Checkpoint Defects Elicit a WRNIP1-Mediated Response to Counteract R-Loop-Associated Genomic Instability

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
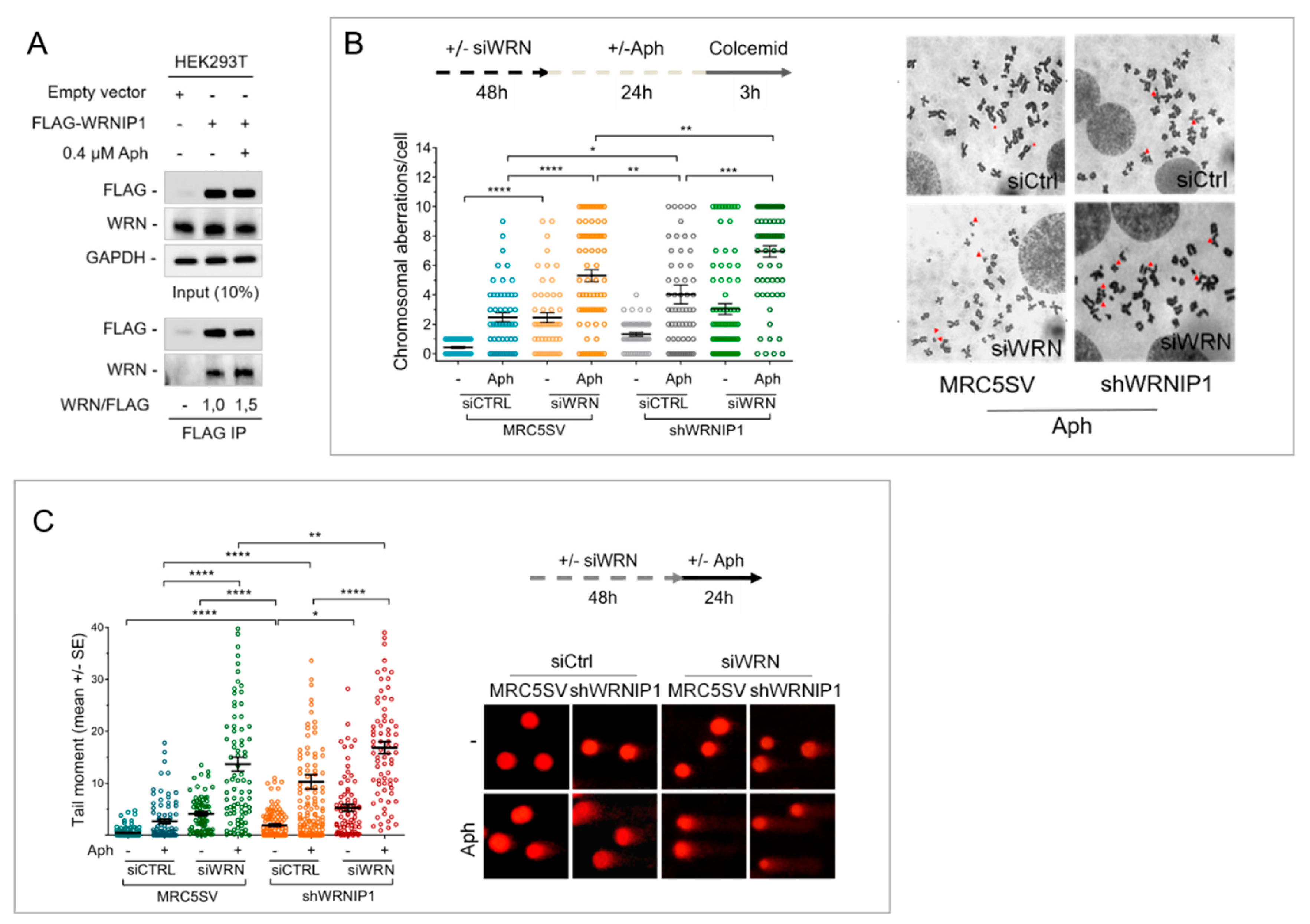
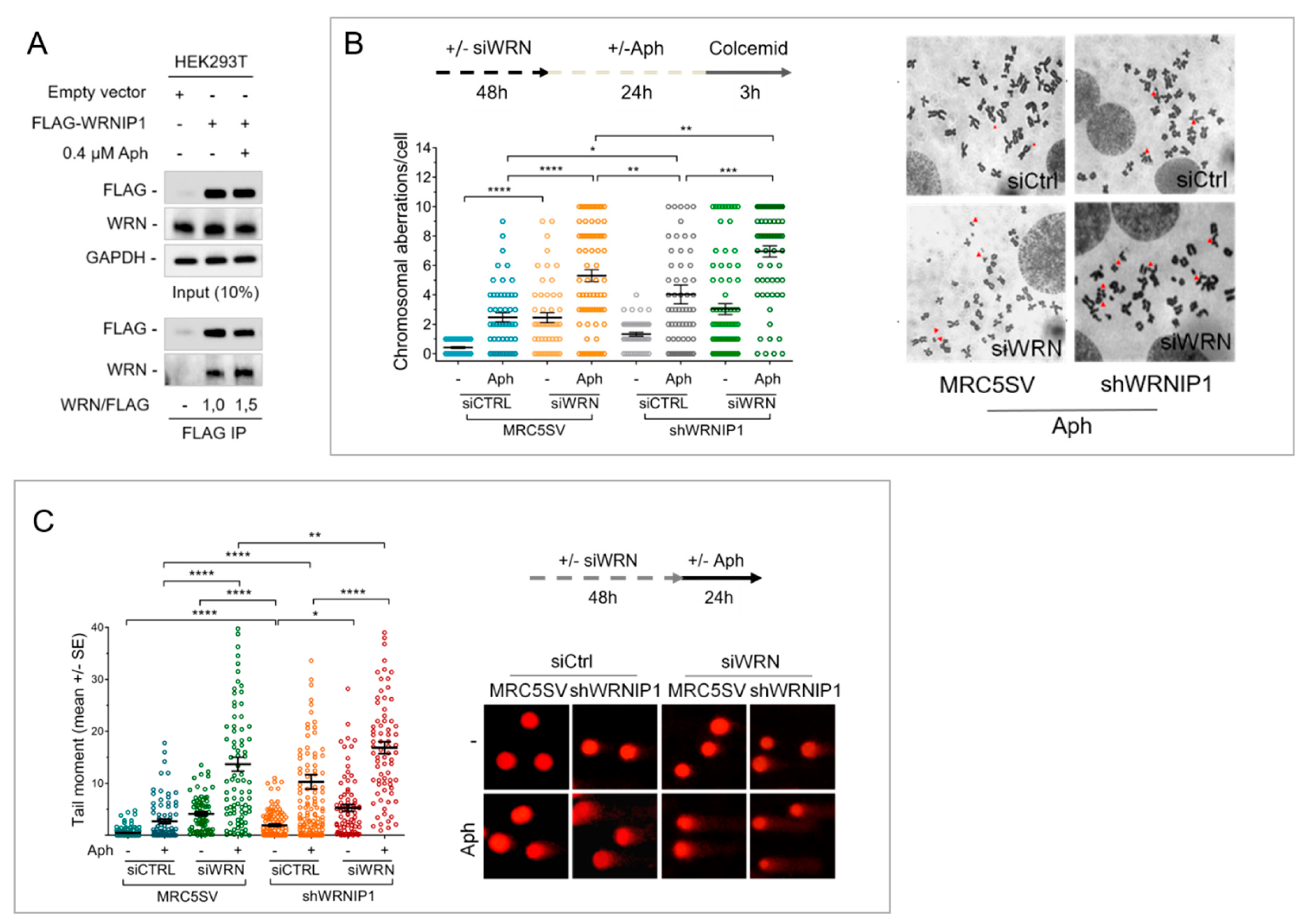
2.1. Combined Loss of WRNIP1 and WRN Results in Increased Sensitivity of Cells to MRS
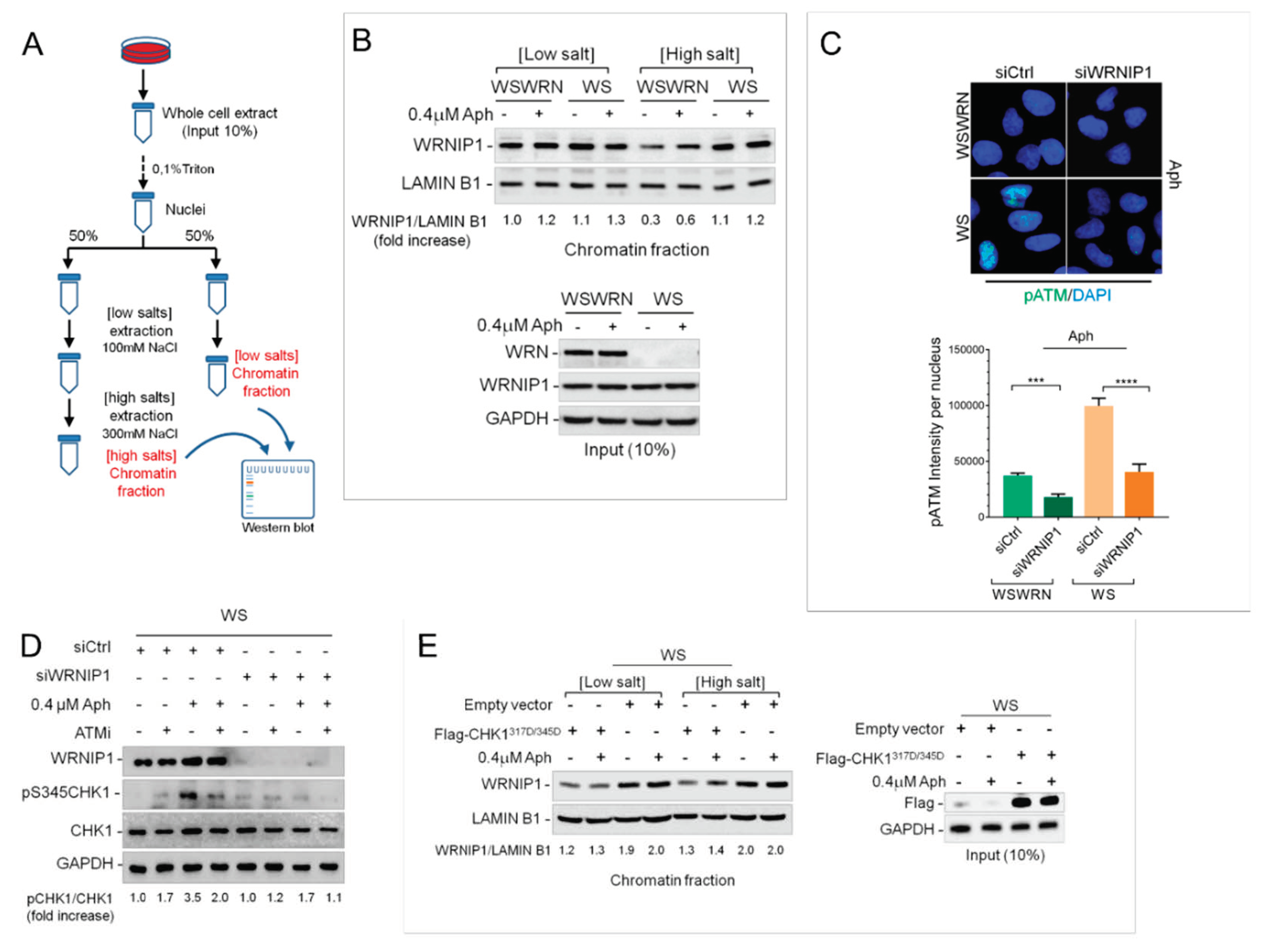
2.2. WRNIP1 is Tightly Associated with Chromatin in WS Cells
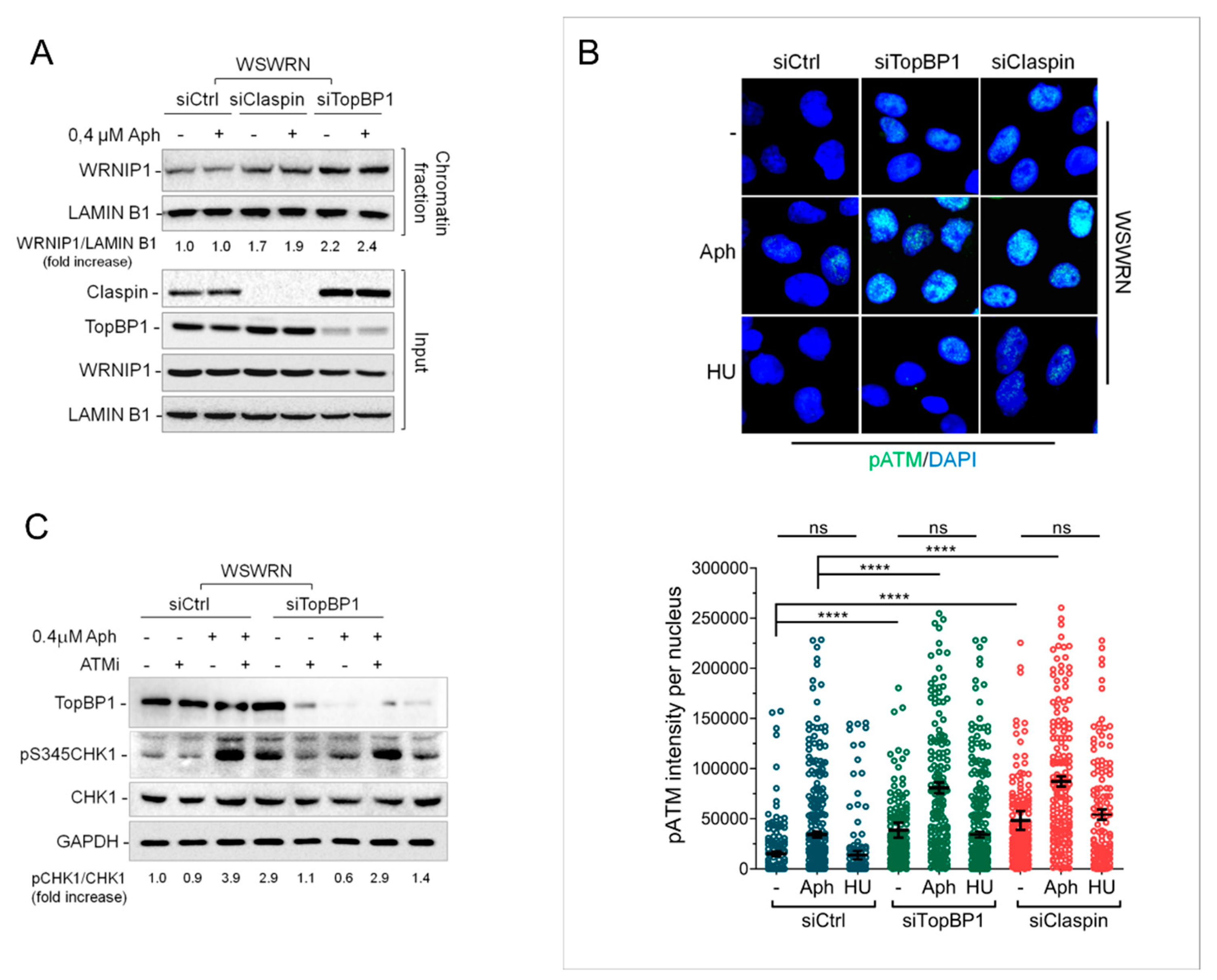
2.3. WRNIP1 Mediates ATM Signaling Activation Leading to CHK1 Phosphorylation in Response to MRS in WS Cells
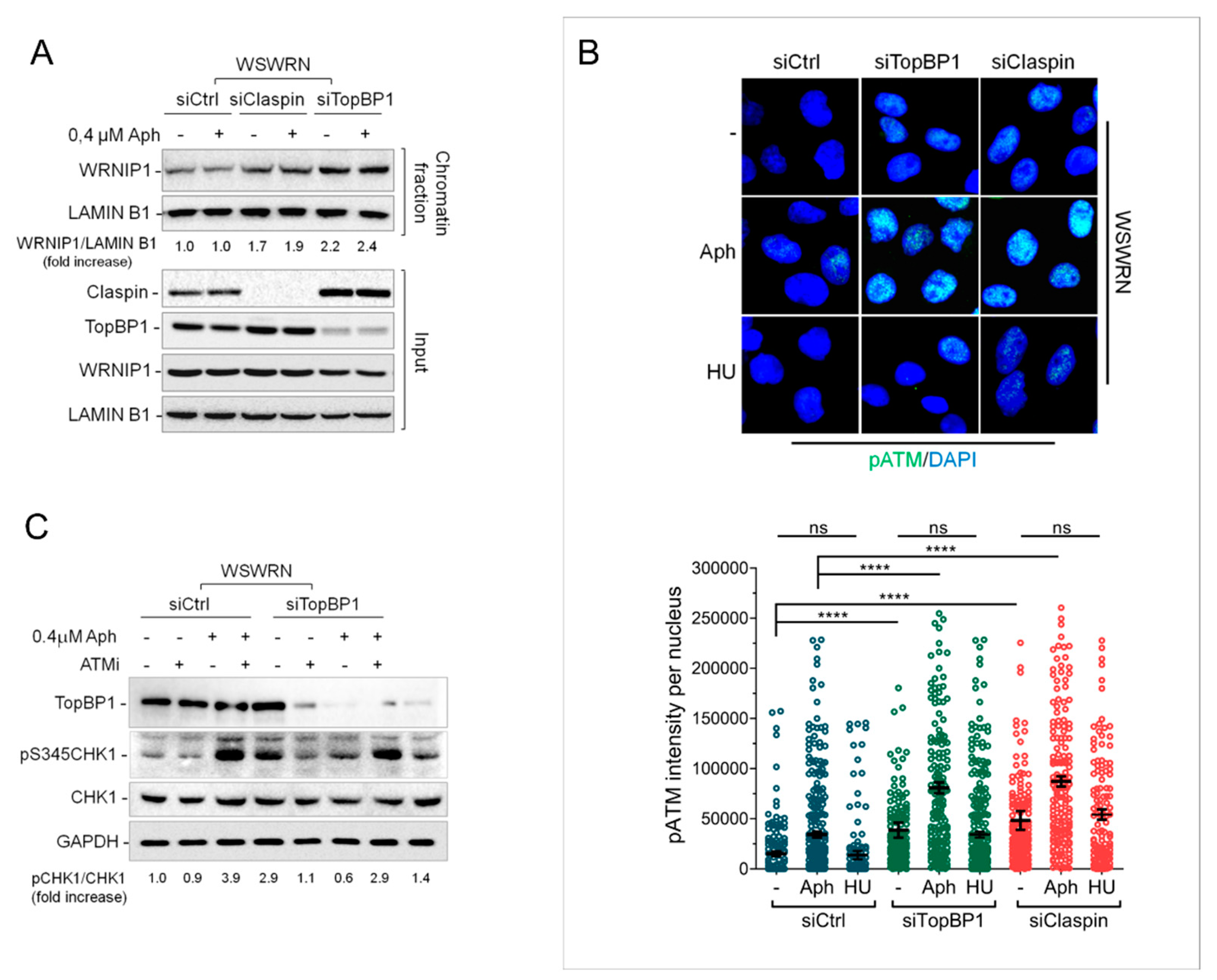
2.4. Depletion of Essential Factors for Activation of ATR-CHK1 Pathway Promotes WRNIP1 Retention in Chromatin
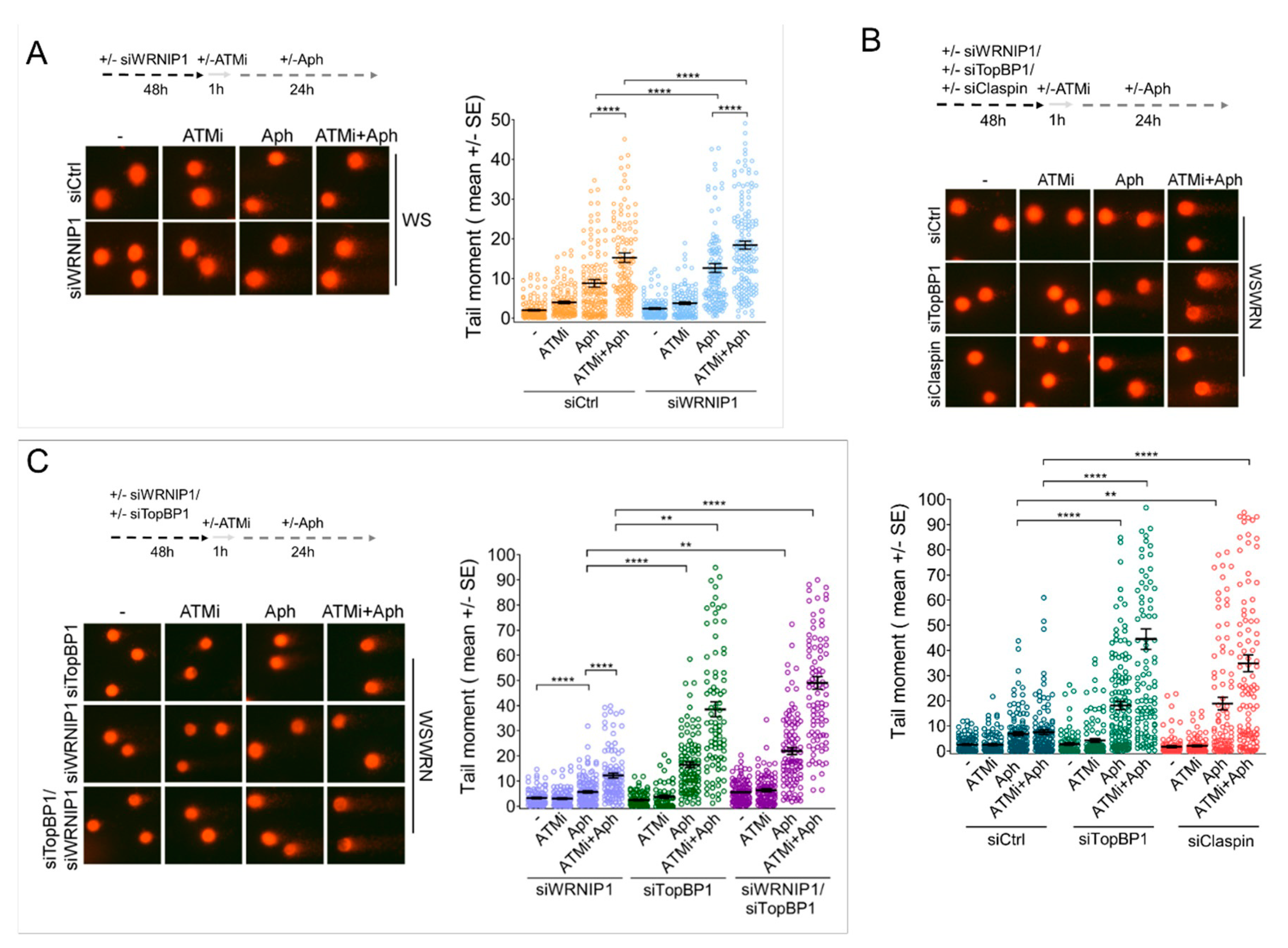
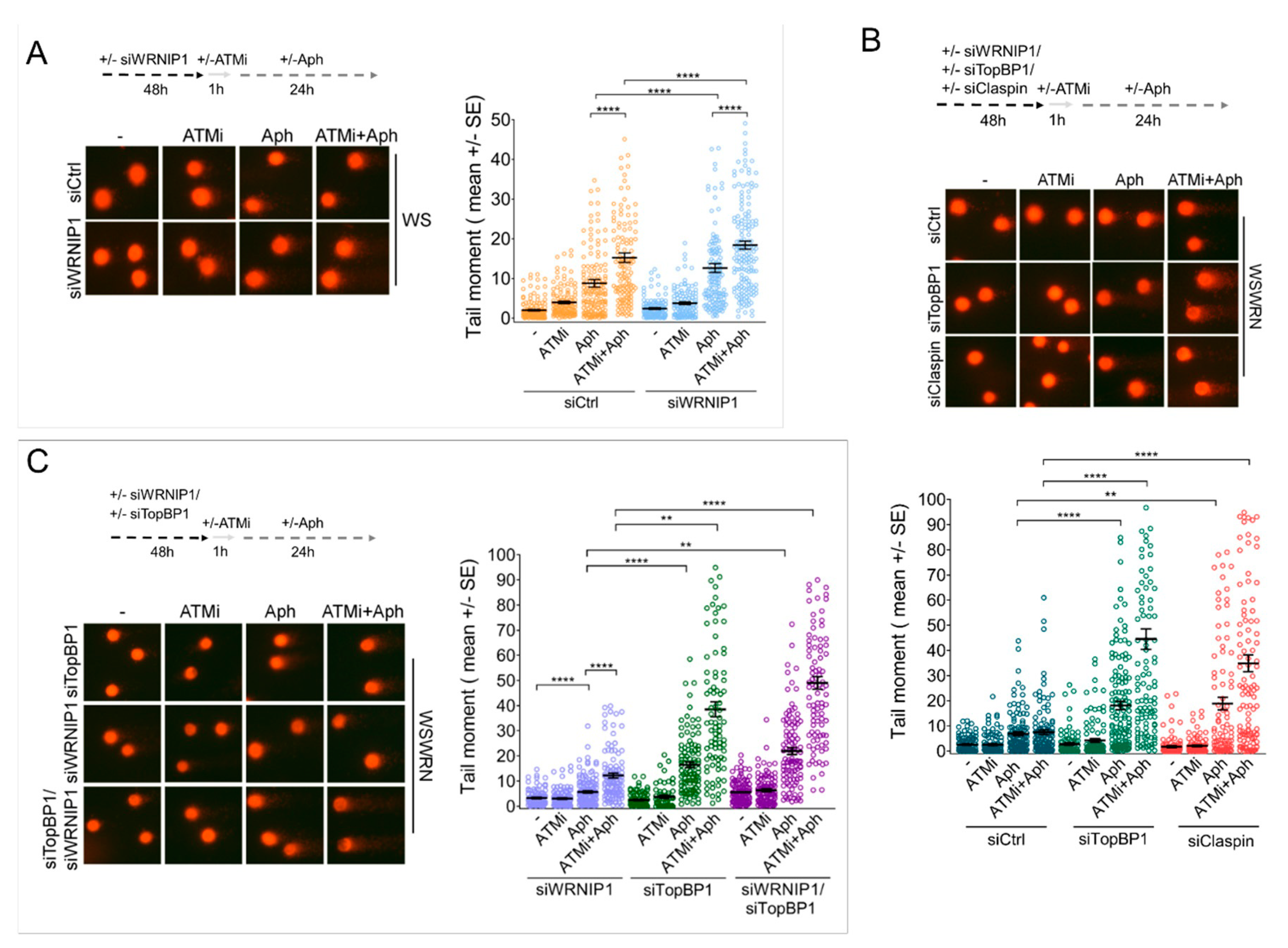
2.5. Combined Abrogation of ATM Activity and WRNIP1 Enhances DNA Damage after MRS in Cells with Defective ATR-CHK1 Signalling
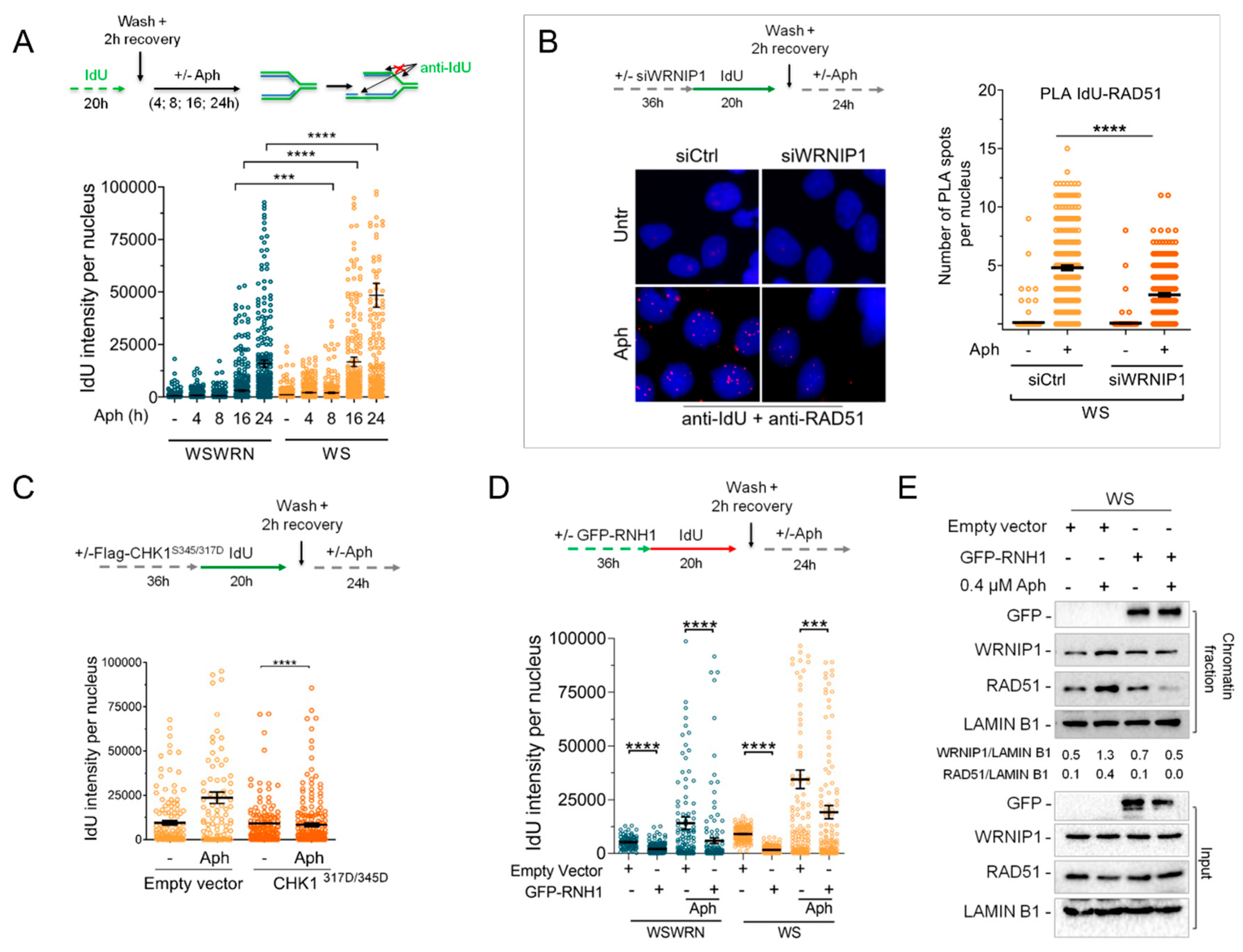
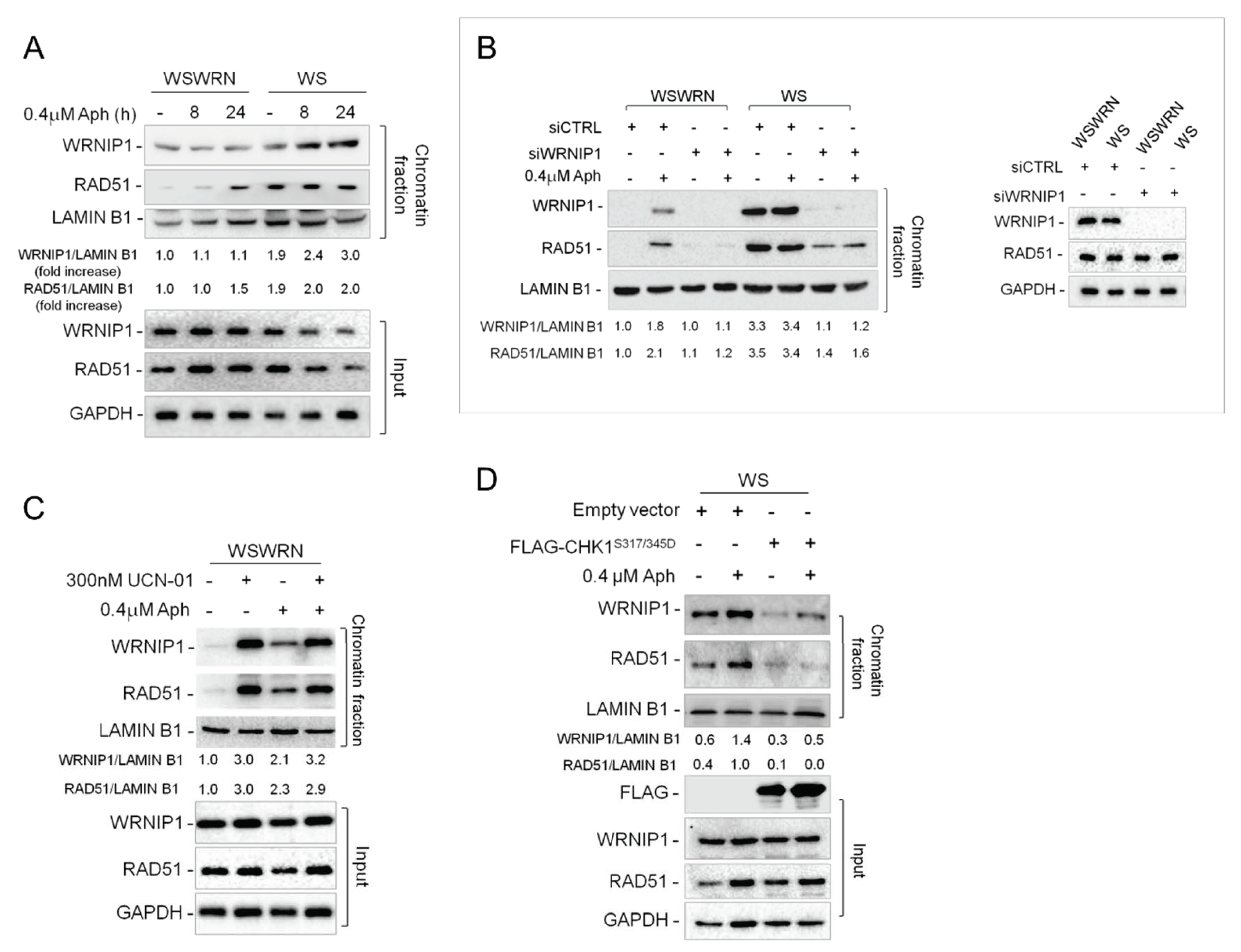
2.6. Retention of WRNIP1 in Chromatin Correlates with the Presence of RAD51 in WS Cells
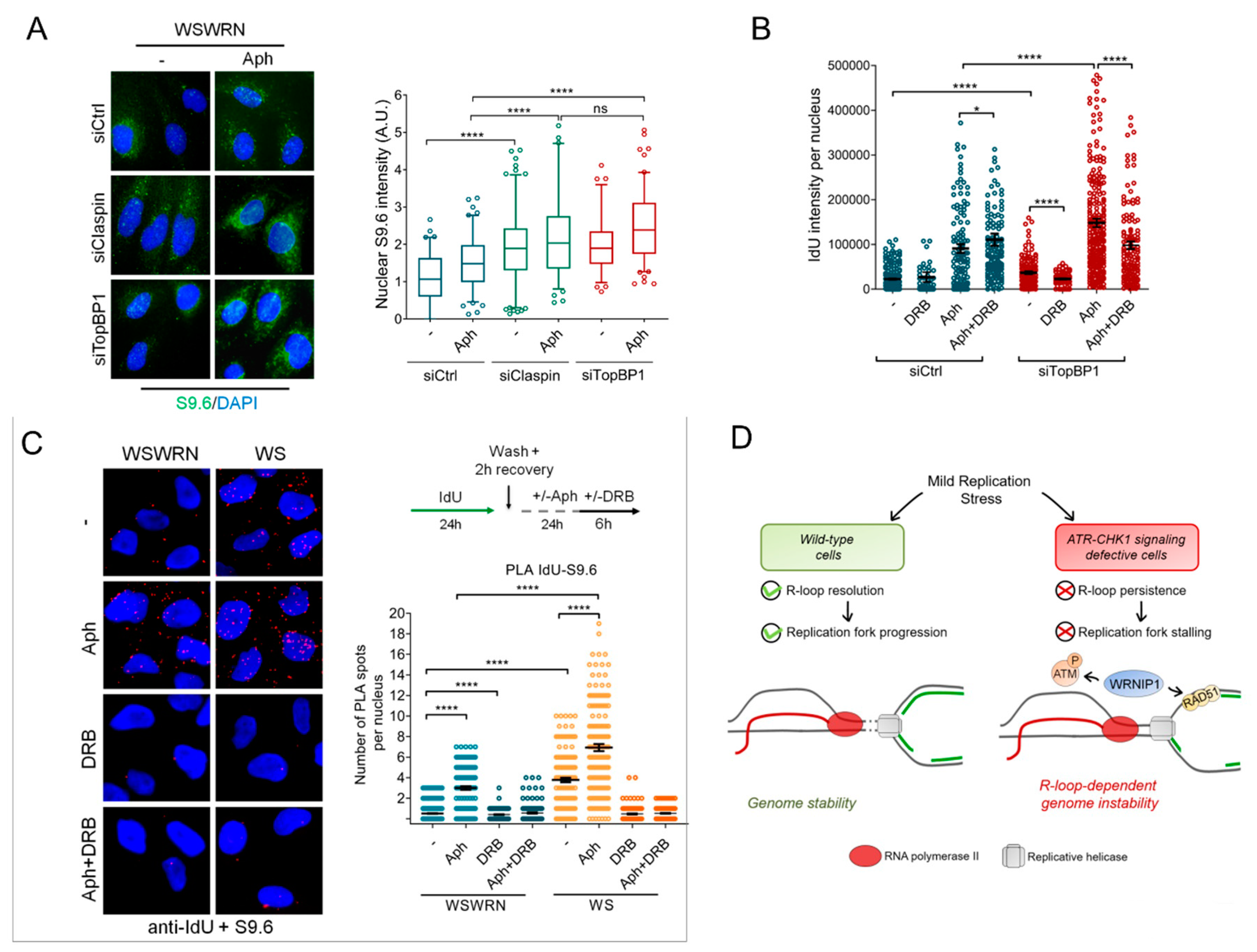
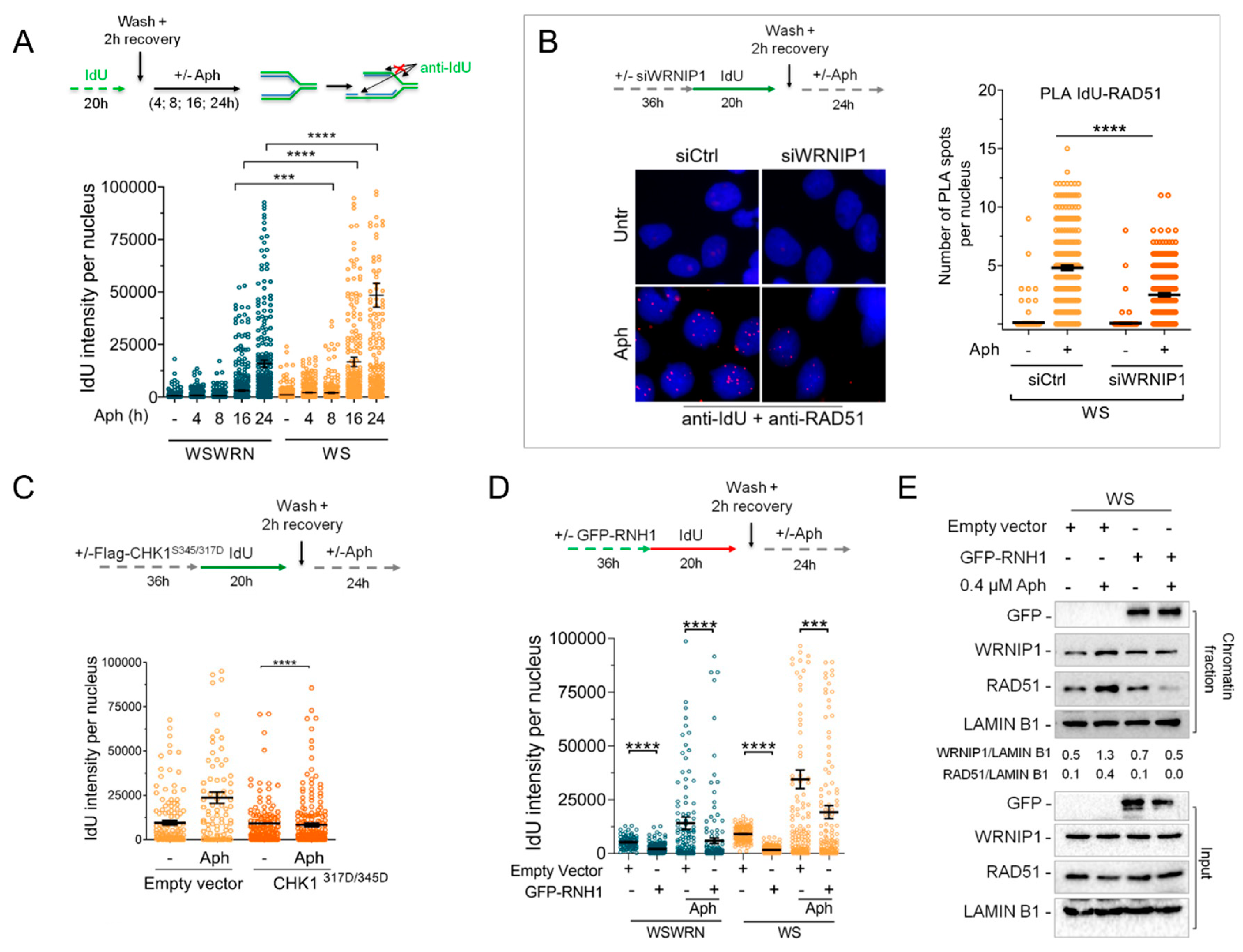
2.7. WRNIP1 Stimulates the Association of RAD51 with ssDNA in Pproximity of R-Loops upon MRS in WS Cells
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Chromatin Fractionation
4.3. Immunofluorescence
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aguilera, A.; García-Muse, T. Causes of Genome Instability. Annu. Rev. Genet. 2013, 47, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Vassiliou, L.-V.F.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; DiTullio, R.A.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef]
