Immunomodulatory Roles of PARP-1 and PARP-2: Impact on PARP-Centered Cancer Therapies

Abstract
1. Introduction
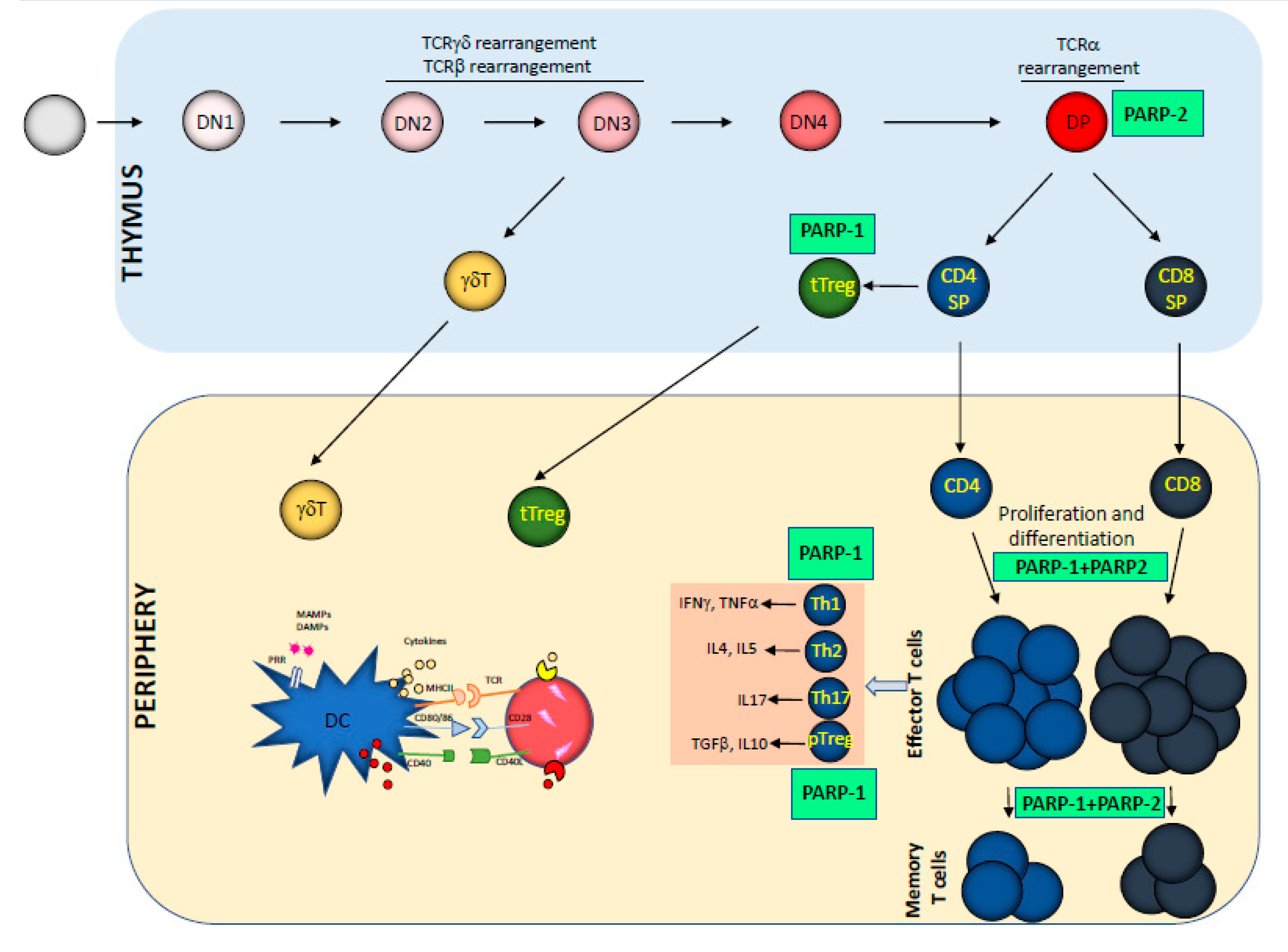
2. Impact of PARP-1 and PARP-2 on T Cell Development and Function
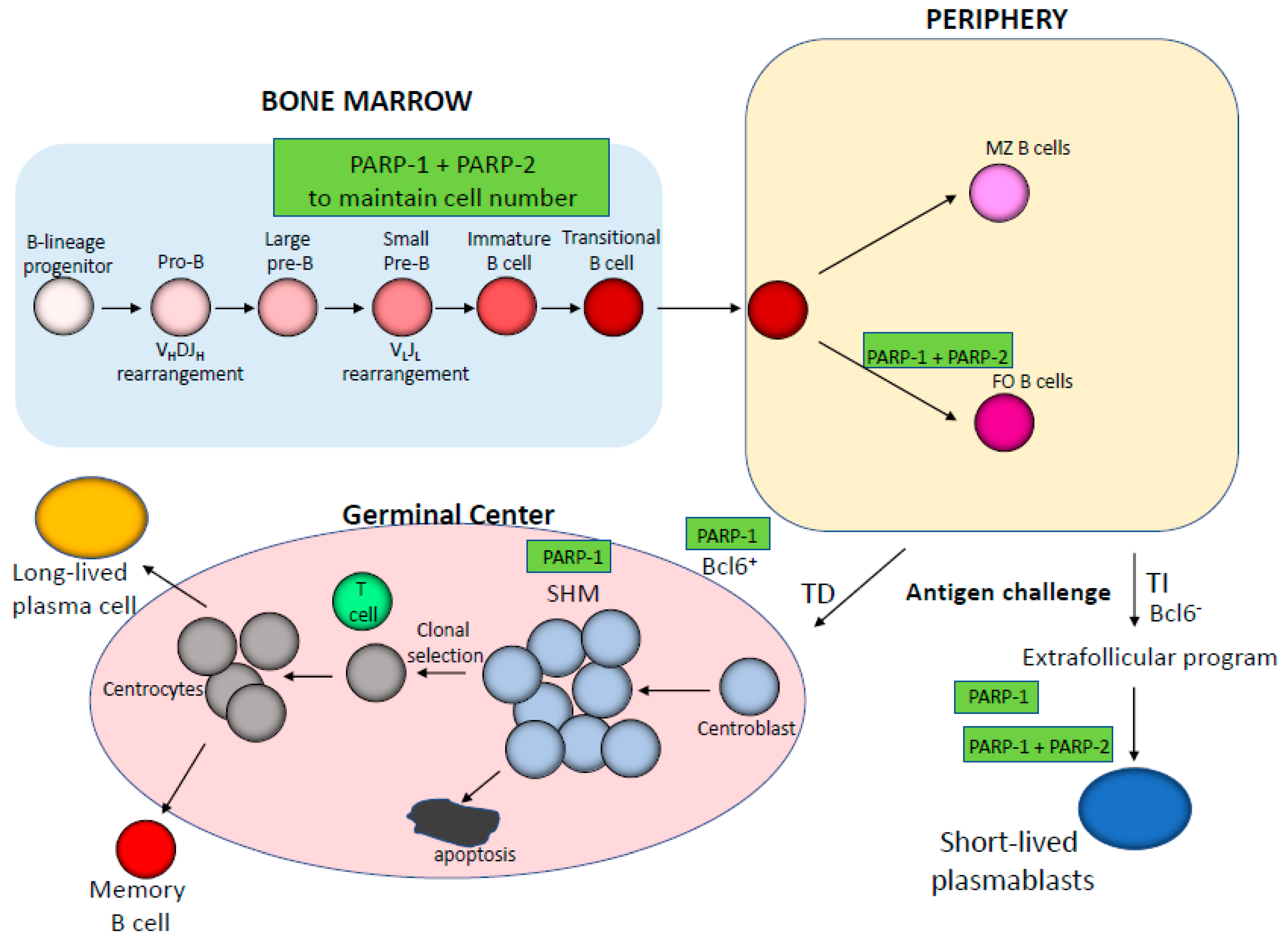
3. Impact of PARP-1 and PARP-2 on B cell Development and Function
4. Role of PARP-1 and PARP-2 in the Cellular Components of the Innate Immune System
5. How Could the Immunomodulatory Roles of PARP-1 and PARP-2 Impact the Immune Response to Tumors?
6. PARP Inhibitors as Immunomodulatory Agents
7. Conclusions and Future Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Daniels, C.M.; Ong, S.-E.; Leung, A.K.L. The Promise of Proteomics for the Study of ADP-Ribosylation. Mol. Cell 2015, 58, 911–924. [Google Scholar] [CrossRef]
- Yelamos, J.; Farres, J.; Llacuna, L.; Ampurdanes, C.; Martin-Caballero, J. PARP-1 and PARP-2: New players in tumour development. Am. J. Cancer Res. 2011, 1, 328–346. [Google Scholar] [PubMed]
- Alemasova, E.E.; Lavrik, O.I. Poly(ADP-ribosyl)ation by PARP1: Reaction mechanism and regulatory proteins. Nucleic Acids Res. 2019, 47, 3811–3827. [Google Scholar] [CrossRef] [PubMed]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Ménissier de Murcia, J.; Ricoul, M.; Tartier, L.; Niedergang, C.; Huber, A.; Dantzer, F.; Schreiber, V.; Amé, J.-C.; Dierich, A.; LeMeur, M.; et al. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003, 22, 2255–2263. [Google Scholar] [CrossRef]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; de Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef]
- Stewart, R.A.; Pilie, P.G.; Yap, T.A. Development of PARP and immune-checkpoint inhibitor combinations. Cancer Res. 2018, 78, 6717–6725. [Google Scholar] [CrossRef]
- Yélamos, J.; Galindo, M.; Navarro, J.; Albanell, J.; Rovira, A.; Rojo, F.; Oliver, J. Enhancing tumor-targeting monoclonal antibodies therapy by PARP inhibitors. Oncoimmunology 2016, 5, e1065370. [Google Scholar] [CrossRef][Green Version]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: What´s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Radtke, F. Mechanisms of T Cell Development and Transformation. Annu. Rev. Cell Dev. Biol. 2011, 27, 539–562. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, S.; Ishikawa, H. Transcriptional Regulation of Differentiation and Functions of Effector T Regulatory Cells. Cells 2019, 8, 939. [Google Scholar] [CrossRef] [PubMed]
- Yélamos, J.; Monreal, Y.; Saenz, L.; Aguado, E.; Schreiber, V.; Mota, R.; Fuente, T.; Minguela, A.; Parrilla, P.; De Murcia, G.; et al. PARP-2 deficiency affects the survival of CD4 + CD8+ double-positive thymocytes. EMBO J. 2006, 25, 4350–4360. [Google Scholar] [CrossRef]
- Nicolás, L.; Martínez, C.; Baró, C.; Rodríguez, M.; Baroja-Mazo, A.; Sole, F.; Flores, J.M.; Ampurdanés, C.; Dantzer, F.; Martin-Caballero, J.; et al. Loss of poly(ADP-ribose) polymerase-2 leads to rapid development of spontaneous T-cell lymphomas in p53-deficient mice. Oncogene 2010, 29, 2877–2883. [Google Scholar] [CrossRef]
- Nasta, F.; Laudisi, F.; Sambucci, M.; Rosado, M.M.; Pioli, C. Increased Foxp3 + Regulatory T Cells in Poly(ADP-Ribose) Polymerase-1 Deficiency. J. Immunol. 2010, 184, 3470–3477. [Google Scholar] [CrossRef]
- Navarro, J.; Gozalbo-López, B.; Méndez, A.C.; Dantzer, F.; Schreiber, V.; Martínez, C.; Arana, D.M.; Farrés, J.; Revilla-Nuin, B.; Bueno, M.F.; et al. PARP-1/PARP-2 double deficiency in mouse T cells results in faulty immune responses and T lymphomas. Sci. Rep. 2017, 7, 41962. [Google Scholar] [CrossRef]
