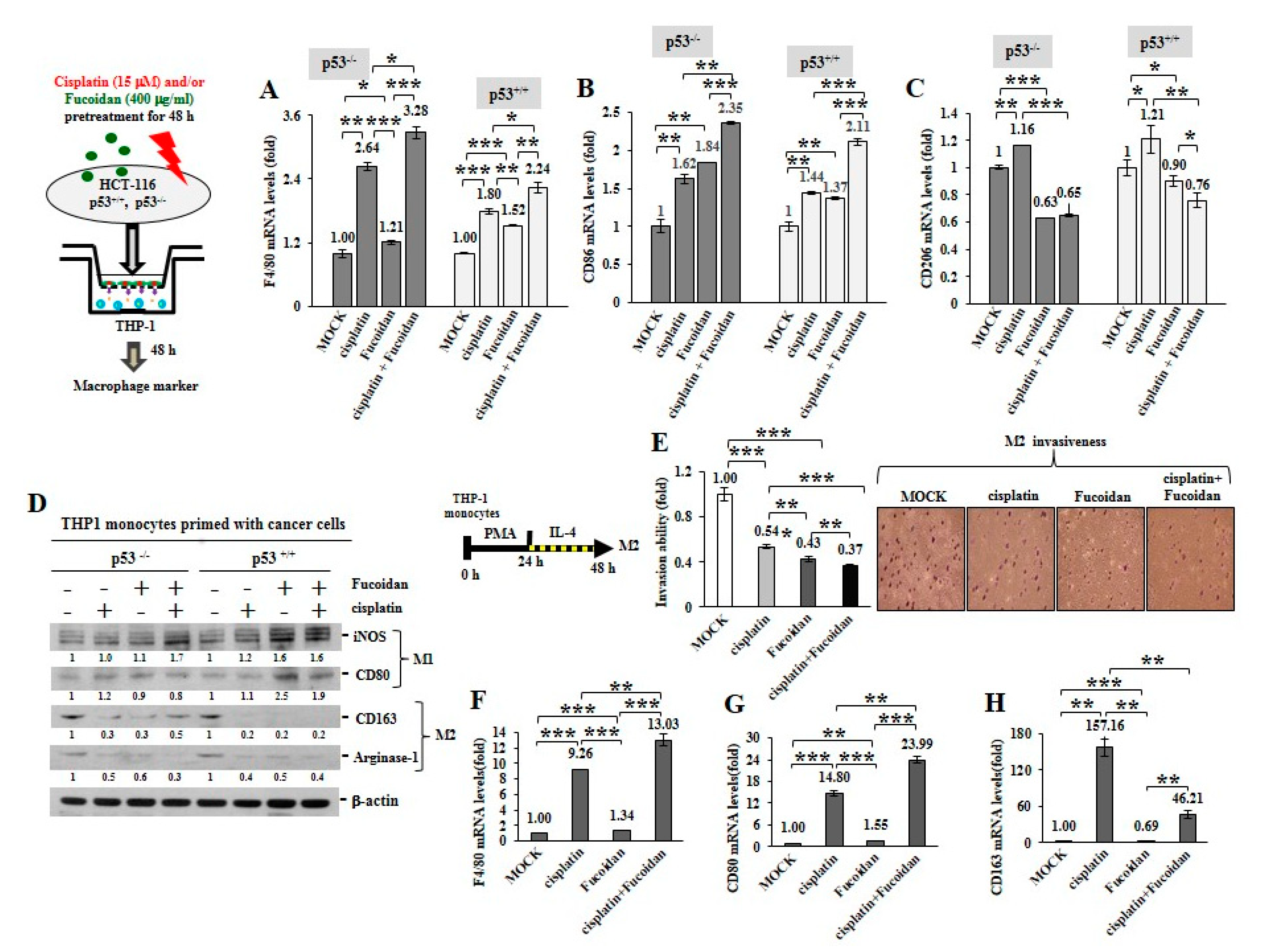
2.3. Cisplatin and Oligo-Fucoidan Cooperatively Promote the Polarization of M1 Macrophages
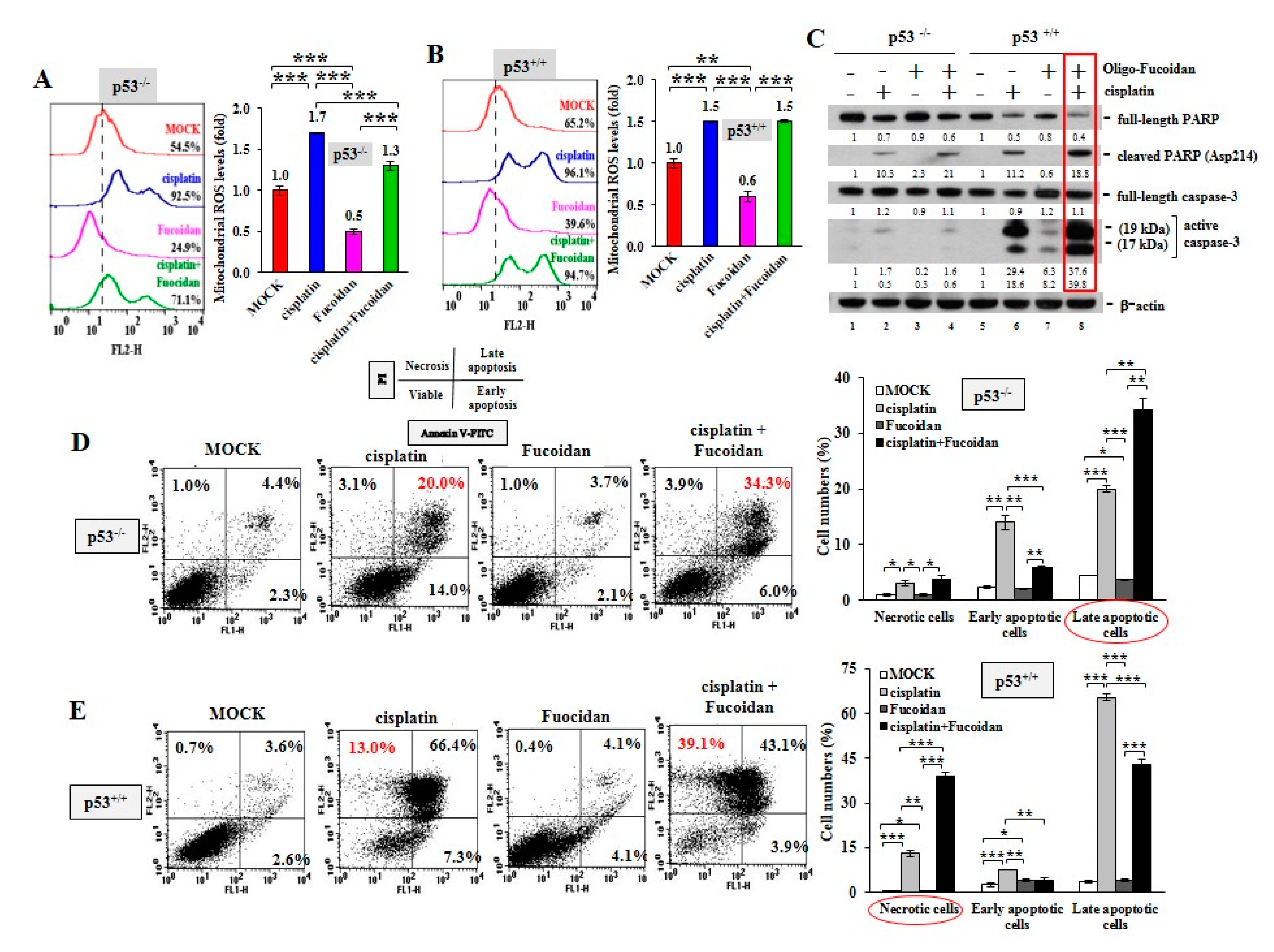
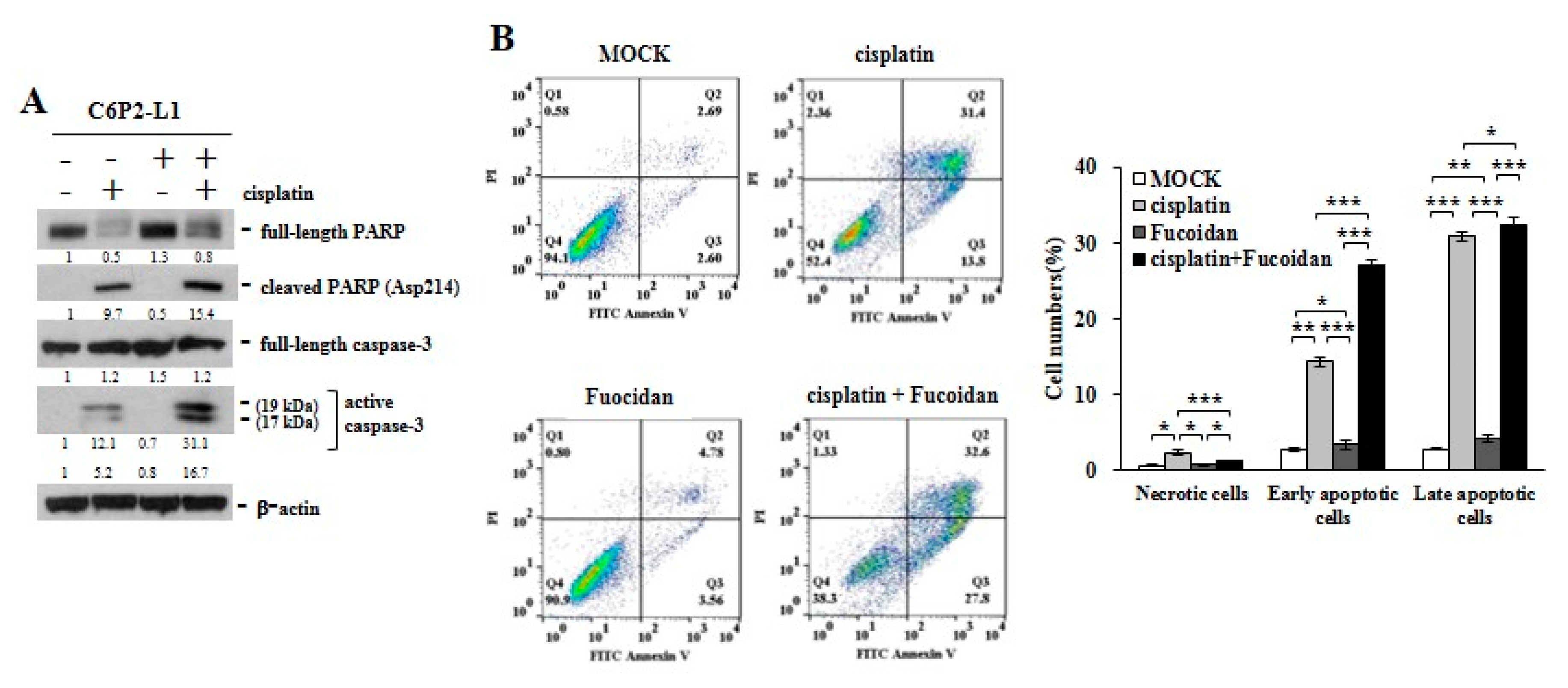
2.4. Cisplatin and Oligo-Fucoidan Additively Enhance Cancer Cell Death and Polarization of M1 Macrophages
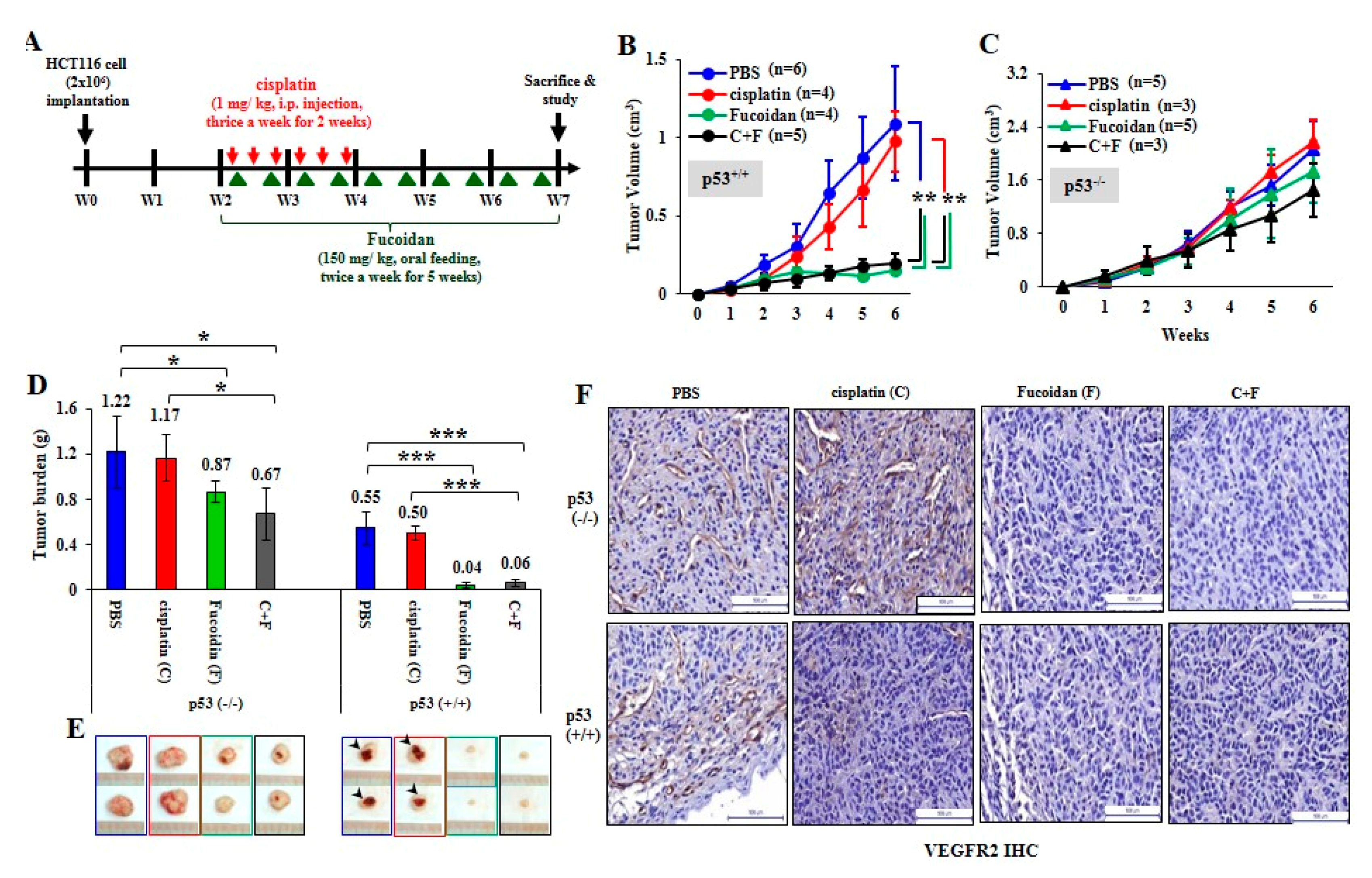
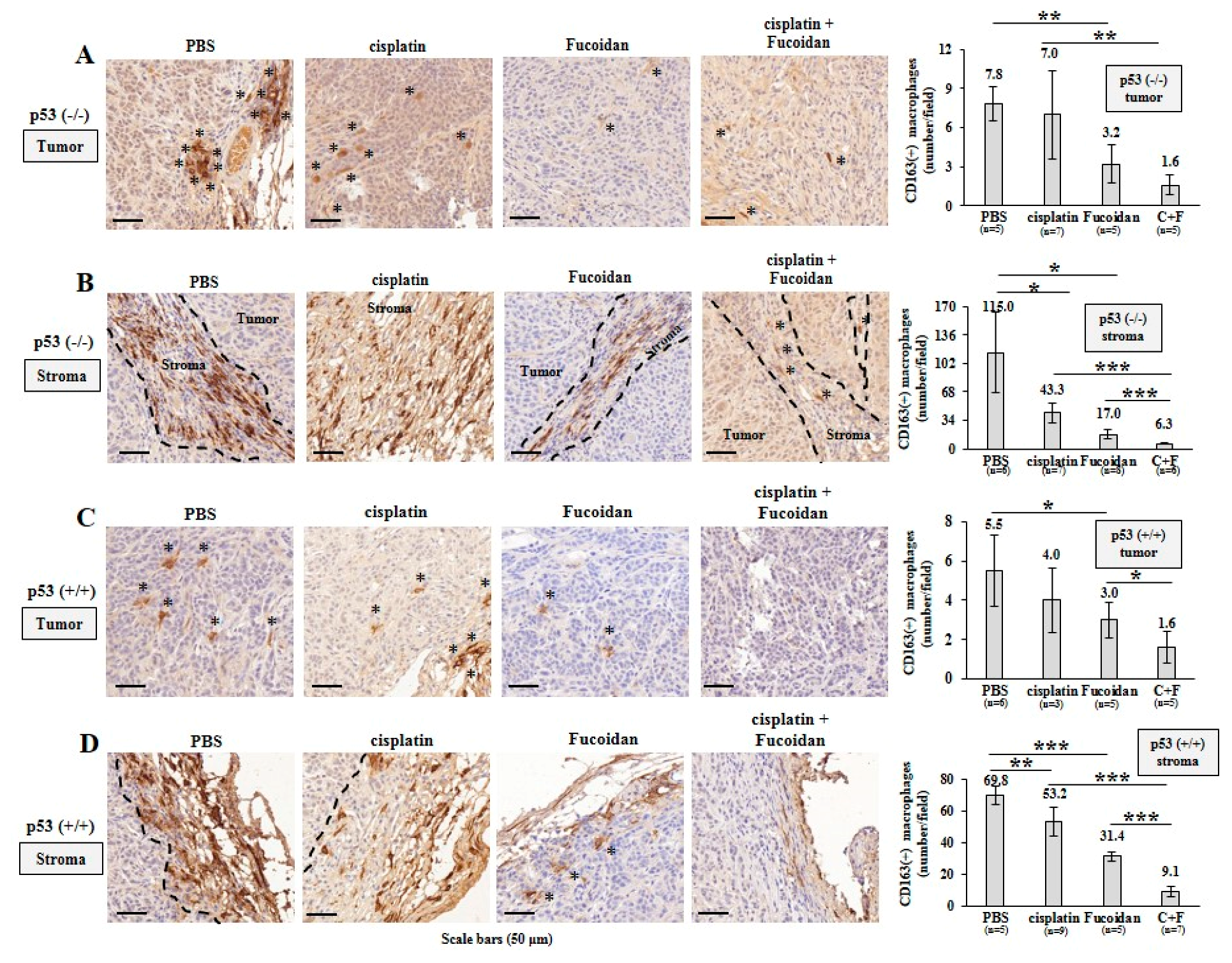
2.5. Oligo-Fucoidan Inhibits Tumor Progression and M2 Macrophage Infiltration
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Oligo-Fucoidan (LMF) and High Molecular Weight Fucoidan (HMF)
4.3. Antibodies (Abs)
4.4. Activation of Macrophage Polarization
4.5. Mitochondrial Superoxide and Intracellular ROS Levels
4.6. Cell Cycle Analysis
4.7. Cytotoxicity Effect and Cell Viability Assay
4.8. Analysis of CD80(+), CD86(+), CD163(+) and CD206(+) Macrophage Populations
4.9. The Invasion Ability of M2 Macrophages and Cancer Cells
4.10. Expression Levels of Macrophage Markers Detected by Quantitative RT-PCR
4.11. Tumor Growth in Xenograft Mice and a Tumor Immunohistochemistry Study
4.12. Disclosure
4.13. Statistics
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Centurione, L.; Aiello, F.B. DNA repair and cytokines: Tgf-beta, il-6, and thrombopoietin as different biomarkers of radioresistance. Front. Oncol. 2016, 6, 175. [Google Scholar] [CrossRef]
- Ho, E.A.; Piquette-Miller, M. Regulation of multidrug resistance by pro-inflammatory cytokines. Curr. Cancer Drug Targets 2006, 6, 295–311. [Google Scholar] [CrossRef]
- Jones, V.S.; Huang, R.Y.; Chen, L.P.; Chen, Z.S.; Fu, L.; Huang, R.P. Cytokines in cancer drug resistance: Cues to new therapeutic strategies. Biochim. Biophys. Acta 2016, 1865, 255–265. [Google Scholar] [CrossRef]