- Allison, D.F.; Wang, G.G. R-loops: Formation, function, and relevance to cell stress. Cell Stress 2019, 3, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaillard, H.; Aguilera, A. Transcription as a Threat to Genome Integrity. Annu. Rev. Biochem. 2016, 85, 291–317. [Google Scholar] [CrossRef] [PubMed]
- Aguilera, A.; García-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-González, B.; Aguilera, A. Transcription-mediated replication hindrance: A major driver of genome instability. Genes Dev. 2019, 33, 1008–1026. [Google Scholar] [CrossRef] [Green Version]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, V.; Barroso, S.I.; García-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef]
- Shivji, M.K.K.; Renaudin, X.; Williams, C.H.; Venkitaraman, A.R. BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep. 2018, 22, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [Green Version]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.-C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Rubio, M.L.; Pérez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi Anemia Pathway Protects Genome Integrity from R-loops. PLoS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.; Liang, F.; Teng, Y.; Chen, X.; Liu, J.; Longerich, S.; Rao, T.; Green, A.M.; Collins, N.B.; Xiong, Y.; et al. Binding of FANCI-FANCD2 Complex to RNA and R-Loops Stimulates Robust FANCD2 Monoubiquitination. Cell Rep. 2019, 26, 564–572.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban, V.; Dobrovolna, J.; Hühn, D.; Fryzelkova, J.; Bartek, J.; Janscak, P. RECQ5 helicase promotes resolution of conflicts between replication and transcription in human cells. J. Cell Biol. 2016, 214, 401–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.Y.