- Grossman, Z.; Paul, W.E. Dynamic Tuning of Lymphocytes: Physiological Basis, Mechanisms, and Function. Annu. Rev. Immunol. 2015, 33, 677–713. [Google Scholar] [CrossRef]
- Jameson, S.C. Maintaining the norm: T-cell homeostasis. Nat. Rev. Immunol. 2002, 2, 547–556. [Google Scholar] [CrossRef]
- Baek, K.-H.; Shin, H.-J.; Yoo, J.-K.; Cho, J.-H.; Choi, Y.-H.; Sung, Y.-C.; McKeon, F.; Lee, C.-W. p53 deficiency and defective mitotic checkpoint in proliferating T lymphocytes increase chromosomal instability through aberrant exit from mitotic arrest. J. Leukoc. Biol. 2003, 73, 850–861. [Google Scholar] [CrossRef]
- Prochazkova, J.; Sakaguchi, S.; Owusu, M.; Mazouzi, A.; Wiedner, M.; Velimezi, G.; Moder, M.; Turchinovich, G.; Hladik, A.; Gurnhofer, E.; et al. DNA Repair Cofactors ATMIN and NBS1 Are Required to Suppress T Cell Activation. PloS Genet. 2015, 11, e1005645. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H.; Szankasi, P.; Roberts, C.J.; Murray, A.W.; Friend, S.H. Integrating genetic approaches into the discovery of anticancer drugs. Science 1997, 278, 1064–1068. [Google Scholar] [CrossRef]
- Moreno-Lama, L.; Galindo-Campos, M.A.; Martínez, C.; Comerma, L.; Vazquez, I.; Vernet-Tomas, M.; Ampurdanés, C.; Lutfi, N.; Martin-Caballero, J.; Dantzer, F.; et al. Coordinated signals from PARP-1 and PARP-2 are required to establish a proper T cell immune response to breast tumors in mice. Oncogene 2020. [Google Scholar] [CrossRef] [PubMed]
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Valdor, R.; Schreiber, V.; Saenz, L.; Martínez, T.; Muñoz-Suano, A.; Dominguez-Villar, M.; Ramírez, P.; Parrilla, P.; Aguado, E.; García-Cózar, F.; et al. Regulation of NFAT by poly(ADP-ribose) polymerase activity in T cells. Mol. Immunol. 2008, 45, 1863–1871. [Google Scholar] [CrossRef]
- Saenz, L.; Lozano, J.J.; Valdor, R.; Baroja-Mazo, A.; Ramirez, P.; Parrilla, P.; Aparicio, P.; Sumoy, L.; Yélamos, J. Transcriptional regulation by poly(ADP-ribose) polymerase-1 during T cell activation. BMC Genom. 2008, 9, 171. [Google Scholar] [CrossRef]
- Sambucci, M.; Laudisi, F.; Novelli, F.; Bennici, E.; Rosado, M.M.; Pioli, C. Effects of PARP-1 deficiency on Th1 and Th2 cell differentiation. Sci. World J. 2013, 2013, 375024. [Google Scholar] [CrossRef]
- Ghonim, M.A.; Pyakurel, K.; Ibba, S.V.; Al-Khami, A.A.; Wang, J.; Rodriguez, P.; Rady, H.F.; El-Bahrawy, A.H.; Lammi, M.R.; Mansy, M.S.; et al. PARP inhibition by olaparib or gene knockout blocks asthma-like manifestation in mice by modulating CD4+ T cell function. J. Transl. Med. 2015, 13, 225. [Google Scholar] [CrossRef]
- Gonzalez-Rey, E.; Martínez-Romero, R.; O’Valle, F.; Aguilar-Quesada, R.; Conde, C.; Delgado, M.; Oliver, F.J. Therapeutic effect of a poly(ADP-ribose) polymerase-1 inhibitor on experimental arthritis by downregulating inflammation and Th1 response. PLoS ONE 2007, 2, e1071. [Google Scholar] [CrossRef]
- Luo, X.; Nie, J.; Wang, S.; Chen, Z.; Chen, W.; Li, D.; Hu, H.; Li, B. Poly(ADP-ribosyl)ation of FOXP3 Protein Mediated by PARP-1 Protein regulates the function of regulatory t cells. J. Biol. Chem. 2015, 290, 28675–28682. [Google Scholar] [CrossRef]