- Mitsunaga, S.; Ikeda, M.; Shimizu, S.; Ohno, I.; Furuse, J.; Inagaki, M.; Higashi, S.; Kato, H.; Terao, K.; Ochiai, A. Serum levels of il-6 and il-1beta can predict the efficacy of gemcitabine in patients with advanced pancreatic cancer. Br. J. Cancer 2013, 108, 2063–2069. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Covarrubias, A.; Byles, V.; Horng, T. Ros sets the stage for macrophage differentiation. Cell Res. 2013, 23, 984–985. [Google Scholar] [CrossRef]
- De Santa, F.; Vitiello, L.; Torcinaro, A.; Ferraro, E. The role of metabolic remodeling in macrophage polarization and its effect on skeletal muscle regeneration. Antioxid. Redox Signal. 2018. [Google Scholar] [CrossRef]
- Santoni, M.; Romagnoli, E.; Saladino, T.; Foghini, L.; Guarino, S.; Capponi, M.; Giannini, M.; Cognigni, P.D.; Ferrara, G.; Battelli, N. Triple negative breast cancer: Key role of tumor-associated macrophages in regulating the activity of anti-pd-1/pd-l1 agents. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 78–84. [Google Scholar] [CrossRef]
- Solinas, G.; Schiarea, S.; Liguori, M.; Fabbri, M.; Pesce, S.; Zammataro, L.; Pasqualini, F.; Nebuloni, M.; Chiabrando, C.; Mantovani, A.; et al. Tumor-conditioned macrophages secrete migration-stimulating factor: A new marker for m2-polarization, influencing tumor cell motility. J. Immunol. 2010, 185, 642–652. [Google Scholar] [CrossRef]
- He, C.; Ryan, A.J.; Murthy, S.; Carter, A.B. Accelerated development of pulmonary fibrosis via cu,zn-superoxide dismutase-induced alternative activation of macrophages. J. Biol. Chem. 2013, 288, 20745–20757. [Google Scholar] [CrossRef]
- Oh, J.; Riek, A.E.; Weng, S.; Petty, M.; Kim, D.; Colonna, M.; Cella, M.; Bernal-Mizrachi, C. Endoplasmic reticulum stress controls m2 macrophage differentiation and foam cell formation. J. Biol. Chem. 2012, 287, 11629–11641. [Google Scholar] [CrossRef]
- Chen, L.M.; Liu, P.Y.; Chen, Y.A.; Tseng, H.Y.; Shen, P.C.; Hwang, P.A.; Hsu, H.L. Oligo-fucoidan prevents il-6 and ccl2 production and cooperates with p53 to suppress atm signaling and tumor progression. Sci. Rep. 2017, 7, 11864. [Google Scholar] [CrossRef]