-C.; Novoa, C.A.; Aristizabal, M.J.; Coulombe, Y.; Segovia, R.; Chaturvedi, R.; Shen, Y.; Keong, C.; Tam, A.S.; Jones, S.J.M.; et al. RECQ-like helicases Sgs1 and BLM regulate R-loop–associated genome instability. J. Cell Biol. 2017, 216, 3991–4005. [Google Scholar] [CrossRef] [Green Version]
- Tresini, M.; Warmerdam, D.O.; Kolovos, P.; Snijder, L.; Vrouwe, M.G.; Demmers, J.A.A.; Van Ijcken, W.F.J.; Grosveld, F.G.; Medema, R.H.; Hoeijmakers, J.H.J.; et al. The core spliceosome as target and effector of non-canonical ATM signalling. Nature 2015, 523, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Barroso, S.; Herrera-Moyano, E.; Muñoz, S.; García-Rubio, M.; Gómez-González, B.; Aguilera, A. The DNA damage response acts as a safeguard against harmful DNA–RNA hybrids of different origins. EMBO Rep. 2019, 20. [Google Scholar] [CrossRef]
- Marabitti, V.; Lillo, G.; Malacaria, E.; Palermo, V.; Sanchez, M.; Pichierri, P.; Franchitto, A. ATM pathway activation limits R-loop-associated genomic instability in Werner syndrome cells. Nucleic Acids Res. 2019, 47, 3485–3502. [Google Scholar] [CrossRef]
- Bayersdorf, R.; Schumacher, B. Recent advances in understanding the mechanisms determining longevity [version 1; peer review: 3 approved]. F1000Research 2019, 8, 1–12. [Google Scholar] [CrossRef]
- Lauper, J.M.; Krause, A.; Vaughan, T.L.; Monnat, R.J. Spectrum and Risk of Neoplasia in Werner Syndrome: A Systematic Review. PLoS ONE 2013, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Pichierri, P.; Ammazzalorso, F.; Bignami, M.; Franchitto, A. The Werner syndrome protein: Linking the replication checkpoint response to genome stability. Aging 2011, 3, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, M.L.; Ghosh, A.K.; Bohr, V. A Roles of Werner syndrome protein in protection of genome integrity. DNA Repair 2010, 9, 331–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basile, G.; Leuzzi, G.; Pichierri, P.; Franchitto, A. Checkpoint-dependent and independent roles of the Werner syndrome protein in preserving genome integrity in response to mild replication stress. Nucleic Acids Res. 2014, 42, 12628–12639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawabe, Y.; Branzei, D.; Hayashi, T.; Suzuki, H.; Masuko, T.; Onoda, F.; Heo, S.J.; Ikeda, H.; Shimamoto, A.; Furuichi, Y.; et al. A novel protein interacts with the Werner’s syndrome gene product physically and functionally. J. Biol. Chem. 2001, 276, 20364–20369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawabe, Y.; Seki, M.; Yoshimura, A.; Nishino, K.; Hayashi, T.; Takeuchi, T.; Iguchi, S.; Kusa, Y.; Ohtsuki, M.; Tsuyama, T.; et al. Analyses of the interaction of WRNIP1 with Werner syndrome protein (WRN) in vitro and in the cell. DNA Repair 2006, 5, 816–828. [Google Scholar] [CrossRef] [PubMed]
- Hishida, T.; Ohno, T.; Iwasaki, H.; Shinagawa, H. Saccharomyces cerevisiae MGS1 is essential in strains deficient in the RAD6-dependent DNA damage tolerance pathway. EMBO J. 2002, 21, 2019–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branzei, D.; Seki, M.; Onoda, F.; Yagi, H.; Kawabe, Y.; Enomoto, T. Characterization of the slow-growth phenotype of S. cerevisiae whip/mgs1 sgs1 double deletion mutants. DNA Repair 2002, 1, 671–682. [Google Scholar] [CrossRef]