- Zhang, P.; Nakatsukasa, H.; Tu, E.; Kasagi, S.; Cui, K.; Ishikawa, M.; Konkel, J.E.; Maruyama, T.; Wei, G.; Abbatiello, B.; et al. PARP-1 regulates expression of TGF-β receptors in T cells. Blood 2013, 122, 2224–2232. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hardy, R.R.; Hayakawa, K. B C ELL D EVELOPMENT P ATHWAYS. Annu. Rev. Immunol. 2001, 19, 595–621. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Q.; Stingl, L.; Morrison, C.; Jantsch, M.; Los, M.; Schulze-Osthoff, K.; Wagner, E.F. PARP is important for genomic stability but dispensable in apoptosis. Genes Dev. 1997, 11, 2347–2358. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.; Smith, G.C.M.; Stingl, L.; Jackson, S.P.; Wagner, E.F.; Wang, Z.Q. Genetic interaction between PARP and DNA-PK in V(D)J recombination and tumorigenesis. Nat. Genet. 1997, 17, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.L.; Franco, D.; Burkle, A.; Chang, Y. Role of poly(ADP-ribosyl)ation in DNA-PKcs- independent V(D)J recombination. Proc. Natl. Acad. Sci. USA 2002, 99, 4532–4537. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Campos, M.A.; Bedora-Faure, M.; Farrés, J.; Lescale, C.; Moreno-Lama, L.; Martínez, C.; Martín-Caballero, J.; Ampurdanés, C.; Aparicio, P.; Dantzer, F.; et al. Coordinated signals from the DNA repair enzymes PARP-1 and PARP-2 promotes B-cell development and function. Cell Death Differ. 2019, 26, 2667–2681. [Google Scholar] [CrossRef]
- Ronson, G.E.; Piberger, A.L.; Higgs, M.R.; Olsen, A.L.; Stewart, G.S.; McHugh, P.J.; Petermann, E.; Lakin, N.D. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 2018, 9, 746. [Google Scholar] [CrossRef]
- García de Vinuesa, C.; O’Leary, P.; Sze, D.M.-Y.; Toellner, K.-M.; MacLennan, I.C.M. T-independent type 2 antigens induce B cell proliferation in multiple splenic sites, but exponential growth is confined to extrafollicular foci. Eur. J. Immunol. 1999, 29, 1314–1323. [Google Scholar] [CrossRef]
- Inoue, T.; Moran, I.; Shinnakasu, R.; Phan, T.G.; Kurosaki, T. Generation of memory B cells and their reactivation. Immunol. Rev. 2018, 283, 138–149. [Google Scholar] [CrossRef]
- Tas, J.M.J.; Mesin, L.; Pasqual, G.; Targ, S.; Jacobsen, J.T.; Mano, Y.M.; Chen, C.S.; Weill, J.-C.; Reynaud, C.-A.; Browne, E.P.; et al. Visualizing antibody affinity maturation in germinal centers. Science 2016, 351, 1048–1054. [Google Scholar] [CrossRef]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef]
- Ambrose, H.E.; Willimott, S.; Beswick, R.W.; Dantzer, F.; de Murcia, J.M.; Yelamos, J.; Wagner, S.D. Poly(ADP-ribose) polymerase-1 (Parp-1)-deficient mice demonstrate abnormal antibody responses. Immunology 2009, 127, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Robert, I.; Dantzer, F.; Reina-San-Martin, B. Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. J. Exp. Med. 2009, 206, 1047–1056. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, H.E.; Papadopoulou, V.; Beswick, R.W.; Wagner, S.D. Poly-(ADP-ribose) polymerase-1 (Parp-1) binds in a sequence-specific manner at the Bcl-6 locus and contributes to the regulation of Bcl-6 transcription. Oncogene 2007, 26, 6244–6252. [Google Scholar] [CrossRef]
- Dent, A.L.; Shaffer, A.L.; Yu, X.; Allman, D.; Staudt, L.M. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science 1997, 276, 589–592. [Google Scholar] [CrossRef]
- Ye, B.H.; Cattoretti, G.; Shen, Q.; Zhang, J.; Hawe, N.; De Waard, R.; Leung, C.; Nouri-Shirazi, M.; Orazi, A.; Chaganti, R.S.K.; et al. The BCL-6 proto-oncogene controls germinal-centre formation and Th2- type inflammation. Nat. Genet. 1997, 16, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, H.; Fukita, Y.; Van Der Horst, G.T.J.; De Boer, J.; Weeda, G.; Essers, J.; De Wind, N.; Engelward, B.P.; Samson, L.; Verbeek, S.; et al. Hypermutation of immunoglobulin genes in memory B cells of DNA repair- deficient mice. J. Exp. Med. 1998, 187, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Tepper, S.; Mortusewicz, O.; Członka, E.; Bello, A.; Schmidt, A.; Jeschke, J.; Fischbach, A.; Pfeil, I.; Petersen-Mahrt, S.K.; Mangerich, A.; et al. Restriction of AID activity and somatic hypermutation by PARP-1. Nucleic Acids Res. 2019, 47, 7418–7429. [Google Scholar] [CrossRef]