- Yun, U.J.; Park, S.E.; Jo, Y.S.; Kim, J.; Shin, D.Y. DNA damage induces the il-6/stat3 signaling pathway, which has anti-senescence and growth-promoting functions in human tumors. Cancer Lett. 2012, 323, 155–160. [Google Scholar] [CrossRef]
- Borsellino, N.; Belldegrun, A.; Bonavida, B. Endogenous interleukin 6 is a resistance factor for cis-diamminedichloroplatinum and etoposide-mediated cytotoxicity of human prostate carcinoma cell lines. Cancer Res. 1995, 55, 4633–4639. [Google Scholar]
- Ara, T.; Nakata, R.; Sheard, M.A.; Shimada, H.; Buettner, R.; Groshen, S.G.; Ji, L.; Yu, H.; Jove, R.; Seeger, R.C.; et al. Critical role of stat3 in il-6-mediated drug resistance in human neuroblastoma. Cancer Res. 2013, 73, 3852–3864. [Google Scholar] [CrossRef]
- Chen, W.; Gao, Q.; Han, S.; Pan, F.; Fan, W. The ccl2/ccr2 axis enhances il-6-induced epithelial-mesenchymal transition by cooperatively activating stat3-twist signaling. Tumour Biol. 2015, 36, 973–981. [Google Scholar] [CrossRef]
- Roca, H.; Varsos, Z.S.; Sud, S.; Craig, M.J.; Ying, C.; Pienta, K.J. Ccl2 and interleukin-6 promote survival of human cd11b+ peripheral blood mononuclear cells and induce m2-type macrophage polarization. J. Biol. Chem. 2009, 284, 34342–34354. [Google Scholar] [CrossRef]
- Huang, T.H.; Chiu, Y.H.; Chan, Y.L.; Chiu, Y.H.; Wang, H.; Huang, K.C.; Li, T.L.; Hsu, K.H.; Wu, C.J. Prophylactic administration of fucoidan represses cancer metastasis by inhibiting vascular endothelial growth factor (vegf) and matrix metalloproteinases (mmps) in lewis tumor-bearing mice. Mar. Drugs 2015, 13, 1882–1900. [Google Scholar] [CrossRef]
- Hsu, H.Y.; Lin, T.Y.; Hwang, P.A.; Tseng, L.M.; Chen, R.H.; Tsao, S.M.; Hsu, J. Fucoidan induces changes in the epithelial to mesenchymal transition and decreases metastasis by enhancing ubiquitin-dependent tgfbeta receptor degradation in breast cancer. Carcinogenesis 2013, 34, 874–884. [Google Scholar] [CrossRef]