- Yoshimura, A.; Seki, M.; Kanamori, M.; Tateishi, S.; Tsurimoto, T.; Tada, S.; Enomoto, T. Physical and functional interaction between WRNIP1 and RAD18. Genes Genet. Syst. 2009, 84, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Leuzzi, G.; Marabitti, V.; Pichierri, P.; Franchitto, A. WRNIP 1 protects stalled forks from degradation and promotes fork restart after replication stress. EMBO J. 2016, 35, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kanu, N.; Zhang, T.; Burrell, R.A.; Chakraborty, A.; Cronshaw, J.; DaCosta, C.; Grönroos, E.; Pemberton, H.N.; Anderton, E.; Gonzalez, L.; et al. RAD18, WRNIP1 and ATMIN promote ATM signalling in response to replication stress. Oncogene 2016, 35, 4009–4019. [Google Scholar] [CrossRef] [Green Version]
- Pirzio, L.M.; Pichierri, P.; Bignami, M.; Franchitto, A. Werner syndrome helicase activity is essential in maintaining fragile site stability. J. Cell Biol. 2008, 180, 305–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Gatei, M.; Sloper, K.; Sörensen, C.; Syljuäsen, R.; Falck, J.; Hobson, K.; Savage, K.; Lukas, J.; Zhou, B.-B.; Bartek, J.; et al. Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent Phosphorylation of Chk1 on Ser-317 in Response to Ionizing Radiation. J. Biol. Chem. 2003, 278, 14806–14811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, Z.; Helen, P.-W. ATR-Mediated Checkpoint Pathways Regulate Phosphorylation and Activation of Human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [Google Scholar]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chini, C.C.S.; Chen, J. Human claspin is required for replication checkpoint control. J. Biol. Chem. 2003, 278, 30057–30062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumagai, A.; Kim, S.