- Thompson, E.C. Innate immune cells in motion. Trends Immunol. 2011, 32, 451. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Szabó, C.; Lim, L.H.; Cuzzocrea, S.; Getting, S.J.; Zingarelli, B.; Flower, R.J.; Salzman, A.L.; Perretti, M. Inhibition of poly (ADP-ribose) synthetase attenuates neutrophil recruitment and exerts antiinflammatory effects. J. Exp. Med. 1997, 186, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Dharwal, V.; Naura, A.S. PARP-1 inhibition ameliorates elastase induced lung inflammation and emphysema in mice. Biochem. Pharm. 2018, 150, 24–34. [Google Scholar] [CrossRef]
- Wang, S.; Yang, F.-J.; Wang, X.; Zhou, Y.; Dai, B.; Han, B.; Ma, H.-C.; Ding, Y.-T.; Shi, X.-L. PARP-1 promotes tumor recurrence after warm ischemic liver graft transplantation via neutrophil recruitment and polarization. Oncotarget 2017, 8, 88918–88933. [Google Scholar] [CrossRef]
- Mota, R.A.; Sánchez-Bueno, F.; Saenz, L.; Hernández-Espinosa, D.; Jimeno, J.; Tornel, P.L.; Martínez-Torrano, A.; Ramírez, P.; Parrilla, P.; Yélamos, J. Inhibition of poly(ADP-ribose) polymerase attenuates the severity of acute pancreatitis and associated lung injury. Lab. Investig. 2005, 85, 1250–1262. [Google Scholar] [CrossRef] [PubMed]
- Lavin, Y.; Mortha, A.; Rahman, A.; Merad, M. Regulation of macrophage development and function in peripheral tissues. Nat. Rev. Immunol. 2015, 15, 731–744. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Hauschildt, S.; Scheipers, P.; Bessler, W.; Schwarz, K.; Ullmer, A.; Flad, H.D.; Heine, H. Role of ADP-ribosylation in activated monocytes/macrophages. Adv. Exp. Med. Biol. 1997, 419, 249–252. [Google Scholar]
- Kunze, F.A.; Bauer, M.; Komuczki, J.; Lanzinger, M.; Gunasekera, K.; Hopp, A.-K.; Lehmann, M.; Becher, B.; Müller, A.; Hottiger, M.O. ARTD1 in Myeloid Cells Controls the IL-12/18-IFN-γ Axis in a Model of Sterile Sepsis, Chronic Bacterial Infection, and Cancer. J. Immunol. 2019, 202, 1406–1416. [Google Scholar] [CrossRef]
- Aguilar-Quesada, R.; Muñoz-Gámez, J.A.; Martín-Oliva, D.; Peralta-Leal, A.; Quiles-Pérez, R.; Rodríguez-Vargas, J.M.; Ruiz de Almodóvar, M.; Conde, C.; Ruiz-Extremera, A.; Oliver, F.J. Modulation of transcription by PARP-1: Consequences in carcinogenesis and inflammation. Curr. Med. Chem. 2007, 14, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Tokarz, P.; Płoszaj, T.; Regdon, Z.; Virág, L.; Robaszkiewicz, A. PARP1-LSD1 functional interplay controls transcription of SOD2 that protects human pro-inflammatory macrophages from death under an oxidative condition. Free Radic. Biol. Med. 2019, 131, 218–224. [Google Scholar] [CrossRef]
- Zerfaoui, M.; Naura, A.S.; Errami, Y.; Hans, C.P.; Rezk, B.M.; Park, J.; Elsegeiny, W.; Kim, H.; Lord, K.; Kim, J.G.; et al. Effects of PARP-1 deficiency on airway inflammatory cell recruitment in response to LPS or TNF: Differential effects on CXCR2 ligands and Duffy Antigen Receptor for Chemokines. J. Leukoc. Biol. 2009, 86, 1385–1392. [Google Scholar] [CrossRef]
- Murphy, T.L.; Grajales-Reyes, G.E.; Wu, X.; Tussiwand, R.; Briseño, C.G.; Iwata, A.; Kretzer, N.M.; Durai, V.; Murphy, K.M.; Edu, T. Transcriptional Control of Dendritic Cell Development. Annu. Rev. Immunol. 2016, 34, 93–119. [Google Scholar] [CrossRef]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Echeverri Tirado, L.C.; Ghonim, M.A.; Wang, J.; Al-Khami, A.A.; Wyczechowska, D.; Luu, H.H.; Kim, H.; Sanchez-Pino, M.D.; Yélamos, J.; Yassin, L.M.; et al. PARP-1 is critical for recruitment of dendritic cells to the lung in a mouse model of asthma but dispensable for their differentiation and function. Mediat. Inflamm. 2019, 2019, 1656484. [Google Scholar] [CrossRef]