- Chen, M.C.; Hsu, W.L.; Hwang, P.A.; Chou, T.C. Low molecular weight fucoidan inhibits tumor angiogenesis through downregulation of hif-1/vegf signaling under hypoxia. Mar. Drugs 2015, 13, 4436–4451. [Google Scholar] [CrossRef]
- Chen, M.C.; Hsu, W.L.; Hwang, P.A.; Chen, Y.L.; Chou, T.C. Combined administration of fucoidan ameliorates tumor and chemotherapy-induced skeletal muscle atrophy in bladder cancer-bearing mice. Oncotarget 2016, 7, 51608–51618. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.; Teng, L.; Legerski, R.J. DNA damage checkpoint recovery and cancer development. Exp. Cell Res. 2015, 334, 350–358. [Google Scholar] [CrossRef]
- Hsu, H.Y.; Lin, T.Y.; Hu, C.H.; Shu, D.T.F.; Lu, M.K. Fucoidan upregulates tlr4/chop-mediated caspase-3 and parp activation to enhance cisplatin-induced cytotoxicity in human lung cancer cells. Cancer Lett. 2018, 432, 112–120. [Google Scholar] [CrossRef]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z.G. Ros play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013, 23, 898–914. [Google Scholar] [CrossRef]
- Bresciani, G.; da Cruz, I.B.; Gonzalez-Gallego, J. Manganese superoxide dismutase and oxidative stress modulation. Adv. Clin. Chem. 2015, 68, 87–130. [Google Scholar]
- Belambri, S.A.; Rolas, L.; Raad, H.; Hurtado-Nedelec, M.; Dang, P.M.; El-Benna, J. Nadph oxidase activation in neutrophils: Role of the phosphorylation of its subunits. Eur. J. Clin. Invest. 2018, 48, e12951. [Google Scholar] [CrossRef]
- Bae, Y.S.; Oh, H.; Rhee, S.G.; Yoo, Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 2011, 32, 491–509. [Google Scholar] [CrossRef]
- Li, Y.; Trush, M.A. Diphenyleneiodonium, an nad(p)h oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem. Biophys. Res. Commun. 1998, 253, 295–299. [Google Scholar] [CrossRef]
- Taylor, W.R.; Stark, G.R. Regulation of the g2/m transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef]