-M.; Dunphy, W.G. Claspin and the activated form of ATR-ATRIP collaborate in the activation of Chk1. J. Biol. Chem. 2004, 279, 49599–49608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malacaria, E.; Pugliese, G.M.; Honda, M.; Marabitti, V.; Aiello, F.A.; Spies, M.; Franchitto, A.; Pichierri, P. Rad52 prevents excessive replication fork reversal and protects from nascent strand degradation. Nat. Commun. 2019, 10, 1412. [Google Scholar] [CrossRef]
- Iannascoli, C.; Palermo, V.; Murfuni, I.; Franchitto, A.; Pichierri, P. The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1-dependent degradation. Nucleic Acids Res. 2015, 43, 9788–9803. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.D.; Yadav, T.; Giri, S.; Saez, B.; Graubert, T.A.; Zou, L. Functions of Replication Protein A as a Sensor of R Loops and a Regulator of RNaseH1. Mol. Cell 2017, 65, 832–847. [Google Scholar] [CrossRef] [Green Version]
- Salas-Armenteros, I.; Pérez-Calero, C.; Bayona-Feliu, A.; Tumini, E.; Luna, R.; Aguilera, A. Human THO–Sin3A interaction reveals new mechanisms to prevent R-loops that cause genome instability. EMBO J. 2017, 36, 3532–3547. [Google Scholar] [CrossRef] [PubMed]
- García-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Matos, D.A.; Zhang, J.-M.; Ouyang, J.; Nguyen, H.D.; Genois, M.-M.; Zou, L. ATR Protects the Genome against R Loops through a MUS81-Triggered Feedback Loop. Mol. Cell 2019. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 2017, 170, 774–786.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellinger, R.E.; Prado, F.; Aguilera, A. Replication Fork Progression Is Impaired by Transcription in Hyperrecombinant Yeast Cells Lacking a Functional THO Complex. Mol. Cell. Biol. 2006, 26, 3327–3334. [Google Scholar] [CrossRef] [Green Version]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef] [Green Version]