- Cavone, L.; Aldinucci, A.; Ballerini, C.; Biagioli, T.; Moroni, F.; Chiarugi, A. PARP-1 inhibition prevents CNS migration of dendritic cells during EAE, suppressing the encephalitogenic response and relapse severity. Mult. Scler. 2011, 17, 794–807. [Google Scholar] [CrossRef]
- Aldinucci, A.; Gerlini, G.; Fossati, S.; Cipriani, G.; Ballerini, C.; Biagioli, T.; Pimpinelli, N.; Borgognoni, L.; Massacesi, L.; Moroni, F.; et al. A key role for poly(ADP-ribose) polymerase-1 activity during human dendritic cell maturation. J. Immunol. 2007, 179, 305–312. [Google Scholar] [CrossRef]
- Wang, J.-Q.; Tang, Y.; Li, Q.-S.; Xiao, M.; Li, M.; Sheng, Y.-T.; Yang, Y.; Wang, Y.-L. PARG regulates the proliferation and differentiation of DCs and T cells via PARP/NF-κB in tumour metastases of colon carcinoma. Oncol. Rep. 2019, 41, 2657–2666. [Google Scholar] [CrossRef]
- Cerwenka, A.; Lanier, L.L. Natural killer cell memory in infection, inflammation and cancer. Nat. Rev. Immunol. 2016, 16, 112–123. [Google Scholar] [CrossRef]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Shou, Q.; Fu, H.; Huang, X.; Yang, Y. PARP-1 controls NK cell recruitment to the site of viral infection. Jci Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Mocchegiani, E.; Muzzioli, M.; Giacconi, R.; Cipriano, C.; Gasparini, N.; Franceschi, C.; Gaetti, R.; Cavalieri, E.; Suzuki, H. Metallothioneins/PARP-1/IL-6 interplay on natural killer cell activity in elderly: Parallelism with nonagenarians and old infected humans. Effect of zinc supply. Mech. Ageing Dev. 2003, 124, 459–468. [Google Scholar] [CrossRef]
- Heyman, B.; Jamieson, C. To PARP or not to PARP?-Toward sensitizing acute myeloid leukemia stem cells to immunotherapy. EMBO J. 2019, 38, e103479. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Chacon-Cabrera, A.; Fermoselle, C.; Salmela, I.; Yelamos, J.; Barreiro, E. MicroRNA expression and protein acetylation pattern in respiratory and limb muscles of Parp-1-/- and Parp-2-/- mice with lung cancer cachexia. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 2530–2543. [Google Scholar] [CrossRef]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.-S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef]
- Kalams, S.A.; Walker, B.D. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J. Exp. Med. 1998, 188, 2199–2204. [Google Scholar] [CrossRef]
- Pardoll, D.M.; Topalian, S.L. The role of CD4+ T cell responses in antitumor immunity. Curr. Opin. Immunol. 1998, 10, 588–594. [Google Scholar] [CrossRef]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNγ, and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef]
- Muntasell, A.; Rojo, F.; Servitja, S.; Rubio-Perez, C.; Cabo, M.; Tamborero, D.; Costa-García, M.; Martínez-Garcia, M.; Menéndez, S.; Vazquez, I.; et al. NK Cell Infiltrates and HLA Class I Expression in Primary HER2+ Breast Cancer Predict and Uncouple Pathological Response and Disease-free Survival. Clin. Cancer Res. 2019, 25, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol. 2012, 24, 207–212. [Google Scholar] [CrossRef]
- Böger, C.; Behrens, H.-M.; Krüger, S.; Röcken, C. The novel negative checkpoint regulator VISTA is expressed in gastric carcinoma and associated with PD-L1/PD-1: A future perspective for a combined gastric cancer therapy? Oncoimmunology 2017, 6, e1293215. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [PubMed]