- Gillis, L.D.; Leidal, A.M.; Hill, R.; Lee, P.W. P21cip1/waf1 mediates cyclin b1 degradation in response to DNA damage. Cell Cycle 2009, 8, 253–256. [Google Scholar] [CrossRef]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a dream. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef]
- Hart, P.C.; Mao, M.; de Abreu, A.L.; Ansenberger-Fricano, K.; Ekoue, D.N.; Ganini, D.; Kajdacsy-Balla, A.; Diamond, A.M.; Minshall, R.D.; Consolaro, M.E.; et al. Mnsod upregulation sustains the warburg effect via mitochondrial ros and ampk-dependent signalling in cancer. Nat. Commun. 2015, 6, 6053. [Google Scholar] [CrossRef]
- Fernandez-Sanchez, A.; Madrigal-Santillan, E.; Bautista, M.; Esquivel-Soto, J.; Morales-Gonzalez, A.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sanchez-Rivera, G.; Valadez-Vega, C.; Morales-Gonzalez, J.A. Inflammation, oxidative stress, and obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef]
- Tseng, H.Y.; Chen, Y.A.; Jen, J.; Shen, P.C.; Chen, L.M.; Lin, T.D.; Wang, Y.C.; Hsu, H.L. Oncogenic mct-1 activation promotes yy1-egfr-mnsod signaling and tumor progression. Oncogenesis 2017, 6, e313. [Google Scholar] [CrossRef]
- Orrenius, S. Reactive oxygen species in mitochondria-mediated cell death. Drug Metab. Rev. 2007, 39, 443–455. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ros in cancer: Initiators, amplifiers or an achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef]
- Gu, H.; Huang, T.; Shen, Y.; Liu, Y.; Zhou, F.; Jin, Y.; Sattar, H.; Wei, Y. Reactive oxygen species-mediated tumor microenvironment transformation: The mechanism of radioresistant gastric cancer. Oxid. Med. Cell Longev. 2018, 2018, 5801209. [Google Scholar] [CrossRef]
- Lee, C.C.; Lin, J.C.; Hwang, W.L.; Kuo, Y.J.; Chen, H.K.; Tai, S.K.; Lin, C.C.; Yang, M.H. Macrophage-secreted interleukin-35 regulates cancer cell plasticity to facilitate metastatic colonization. Nat. Commun. 2018, 9, 3763. [Google Scholar] [CrossRef]
- Canli, O.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Lower, M.; Sahin, U.; et al. Myeloid cell-derived reactive oxygen species induce epithelial mutagenesis. Cancer Cell 2017, 32, 869–883. [Google Scholar] [CrossRef]