- Tuduri, S.; Crabbé, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef]
- Schmidt, L.; Wiedner, M.; Velimezi, G.; Prochazkova, J.; Owusu, M.; Bauer, S.; Loizou, J.I. ATMIN is required for the ATM-mediated signaling and recruitment of 53BP1 to DNA damage sites upon replication stress. DNA Repair 2014, 24, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Pichierri, P.; Franchitto, A.; Mosesso, P.; Palitti, F.; Molecolare, C. Werner’s Syndrome Protein Is Required for Correct Recovery after Replication Arrest and DNA Damage Induced in S-Phase of Cell Cycle. Mol. Biol. Cell 2001, 12, 2412–2421. [Google Scholar] [CrossRef] [Green Version]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Doty, T.; Gibson, B.; Heyer, W.-D. Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat. Struct. Mol. Biol. 2010, 17, 1260–1262. [Google Scholar] [CrossRef] [PubMed]
- Stirling, P.C.; Hieter, P. Canonical DNA Repair Pathways Influence R-Loop-Driven Genome Instability. J. Mol. Biol. 2017, 429, 3132–3138. [Google Scholar] [CrossRef] [PubMed]
- Taglialatela, A.; Alvarez, S.; Leuzzi, G.; Sannino, V.; Ranjha, L.; Huang, J.-W.; Madubata, C.; Anand, R.; Levy, B.; Rabadan, R.; et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol. Cell 2017, 68, 414–430.e8. [Google Scholar] [CrossRef] [Green Version]
- Sollier, J.; Stork, C.T.; García-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-Coupled Nucleotide Excision Repair Factors Promote R-Loop-Induced Genome Instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Chappidi, N.; Nascakova, Z.; Boleslavska, B.; Zellweger, R.; Isik, E.; Andrs, M.; Menon, S.; Dobrovolna, J.; Balbo Pogliano, C.; Matos, J.; et al. Fork Cleavage-Religation Cycle and Active Transcription Mediate Replication Restart after Fork Stalling at Co-transcriptional R-Loops. Mol. Cell 2019. [Google Scholar] [CrossRef]