- Van Der Burg, S.H.; Arens, R.; Ossendorp, F.; Van Hall, T.; Melief, C.J.M. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Adams, J.L.; Smothers, J.; Srinivasan, R.; Hoos, A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discov. 2015, 14, 603–622. [Google Scholar] [CrossRef]
- Jiao, S.; Xia, W.; Yamaguchi, H.; Wei, Y.; Chen, M.-K.; Hsu, J.-M.; Hsu, J.L.; Yu, W.-H.; Du, Y.; Lee, H.-H.; et al. PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin. Cancer Res. 2017, 23, 3711–3720. [Google Scholar] [CrossRef]
- Lassen, U. Combining PARP inhibition with PD-1 inhibitors. Lancet. Oncol. 2019, 20, 1196–1198. [Google Scholar] [CrossRef]
- Li, A.; Yi, M.; Qin, S.; Chu, Q.; Luo, S.; Wu, K. Prospects for combining immune checkpoint blockade with PARP inhibition. J. Hematol. Oncol. 2019, 12, 98. [Google Scholar] [CrossRef]
- Higuchi, T.; Flies, D.B.; Marjon, N.A.; Mantia-Smaldone, G.; Ronner, L.; Gimotty, P.A.; Adams, S.F. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Li, C.-W.; Lim, S.-O.; Sun, L.; Lai, Y.-J.; Hou, J.; Liu, C.; Chang, C.-W.; Qiu, Y.; Hsu, J.-M.; et al. Deglycosylation of PD-L1 by 2-deoxyglucose reverses PARP inhibitor-induced immunosuppression in triple-negative breast cancer. Am. J. Cancer Res. 2018, 8, 1837–1846. [Google Scholar] [PubMed]
- Thomas, A.; Vilimas, R.; Trindade, C.; Erwin-Cohen, R.; Roper, N.; Xi, L.; Krishnasamy, V.; Levy, E.; Mammen, A.; Nichols, S.; et al. Durvalumab in Combination with Olaparib in Patients with Relapsed SCLC: Results from a Phase II Study. J. Thorac. Oncol. 2019, 14, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, K.; Xiao, Y.; Feng, B.; Mikule, K.; Ma, X.; Feng, N.; Vellano, C.P.; Federico, L.; Marszalek, J.R.; et al. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci. Rep. 2019, 9, 1853. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.W.; Koh, B.D.; Zhang, J.-S.; Flatten, K.S.; Schneider, P.A.; Billadeau, D.D.; Hess, A.D.; Smith, B.D.; Karp, J.E.; Kaufmann, S.H. Poly(ADP-ribose) polymerase inhibitors sensitize cancer cells to death receptor-mediated apoptosis by enhancing death receptor expression. J. Biol. Chem. 2014, 289, 20543–20558. [Google Scholar] [CrossRef] [PubMed]
- Fenerty, K.E.; Padget, M.; Wolfson, B.; Gameiro, S.R.; Su, Z.; Lee, J.H.; Rabizadeh, S.; Soon-Shiong, P.; Hodge, J.W. Immunotherapy utilizing the combination of natural killer- and antibody dependent cellular cytotoxicity (ADCC)-mediating agents with poly (ADP-ribose) polymerase (PARP) inhibition. J. Immunother. Cancer 2018, 6, 133. [Google Scholar] [CrossRef] [PubMed]
- Aurelius, J.; Martner, A.; Riise, R.E.; Romero, A.I.; Palmqvist, L.; Brune, M.; Hellstrand, K.; Thorén, F.B. Chronic myeloid leukemic cells trigger poly(ADP-ribose) polymerase-dependent inactivation and cell death in lymphocytes. J. Leukoc. Biol. 2013, 93, 155–160. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Cantley, L.C. The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 2018, 174, 1347–1360. [Google Scholar] [CrossRef]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPI triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCANEss. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef]
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Muirhead, G.; Krastev, D.B.; Adam, J.; Morel, D.; Garrido, M.; Lamb, A.; Hénon, C.; Dorvault, N.; Rouanne, M.; et al. PARP inhibition enhances tumor cell–intrinsic immunity in ERCC1-deficient non–small cell lung cancer. J. Clin. Invest. 2019, 129, 1211–1228. [Google Scholar] [CrossRef] [PubMed]