- Hongo, F.; Garban, H.; Huerta-Yepez, S.; Vega, M.; Jazirehi, A.R.; Mizutani, Y.; Miki, T.; Bonavida, B. Inhibition of the transcription factor yin yang 1 activity by s-nitrosation. Biochem. Biophys. Res. Commun. 2005, 336, 692–701. [Google Scholar] [CrossRef]
- Truong, T.H.; Ung, P.M.; Palde, P.B.; Paulsen, C.E.; Schlessinger, A.; Carroll, K.S. Molecular basis for redox activation of epidermal growth factor receptor kinase. Cell Chem. Biol. 2016, 23, 837–848. [Google Scholar] [CrossRef]
- Liu, W.; Guo, Q.; Zhao, H. Oxidative stress-elicited yy1 potentiates antioxidative response via enhancement of nrf2-driven transcriptional activity: A potential neuronal defensive mechanism against ischemia/reperfusion cerebral injury. Biomed Pharm. 2018, 108, 698–706. [Google Scholar] [CrossRef]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Ohlund, D.; et al. Nrf2 promotes tumor maintenance by modulating mrna translation in pancreatic cancer. Cell 2016, 166, 963–976. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced nrf2 transcription promotes ros detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Klettner, A. Fucoidan as a potential therapeutic for major blinding diseases–a hypothesis. Mar. Drugs 2016, 14, E31. [Google Scholar] [CrossRef]
- Zhang, W.; Oda, T.; Yu, Q.; Jin, J.O. Fucoidan from macrocystis pyrifera has powerful immune-modulatory effects compared to three other fucoidans. Mar. Drugs 2015, 13, 1084–1104. [Google Scholar] [CrossRef]
- Ale, M.T.; Maruyama, H.; Tamauchi, H.; Mikkelsen, J.D.; Meyer, A.S. Fucoidan from sargassum sp. And fucus vesiculosus reduces cell viability of lung carcinoma and melanoma cells in vitro and activates natural killer cells in mice in vivo. Int. J. Biol. Macromol. 2011, 49, 331–336. [Google Scholar] [CrossRef]