- Murfuni, I.; De Santis, A.; Federico, M.; Bignami, M.; Pichierri, P.; Franchitto, A. Perturbed replication induced genome wide or at common fragile sites is differently managed in the absence of WRN. Carcinogenesis 2012, 33, 1655–1663. [Google Scholar] [CrossRef] [Green Version]
- Wells, J.P.; White, J.; Stirling, P.C. R Loops and Their Composite Cancer Connections. Trends in Cancer 2019, 5, 619–631. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marabitti, V.; Lillo, G.; Malacaria, E.; Palermo, V.; Pichierri, P.; Franchitto, A. Checkpoint Defects Elicit a WRNIP1-Mediated Response to Counteract R-Loop-Associated Genomic Instability. Cancers 2020, 12, 389. https://doi.org/10.3390/cancers12020389
Marabitti V, Lillo G, Malacaria E, Palermo V, Pichierri P, Franchitto A. Checkpoint Defects Elicit a WRNIP1-Mediated Response to Counteract R-Loop-Associated Genomic Instability. Cancers. 2020; 12(2):389. https://doi.org/10.3390/cancers12020389
Chicago/Turabian StyleMarabitti, Veronica, Giorgia Lillo, Eva Malacaria, Valentina Palermo, Pietro Pichierri, and Annapaola Franchitto. 2020. "Checkpoint Defects Elicit a WRNIP1-Mediated Response to Counteract R-Loop-Associated Genomic Instability" Cancers 12, no. 2: 389. https://doi.org/10.3390/cancers12020389
APA StyleMarabitti, V., Lillo, G., Malacaria, E., Palermo, V., Pichierri, P., & Franchitto, A. (2020). Checkpoint Defects Elicit a WRNIP1-Mediated Response to Counteract R-Loop-Associated Genomic Instability. Cancers, 12(2), 389. https://doi.org/10.3390/cancers12020389