- Reisländer, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 2019, 10, 3143. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Della Corte, C.M.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Pantelidou, C.; Sonzogni, O.; Taveira, M.D.O.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. Parp inhibitor efficacy depends on CD8+ T-cell recruitment via intratumoral sting pathway activation in brca-deficient models of triple-negative breast cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Immunological Process | Parp-1-/- | Parp-2-/- | Parp-1-/-CD4-Parp-2f/f | Parp-1-/-CD19-Parp-2f/f | Artd1DMyel | References |
|---|---|---|---|---|---|---|
| Thymocyte development | Decreased DP survival | Decreased DP survival | [14,15]. | |||
| Peripheral T cell homeostasis | T cell lymphopenia | [17] | ||||
| T cell differentiation | Bias to a Th1 phenotype Increased Treg Increased TGFβR | Compromises antibody production T cell-dependent (TD) antigens | [17] [26] [16,30,31] [31] | |||
| Central and peripheral B cell homeostasis | B cell lymphopenia | [36] | ||||
| B cell function | Increased antibody production to TI antigens Enhanced somatic hypermutation(SHM) | Depletion of follicular (FO) B cells Compromises antibody production to T cell-independent (TI) antigens | [36] [36] [48] | |||
| Neutrophil function | Impaired recruitment | [52,53,54,55] | ||||
| Macrophage function | Impaired recruitment | Inhibition of LPS-induced pro-inflammatory genes | [60] [63] | |||
| Dendritic cell function | Impaired recruitment | [66,67,68,69] | ||||
| Natural killer (NK) cell function | Impaired recruitment | [72] |
| PARPi | IMMUNE CHECK-POINT INHIBITOR | CLINICAL PHASE | CONDITIONS | IDENTIFIER |
|---|---|---|---|---|
| Talazoparib | Avelumab (anti-PD-L1) | II | Advanced or metastatic solid tumors | NCT03330405 |
| Pamiparib | Tislelizumab (anti-PD-1) | I/Ib | Solid tumors | NCT02660034 |
| Rucaparib | Nivolumab (anti-PD-1) | Ib/IIa | Prostate Cancer, Endometrial Cancer | NCT03572478 |
| Olaparib | Tremelimumab (anti-CTLA-4) | I/II | Ovarian Cancer, Fallopian Tube Cancer, Peritoneal Neoplasms | NCT02571725 |
| Talazoparib | Avelumab (anti-PD-L1) | III | Ovarian Cancer | NCT03642132 |
| Rucaparib | Nivolumab (anti-PD-1) | II | Biliary Tract Cancer | NCT03639935 |
| Niraparib | PD-1 Inhibitor | II | Lung Neoplasms | NCT03308942 |
| Talazoparib | Pembrolizumab (anti-PD1) | I/II | Solid Tumor, Epithelial Ovarian Cancer, Fallopian Tube Cancer, Peritoneal Cancer, Triple Negative Breast Cancer, Small Cell Lung Cancer, Metastatic Breast Cancer, Malignant Melanoma, Non-Small Cell Lung Cancer, Urothelial Carcinoma | NCT04158336 |
| Olaparib | Atezolizumab (anti-PD-L1) | II | Locally Advanced Unresectable Breast Carcinoma, Metastatic Breast Carcinoma, Stage III Breast Cancer AJCC v7, Stage IIIA Breast Cancer AJCC v7, Stage IIIB Breast Cancer AJCC v7, Stage IIIC Breast Cancer AJCC v7, Stage IV Breast Cancer AJCC v6 and v7 | NCT02849496 |
| Olaparib | Durvalumab (Anti-PD-L1) | II | Endometrial Neoplasms, Uterine Neoplasms, Endometrium Cancer | NCT03951415 |
| Talazoparib | Avelumab (anti-PD-L1) | I/II | Breast Cancer | NCT03964532 |
| Olaparib | Durvalumab (Anti-PD-L1) | II | Mismatch Repair Proficient Colorectal Cancer, Pancreatic Adenocarcinoma, Leiomyosarcoma | NCT03851614 |
| Olaparib | Durvalumab (Anti-PD-L1) | II | Triple Negative Breast Cancer | NCT03167619 |