- Shimizu, J.; Wada-Funada, U.; Mano, H.; Matahira, Y.; Kawaguchi, M.; Wada, M. Proportion of murine cytotoxic t cells is increased by high molecular-weight fucoidan extracted from okinawa mozuku (cladosiphon okamuranus). J. Health Sci. 2005, 51, 394–397. [Google Scholar] [CrossRef]
- Sun, J.; Sun, J.; Song, B.; Zhang, L.; Shao, Q.; Liu, Y.; Yuan, D.; Zhang, Y.; Qu, X. Fucoidan inhibits ccl22 production through nf-kappab pathway in m2 macrophages: A potential therapeutic strategy for cancer. Sci. Rep. 2016, 6, 35855. [Google Scholar] [CrossRef]
- Hsu, H.Y.; Lin, T.Y.; Lu, M.K.; Leng, P.J.; Tsao, S.M.; Wu, Y.C. Fucoidan induces toll-like receptor 4-regulated reactive oxygen species and promotes endoplasmic reticulum stress-mediated apoptosis in lung cancer. Sci. Rep. 2017, 7, 44990. [Google Scholar] [CrossRef]
- Hsu, H.Y.; Lin, T.Y.; Wu, Y.C.; Tsao, S.M.; Hwang, P.A.; Shih, Y.W.; Hsu, J. Fucoidan inhibition of lung cancer in vivo and in vitro: Role of the smurf2-dependent ubiquitin proteasome pathway in tgfbeta receptor degradation. Oncotarget 2014, 5, 7870–7885. [Google Scholar] [CrossRef]
- van Weelden, G.; Bobinski, M.; Okla, K.; van Weelden, W.J.; Romano, A.; Pijnenborg, J.M.A. Fucoidan structure and activity in relation to anti-cancer mechanisms. Mar. Drugs 2019, 17, E23. [Google Scholar] [CrossRef]
- Cumashi, A.; Ushakova, N.A.; Preobrazhenskaya, M.E.; D’Incecco, A.; Piccoli, A.; Totani, L.; Tinari, N.; Morozevich, G.E.; Berman, A.E.; Bilan, M.I.; et al. A comparative study of the anti-inflammatory, anticoagulant, antiangiogenic, and antiadhesive activities of nine different fucoidans from brown seaweeds. Glycobiology 2007, 17, 541–552. [Google Scholar] [CrossRef]
- Kusaykin, M.; Bakunina, I.; Sova, V.; Ermakova, S.; Kuznetsova, T.; Besednova, N.; Zaporozhets, T.; Zvyagintseva, T. Structure, biological activity, and enzymatic transformation of fucoidans from the brown seaweeds. Biotechnol. J. 2008, 3, 904–915. [Google Scholar] [CrossRef]
- Li, B.; Lu, F.; Wei, X.; Zhao, R. Fucoidan: Structure and bioactivity. Molecules 2008, 13, 1671–1695. [Google Scholar] [CrossRef]
- Patankar, M.S.; Oehninger, S.; Barnett, T.; Williams, R.L.; Clark, G.F. A revised structure for fucoidan may explain some of its biological activities. J. Biol. Chem. 1993, 268, 21770–21776. [Google Scholar]
- Hwang, P.A.; Chien, S.Y.; Chan, Y.L.; Lu, M.K.; Wu, C.H.; Kong, Z.L.; Wu, C.J. Inhibition of lipopolysaccharide (lps)-induced inflammatory responses by sargassum hemiphyllum sulfated polysaccharide extract in Raw264.7 macrophage cells. J. Agric. Food Chem. 2011, 59, 2062–2068. [Google Scholar] [CrossRef]
- Shih, H.J.; Chu, K.L.; Wu, M.H.; Wu, P.H.; Chang, W.W.; Chu, J.S.; Wang, L.H.; Takeuchi, H.; Ouchi, T.; Hsu, H.L. The involvement of mct-1 oncoprotein in inducing mitotic catastrophe and nuclear abnormalities. Cell Cycle 2012, 11, 934–952. [Google Scholar] [CrossRef]
- Kasiappan, R.; Shih, H.J.; Wu, M.H.; Choy, C.; Lin, T.D.; Chen, L.; Hsu, H.L. The antagonism between mct-1 and p53 affects the tumorigenic outcomes. Mol. Cancer 2010, 9, 311. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.-M.; Tseng, H.-Y.; Chen, Y.-A.; Al Haq, A.T.; Hwang, P.-A.; Hsu, H.-L. Oligo-Fucoidan Prevents M2 Macrophage Differentiation and HCT116 Tumor Progression. Cancers 2020, 12, 421. https://doi.org/10.3390/cancers12020421
Chen L-M, Tseng H-Y, Chen Y-A, Al Haq AT, Hwang P-A, Hsu H-L. Oligo-Fucoidan Prevents M2 Macrophage Differentiation and HCT116 Tumor Progression. Cancers. 2020; 12(2):421. https://doi.org/10.3390/cancers12020421
Chicago/Turabian StyleChen, Li-Mei, Hong-Yu Tseng, Yen-An Chen, Aushia Tanzih Al Haq, Pai-An Hwang, and Hsin-Ling Hsu. 2020. "Oligo-Fucoidan Prevents M2 Macrophage Differentiation and HCT116 Tumor Progression" Cancers 12, no. 2: 421. https://doi.org/10.3390/cancers12020421
APA StyleChen, L.-M., Tseng, H.-Y., Chen, Y.-A., Al Haq, A. T., Hwang, P.-A., & Hsu, H.-L. (2020). Oligo-Fucoidan Prevents M2 Macrophage Differentiation and HCT116 Tumor Progression. Cancers, 12(2), 421. https://doi.org/10.3390/cancers12020421