| Olaparib | Durvalumab (Anti-PD-L1) | I | Anatomic Stage IV Breast Cancer AJCC v8, Estrogen Receptor Negative, HER2/Neu Negative, Progesterone Receptor Negative, Prognostic Stage IV Breast Cancer AJCC v8, Triple-Negative Breast Carcinoma | NCT03544125 |
| Niraparib | Dostarlimab (Anti-PD-1) | II/III | Ovarian Carcinosarcoma, Endometrial Carcinosarcoma | NCT03651206 |
| Veliparib | Nivolumab (anti-PD-1) | I | Advanced Solid Neoplasm, Aggressive Non-Hodgkin Lymphoma, Recurrent Solid Neoplasm, Refractory Mantle Cell Lymphoma, T-Cell Non-Hodgkin Lymphoma, Unresectable Solid Neoplasm | NCT03061188 |
| Rucaparib | Nivolumab (anti-PD-1) | II | Epithelial Ovarian Cancer, Fallopian Tube Cancer, Primary Peritoneal Carcinoma, High Grade Serous Carcinoma, Endometrioid Adenocarcinoma | NCT03824704 |
| Talazoparib | Avelumab (anti-PD-L1) | II | Squamous Cell Carcinoma of the Head and Neck (SCCHN), Metastatic Castration Resistant Prostate Cancer (mCRPC) | NCT04052204 |
| Niraparib | Pembrolizumab (anti-PD1) | I/II | Triple Negative Breast Cancer, Ovarian Cancer, Breast Cancer, Metastatic Breast Cancer, Advanced Breast Cancer, Stage IV Breast Cancer, Fallopian Tube Cancer, Peritoneal Cancer | NCT02657889 |
| Olaparib | Durvalumab (anti-PD-L1) | II | Metastatic Triple Negative Breast Cancer, Breast Cancer, ER-Negative PR-Negative HER2-Negative Breast Cancer, ER-Negative PR-Negative HER2-Negative Breast Neoplasms, Triple-Negative Breast Cancer, Triple-Negative Breast Neoplasm | NCT03801369 |
| Olaparib | Durvalumab (anti-PD-L1) | II | Prostate Cancer | NCT03810105 |
| Rucaparib | Nivolumab (anti-PD1) | II | Small Cell Lung Cancer | NCT03958045 |
| veliparib | Nivolumab (anti-PD1) | I | Non-Small Cell Lung Cancer | NCT02944396 |
| Olaparib | Pembrolizumab (anti-PD1) | III | Prostatic Neoplasms | NCT03834519 |
| Olaparib | Durvalumab (anti-PD-L1) and Tremelimumab (anti-CTLA-4) | II | BRCA1 Gene Mutation, BRCA2 Gene Mutation, Ovarian Serous Adenocarcinoma, Recurrent Fallopian Tube Carcinoma, Recurrent Ovarian Carcinoma, Recurrent Primary Peritoneal Carcinoma | NCT02953457 |
| Olaparib | Durvalumab (anti-PD-L1) | II | Squamous Cell Carcinoma of the Head and Neck | NCT02882308 |
| Olaparib | Durvalumab (anti-PD-L1) | II | Glioma, Cholangiocarcinoma, Solid Tumor, IDH Mutation | NCT03991832 |
| Niraparib | Dostarlimab (anti-PD1) | III | Ovarian Cancer | NCT03602859 |
| Olaparib | Durvalumab (anti-PD-L1) | I | Advanced Malignant Solid Neoplasm, Metastatic Malignant Solid Neoplasm, Unresectable Malignant Solid Neoplasm | NCT03842228 |
| Niraparib | Nivolumab (anti-PD1) or Ipilimumab (anti-CTLA4) | I II | Pancreatic Adenocarcinoma | NCT03404960 |
| Niraparib | Atezolizumab (anti-PD-L1) | II | Solid Tumor | NCT04185831 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yélamos, J.; Moreno-Lama, L.; Jimeno, J.; Ali, S.O. Immunomodulatory Roles of PARP-1 and PARP-2: Impact on PARP-Centered Cancer Therapies. Cancers 2020, 12, 392. https://doi.org/10.3390/cancers12020392
Yélamos J, Moreno-Lama L, Jimeno J, Ali SO. Immunomodulatory Roles of PARP-1 and PARP-2: Impact on PARP-Centered Cancer Therapies. Cancers. 2020; 12(2):392. https://doi.org/10.3390/cancers12020392
Chicago/Turabian StyleYélamos, José, Lucia Moreno-Lama, Jaime Jimeno, and Syed O. Ali. 2020. "Immunomodulatory Roles of PARP-1 and PARP-2: Impact on PARP-Centered Cancer Therapies" Cancers 12, no. 2: 392. https://doi.org/10.3390/cancers12020392
APA StyleYélamos, J., Moreno-Lama, L., Jimeno, J., & Ali, S. O. (2020). Immunomodulatory Roles of PARP-1 and PARP-2: Impact on PARP-Centered Cancer Therapies. Cancers, 12(2), 392. https://doi.org/10.3390/cancers12020392